
Bacteriophage Technology and Modern Medicine




Abstract
:1. Brief Story of Bacteriophage as Medicines
2. The Emergence of Antimicrobial-Resistant Bacteria and Phage Therapy
3. The Rise of Phage Engineering Technologies toward Clinical Applications
3.1. Phage Engineering for a Safer Phage Product
3.2. Phage Engineering to Broaden the Host Range and Limit the Emergence of Phage-Resistant Bacteria
3.3. Phage Engineering for Stabilizing Phages in Blood Circulation
3.4. Phage Engineering for Phages That Can Be Easily Commercialized
4. Development of Gene-Specific Antimicrobials
4.1. The Use of a Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)-Cas System as a Gene-Specific Antimicrobial
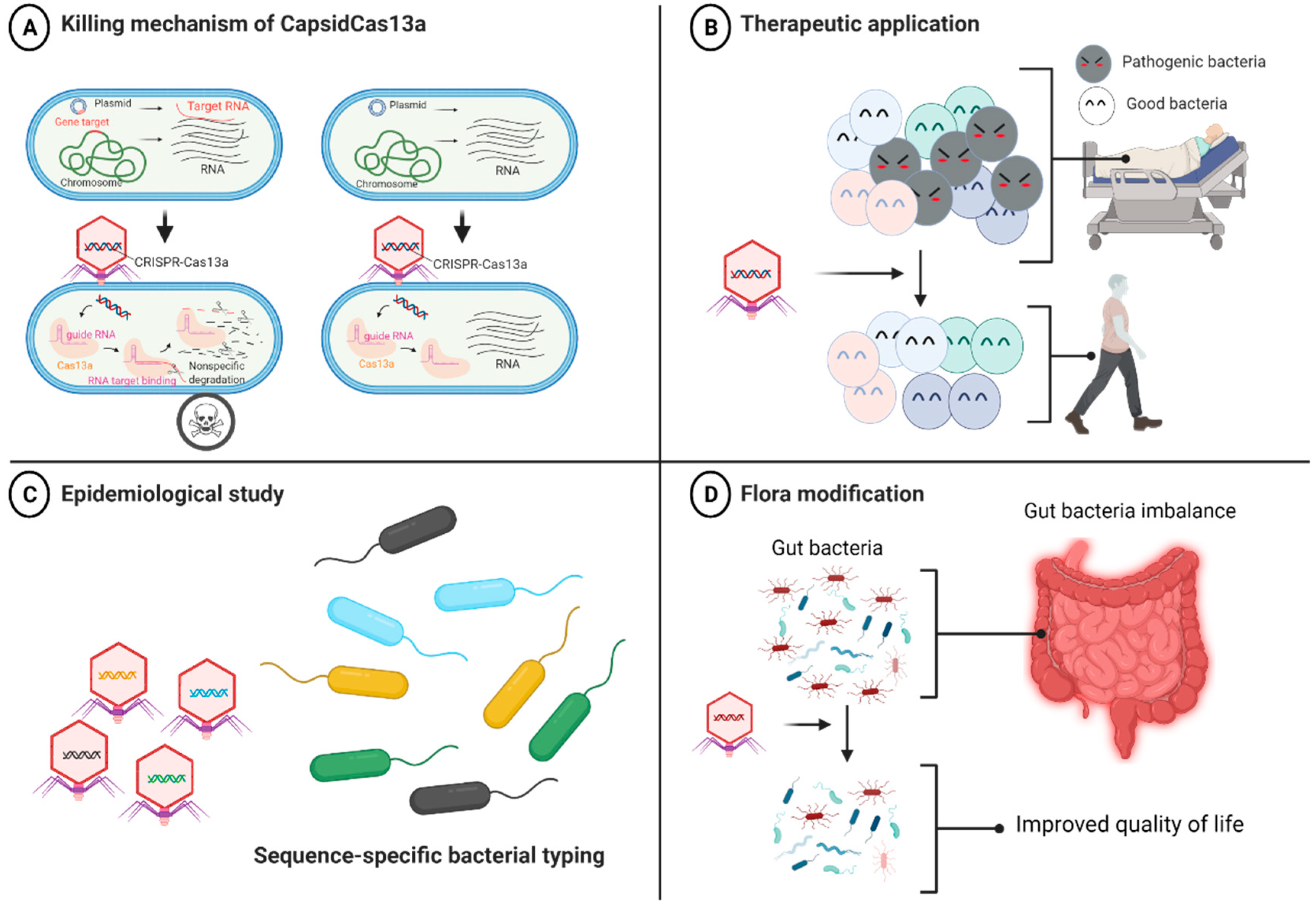
4.2. CRISPR-Cas13a-Based Antibacterial Nucleocapsid
5. Other Applications of Phages
6. Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clokie, M.R.J.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in Nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Adhya, S.; Merril, C. The Road to Phage Therapy. Nature 2006, 443, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Samsygina, G.A.; Boni, E.G. Bacteriophages and phage therapy in pediatric practice. Pediatriia 1984, 4, 67–70. [Google Scholar]
- Duckworth, D.H. Who Discovered Bacteriophage. Bacteriol. Rev. 1976, 40, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Wittebole, X.; De Roock, S.; Opal, S.M. A Historical Overview of Bacteriophage Therapy as an Alternative to Antibiotics for the Treatment of Bacterial Pathogens. Virulence 2013, 5, 226–235. [Google Scholar] [CrossRef]
- D’Herelle, F. Studies Upon Asiatic Cholera. Yale J. Biol. Med. 1929, 1, 195–219. [Google Scholar] [PubMed]
- Babalova, E.G.; Katsitadze, K.T.; Sakvarelidze, L.A.; Imnaishvili, N.S.; Dekanosidze, N.G. O Profilakticheskom Znachenii Dizenteriĭnogo Sukhogo Bakteriofaga Preventive Value of Dried Dysentery Bacteriophage. Zh. Mikrobiol. Epidemiol. Immunobiol. 1968, 45, 143–145. [Google Scholar]
- Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S. Phage Therapy in Clinical Practice: Treatment of Human Infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Kutateladze, M. Experience of the Eliava Institute in Bacteriophage Therapy. Virol. Sin. 2015, 30, 80–81. [Google Scholar] [CrossRef] [PubMed]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails to Treat a Patient with a Disseminated Resistant Acinetobacter Baumannii Infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiología 1996, 12, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Luepke, K.H.; Mohr, J.F. The Antibiotic Pipeline: Reviving Research and Development and Speeding Drugs to Market. Expert Rev. Anti-Infect. Ther. 2017, 15, 425–433. [Google Scholar] [CrossRef]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-Resistant, Extensively Drug-Resistant and Pandrug-Resistant Bacteria: An International Expert Proposal for Interim Standard Definitions for Acquired Resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical Relevance of the ESKAPE Pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- Piddock, L.J.V. Reflecting on the Final Report of the O’Neill Review on Antimicrobial Resistance. Lancet Infect. Dis. 2016, 16, 767–768. [Google Scholar] [CrossRef]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage Cocktails and the Future of Phage Therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.P.; Azam, A.H.; Sasahara, T.; Miyanaga, K.; Tanji, Y. Characterization of Pseudomonas Lytic Phages and Their Application as a Cocktail with Antibiotics in Controlling Pseudomonas Aeruginosa. J. Biosci. Bioeng. 2020, 129, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Kilcher, S.; Studer, P.; Muessner, C.; Klumpp, J.; Loessner, M.J.; Adhya, S. Cross-Genus Rebooting of Custom-Made, Synthetic Bacteriophage Genomes in L-Form Bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Hanawa, T.; Azam, A.H.; LeBlanc, C.; Ung, P.; Matsuda, T.; Onishi, H.; Miyanaga, K.; Tanji, Y. Silviavirus Phage ɸMR003 Displays a Broad Host Range against Methicillin-Resistant Staphylococcus Aureus of Human Origin. Appl. Microbiol. Biotechnol. 2019, 103, 7751–7765. [Google Scholar] [CrossRef]
- Dunne, M.; Rupf, B.; Tala, M.; Qabrati, X.; Ernst, P.; Shen, Y.; Sumrall, E.; Heeb, L.; Plückthun, A.; Loessner, M.J.; et al. Reprogramming Bacteriophage Host Range through Structure-Guided Design of Chimeric Receptor Binding Proteins. Cell Rep. 2019, 29, 1336–1350.e4. [Google Scholar] [CrossRef] [Green Version]
- Libis, V.K.; Bernheim, A.G.; Basier, C.; Jaramillo-Riveri, S.; Deyell, M.; Aghoghogbe, I.; Atanaskovic, I.; Bencherif, A.C.; Benony, M.; Koutsoubelis, N.; et al. Silencing of Antibiotic Resistance in E. Coli with Engineered Phage Bearing Small Regulatory RNAs. ACS Synth. Biol. 2014, 3, 1003–1006. [Google Scholar] [CrossRef]
- Kiga, K.; Tan, X.E.; Ibarra-Chávez, R.; Watanabe, S.; Aiba, Y.; Sato’o, Y.; Li, F.Y.; Sasahara, T.; Cui, B.; Kawauchi, M.; et al. Development of CRISPR-Cas13a-Based Antimicrobials Capable of Sequence-Specific Killing of Target Bacteria. Nat. Commun. 2020, 11, 2934. [Google Scholar] [CrossRef]
- Ronayne, E.A.; Wan, Y.C.S.; Boudreau, B.A.; Landick, R.; Cox, M.M. P1 Ref Endonuclease: A Molecular Mechanism for Phage-Enhanced Antibiotic Lethality. PLoS Genet. 2016, 12, e1005797. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage Therapy: An Alternative to Antibiotics in the Age of Multi-Drug Resistance. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Merril, C.R.; Biswas, B.; Carlton, R.; Jensen, N.C.; Creed, G.J.; Zullo, S.; Adhya, S. Long-Circulating Bacteriophage as Antibacterial Agents. Proc. Natl. Acad. Sci. USA 1996, 93, 3188–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.P.; Cha, J.D.; Jang, E.H.; Klumpp, J.; Hagens, S.; Hardt, W.D.; Lee, K.Y.; Loessner, M.J. PEGylation of Bacteriophages Increases Blood Circulation Time and Reduces T-Helper Type 1 Immune Response. Microb. Biotechnol. 2008, 1, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischetti, V.A. Bacteriophage Endolysins: A Novel Anti-Infective to Control Gram-Positive Pathogens. Int. J. Med. Microbiol. 2010, 300, 357–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizarro-Bauerle, J.; Ando, H. Engineered Bacteriophages for Practical Applications. Biol. Pharm. Bull. 2020, 43, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ram, G.; Ross, H.F.; Novick, R.P.; Rodriguez-Pagan, I.; Jiang, D. Conversion of Staphylococcal Pathogenicity Islands to Crispr-Carrying Antibacterial Agents That Cure Infections in Mice. Nat. Biotechnol. 2018, 36, 971. [Google Scholar] [CrossRef] [PubMed]
- Garenne, D.; Noireaux, V. Cell-Free Transcription–Translation: Engineering Biology from the Nanometer to the Millimeter Scale. Curr. Opin. Biotechnol. 2019, 58, 19–27. [Google Scholar] [CrossRef]
- Oślizło, A.; Miernikiewicz, P.; Piotrowicz, A.; Owczarek, B.; Kopciuch, A.; Figura, G.; Dabrowska, K. Purification of Phage Display-Modified Bacteriophage T4 by Affinity Chromatography. BMC Biotechnol. 2011, 11, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colomer-Lluch, M.; Imamovic, L.; Jofre, J.; Muniesa, M. Bacteriophages Carrying Antibiotic Resistance Genes in Fecal Waste from Cattle, Pigs, and Poultry. Antimicrob. Agents Chemother. 2011, 55, 4908–4911. [Google Scholar] [CrossRef] [Green Version]
- Sumby, P.; Waldor, M.K. Transcription of the Toxin Genes Present within the Staphylococcal Phage ΦSa3ms Is Intimately Linked with the Phage’s Life Cycle. J. Bacteriol. 2003, 185, 6841–6851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, P.L.; Waldor, M.K. Bacteriophage Control of Bacterial Virulence. Infect. Immun. 2002, 70, 3985–3993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirnay, J.P.; De Vos, D.; Verbeken, G. Clinical Application of Bacteriophages in Europe. Microbiol. Aust. 2019, 40, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Xia, G.; Wolz, C. Phages of Staphylococcus Aureus and Their Impact on Host Evolution. Infect. Genet. Evol. 2014, 21, 593–601. [Google Scholar] [CrossRef]
- Azam, A.H.; Tanji, Y. Peculiarities of Staphylococcus Aureus Phages and Their Possible Application in Phage Therapy. Appl. Microbiol. Biotechnol. 2019, 103, 4279–4289. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and Their Genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered Bacteriophages for Treatment of a Patient with a Disseminated Drug-Resistant Mycobacterium Abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Azam, A.H.; Tanji, Y. Bacteriophage-Host Arm Race: An Update on the Mechanism of Phage Resistance in Bacteria and Revenge of the Phage with the Perspective for Phage Therapy. Appl. Microbiol. Biotechnol. 2019, 103, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, J.; Suzuki, M.; Nishifuji, K.; Kato, S.I.; Miyata, R.; Nasukawa, T.; Yamaguchi, K.; Takemura-Uchiyama, I.; Ujihara, T.; Shimakura, H.; et al. Analyses of Short-Term Antagonistic Evolution of Pseudomonas Aeruginosa Strain PAO1 and Phage KPP22 (Myoviridae Family, PB1-like Virus Genus). Appl. Environ. Microbiol. 2016, 82, 4482–4491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A.R.; Scanlan, P.D.; Morgan, A.D.; Buckling, A. Host-Parasite Coevolutionary Arms Races Give Way to Fluctuating Selection. Ecol. Lett. 2011, 14, 635–642. [Google Scholar] [CrossRef]
- Golais, F.; Hollý, J.; Vítkovská, J. Coevolution of Bacteria and Their Viruses. Folia Microbiol. 2013, 58, 177–186. [Google Scholar] [CrossRef]
- Morozova, V.V.; Vlassov, V.V.; Tikunova, N.V. Applications of Bacteriophages in the Treatment of Localized Infections in Humans. Front. Microbiol. 2018, 9, 1969. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, S.; Rashel, M.; Uchiyama, J.; Sakurai, S.; Ujihara, T.; Kuroda, M.; Ikeuchi, M.; Tani, T.; Fujieda, M.; Wakiguchi, H.; et al. Bacteriophage Therapy: A Revitalized Therapy against Bacterial Infectious Diseases. J. Infect. Chemother. 2005, 11, 211–219. [Google Scholar] [CrossRef]
- McCallin, S.; Sarker, S.A.; Sultana, S.; Oechslin, F.; Brüssow, H. Metagenome Analysis of Russian and Georgian Pyophage Cocktails and a Placebo-Controlled Safety Trial of Single Phage versus Phage Cocktail in Healthy Staphylococcus Aureus Carriers. Environ. Microbiol. 2018, 20, 3278–3293. [Google Scholar] [CrossRef]
- Azam, A.H.; Kadoi, K.; Miyanaga, K.; Usui, M.; Tamura, Y.; Cui, L.; Tanji, Y. Analysis Host-Recognition Mechanism of Staphylococcal Kayvirus ɸSA039 Reveals a Novel Strategy That Protects Staphylococcus Aureus against Infection by Staphylococcus Pseudintermedius Siphoviridae Phages. Appl. Microbiol. Biotechnol. 2019, 103, 6809–6823. [Google Scholar] [CrossRef] [PubMed]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.D.T.; de la Fuente-Nunez, C.; Lu, T.K. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell 2019, 179, 459–469.e9. [Google Scholar] [CrossRef]
- Vitiello, C.L.; Merril, C.R.; Adhya, S. An Amino Acid Substitution in a Capsid Protein Enhances Phage Survival in Mouse Circulatory System More than a 1000-Fold. Virus Res. 2005, 114, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.; Hershfield, M.S.; Fernandez-Mejia, C.; Polmar, S.H.; Scudiery, D.; Berger, M.; Sorensen, R.U. Adenosine Deaminase Deficiency with Late Onset of Recurrent Infections: Response to Treatment with Polyethylene Glycol-Modified Adenosine Deaminase. J. Pediatr. 1988, 113, 312–317. [Google Scholar] [CrossRef]
- Rajender Reddy, K.; Modi, M.W.; Pedder, S. Use of Peginterferon Alfa-2a (40 KD) (Pegasys®) for the Treatment of Hepatitis C. Adv. Drug Deliv. Rev. 2002, 54, 571–586. [Google Scholar] [CrossRef]
- Graham, M.L. Pegaspargase: A Review of Clinical Studies. Adv. Drug Deliv. Rev. 2003, 55, 1293–1302. [Google Scholar] [CrossRef]
- Mok, H.; Palmer, D.J.; Ng, P.; Barry, M.A. Evaluation of Polyethylene Glycol Modification of First-Generation and Helper-Dependent Adenoviral Vectors to Reduce Innate Immune Responses. Mol. Ther. 2005, 11, 66–79. [Google Scholar] [CrossRef]
- Croyle, M.A.; Callahan, S.M.; Auricchio, A.; Schumer, G.; Linse, K.D.; Wilson, J.M.; Brunner, L.J.; Kobinger, G.P. PEGylation of a Vesicular Stomatitis Virus G Pseudotyped Lentivirus Vector Prevents Inactivation in Serum. J. Virol. 2004, 78, 912–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, G.; Yang, R.; Fan, K.; Dong, H.; Gao, C.; Wang, S.; Yu, L.; Cheng, Z.; Lei, L. External Lysis of Escherichia Coli by a Bacteriophage Endolysin Modified with Hydrophobic Amino Acids. AMB Express 2019, 9, 106. [Google Scholar] [CrossRef] [Green Version]
- Antonova, N.P.; Vasina, D.V.; Rubalsky, E.O.; Fursov, M.V.; Savinova, A.S.; Grigoriev, I.V.; Usachev, E.V.; Shevlyagina, N.V.; Zhukhovitsky, V.G.; Balabanyan, V.U.; et al. Modulation of Endolysin LysECD7 Bactericidal Activity by Different Peptide Tag Fusion. Biomolecules 2020, 10, 440. [Google Scholar] [CrossRef] [Green Version]
- Lund, P.E.; Hunt, R.C.; Gottesman, M.M.; Kimchi-Sarfaty, C. Pseudovirions as Vehicles for the Delivery of SiRNA. Pharm. Res. 2010, 27, 400–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillol-Salom, A.; Bacarizo, J.; Alqasmi, M.; Ciges-Tomas, J.R.; Martínez-Rubio, R.; Roszak, A.W.; Cogdell, R.J.; Chen, J.; Marina, A.; Penadés, J.R. Hijacking the Hijackers: Escherichia Coli Pathogenicity Islands Redirect Helper Phage Packaging for Their Own Benefit. Mol. Cell 2019, 75, 1020–1030.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Rubio, R.; Quiles-Puchalt, N.; Martí, M.; Humphrey, S.; Ram, G.; Smyth, D.; Chen, J.; Novick, R.P.; Penadés, J.R. Phage-Inducible Islands in the Gram-Positive Cocci. ISME J. 2017, 11, 1029–1042. [Google Scholar] [CrossRef] [Green Version]
- Penadés, J.R.; Chen, J.; Quiles-Puchalt, N.; Carpena, N.; Novick, R.P. Bacteriophage-Mediated Spread of Bacterial Virulence Genes. Curr. Opin. Microbiol. 2015, 23, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Carpena, N.; Manning, K.A.; Dokland, T.; Marina, A.; Penadés, J.R. Convergent Evolution of Pathogenicity Islands in Helper Cos Phage Interference. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.K.; Collins, J.J. Dispersing Biofilms with Engineered Enzymatic Bacteriophage. Proc. Natl. Acad. Sci. USA 2007, 104, 11197–11202. [Google Scholar] [CrossRef] [Green Version]
- Born, Y.; Fieseler, L.; Thöny, V.; Leimer, N.; Duffy, B.; Loessner, M.J. Engineering of Bacteriophages Y2::DpoL1-C and Y2::LuxAB for Efficient Control and Rapid Detection of the Fire Blight Pathogen, Erwinia Amylovora. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [Green Version]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA Maturation by Trans-Encoded Small RNA and Host Factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Gomaa, A.A.; Klumpe, H.E.; Luo, M.L.; Selle, K.; Barrangou, R.; Beisel, C.L. Programmable Removal of Bacterial Strains by Use of Genome- Targeting CRISPR-Cas Systems. MBio 2014, 5, e00928-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Bacteriophage-Based Synthetic Biology for the Study of Infectious Diseases. Curr. Opin. Microbiol. 2014, 19, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-Specific Antimicrobials Using Efficiently Delivered RNA-Guided Nucleases. Nat. Biotechnol. 2014, 32, 1141–1145. [Google Scholar] [CrossRef] [Green Version]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Fischetti, V.A.; Marraffini, L.A. Exploiting CRISPR-Cas Nucleases to Produce Sequence-Specific Antimicrobials. Nat. Biotechnol. 2014, 32, 1146–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yosef, I.; Manor, M.; Kiro, R.; Qimron, U. Temperate and Lytic Bacteriophages Programmed to Sensitize and Kill Antibiotic-Resistant Bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 7267–7272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Moon, B.Y.; Park, J.W.; Thornton, J.A.; Park, Y.H.; Seo, K.S. Genetic Engineering of a Temperate Phage-Based Delivery System for CRISPR/Cas9 Antimicrobials against Staphylococcus Aureus. Sci. Rep. 2017, 7, 44929. [Google Scholar] [CrossRef] [Green Version]
- Selle, K.; Fletcher, J.R.; Tuson, H.; Schmitt, D.S.; McMillan, L.; Vridhambal, G.S.; Rivera, A.J.; Montgomery, S.A.; Fortier, L.C.; Barrangou, R.; et al. In Vivo Targeting of Clostridioides Difficile Using Phage- Delivered CRISPR-Cas3 Antimicrobials. MBio 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San Millan, A. Evolution of Plasmid-Mediated Antibiotic Resistance in the Clinical Context. Trends Microbiol. 2018, 26, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 Is a Single-Component Programmable RNA-Guided RNA-Targeting CRISPR Effector. Science 2016, 353, 6299. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Li, X.; Wang, J.; Wang, M.; Chen, P.; Yin, M.; Li, J.; Sheng, G.; Wang, Y. Two Distant Catalytic Sites Are Responsible for C2c2 RNase Activities. Cell 2017, 168, 121–134.e12. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Cui, B.; Kiga, K.; Aiba, Y.; Tan, X.E.; Sato’o, Y.; Kawauchi, M.; Boonsiri, T.; Thitiananpakorn, K.; Taki, Y.; et al. Composition and Diversity of CRISPR-Cas13a Systems in the Genus Leptotrichia. Front. Microbiol. 2019, 10, 2838. [Google Scholar] [CrossRef] [Green Version]
- Ubeda, C.; Olivarez, N.P.; Barry, P.; Wang, H.; Kong, X.; Matthews, A.; Tallent, S.M.; Christie, G.E.; Novick, R.P. Specificity of Staphylococcal Phage and SaPI DNA Packaging as Revealed by Integrase and Terminase Mutations. Mol. Microbiol. 2009, 72, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational Signature in Colorectal Cancer Caused by Genotoxic Pks + E. Coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P.; Petrenko, V.A. Phage Display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef]
- Aghebati-Maleki, L.; Bakhshinejad, B.; Baradaran, B.; Motallebnezhad, M.; Aghebati-Maleki, A.; Nickho, H.; Yousefi, M.; Majidi, J. Phage Display as a Promising Approach for Vaccine Development. J. Biomed. Sci. 2016, 23, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, L.; Yadav, R.; Kunda, N.K.; Anderson, D.; Bruckner, E.; Miller, E.K.; Basu, R.; Muttil, P.; Tumban, E. Oral Immunization with Bacteriophage MS2-L2 VLPs Protects against Oral and Genital Infection with Multiple HPV Types Associated with Head & Neck Cancers and Cervical Cancer. Antiviral Res. 2019, 166, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Shukla, G.S.; Sun, Y.J.; Pero, S.C.; Sholler, G.S.; Krag, D.N. Immunization with Tumor Neoantigens Displayed on T7 Phage Nanoparticles Elicits Plasma Antibody and Vaccine-Draining Lymph Node B Cell Responses. J. Immunol. Methods 2018, 460, 51–62. [Google Scholar] [CrossRef]
- Cui, Z.; Song, Z.; Wang, Y.; Zeng, L.; Shen, W.; Wang, Z.; Li, Q.; He, P.; Qin, J.; Guo, X. Complete Genome Sequence of Wide-Host-Range Staphylococcus Aureus Phage JD007. J. Virol. 2012, 86, 13880–13881. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.; Li, Y.; Yang, T.; Bao, Q.; Yang, M.; Mao, C. Bacteriophage-Based Biomaterials for Tissue Regeneration. Adv. Drug Deliv. Rev. 2019, 145, 73–95. [Google Scholar] [CrossRef]
- Ma, K.; Wang, D.D.; Lin, Y.; Wang, J.; Petrenko, V.; Mao, C. Synergetic Targeted Delivery of Sleeping-Beauty Transposon System to Mesenchymal Stem Cells Using LPD Nanoparticles Modified with a Phage-Displayed Targeting Peptide. Adv. Funct. Mater. 2013, 23, 1172–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bábíčková, J.; Tóthová, Ľ.; Boor, P.; Celec, P. In Vivo Phage Display—A Discovery Tool in Molecular Biomedicine. Biotechnol. Adv. 2013, 31, 1247–1259. [Google Scholar] [CrossRef]
- Teesalu, T.; Sugahara, K.N.; Ruoslahti, E. Mapping of Vascular ZIP Codes by Phage Display. Methods Enzymol. 2012, 503, 35–56. [Google Scholar] [CrossRef]
- Sanghvi, A.B.; Miller, K.P.H.; Belcher, A.M.; Schmidt, C.E. Biomaterials Functionalization Using a Novel Peptide That Selectively Binds to a Conducting Polymer. Nat. Mater. 2005, 4, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Mao, C. Identification of Microtubule-Binding Domains on Microtubule-Associated Proteins by Major Coat Phage Display Technique. Biomacromolecules 2009, 10, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cao, B.; Yang, M.; Zhu, Y.; Suh, J.; Mao, C. Identification of Novel Short BaTiO3-Binding/Nucleating Peptides for Phage-Templated in Situ Synthesis of BaTiO3 Polycrystalline Nanowires at Room Temperature. ACS Appl. Mater. Interfaces 2016, 8, 30714–30721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, P.; Zhu, J.; Mahalingam, M.; Batra, H.; Rao, V.B. Bacteriophage T4 Nanoparticles for Vaccine Delivery against Infectious Diseases. Adv. Drug Deliv. Rev. 2019, 145, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Subedi, D.; Barr, J.J. Temporal Stability and Genetic Diversity of 48-Year-Old T-Series Phages. mSystems 2021, 6, e00990-20. [Google Scholar] [CrossRef] [PubMed]
- Rajagopala, S.V.; Casjens, S.; Uetz, P. The Protein Interaction Map of Bacteriophage Lambda. BMC Microbiol. 2011, 11, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, H.-W. The Lambda—P22 Problem. Bacteriophage 2015, 5, e1017084. [Google Scholar] [CrossRef] [Green Version]
- Dearborn, A.D.; Laurinmaki, P.; Chandramouli, P.; Rodenburg, C.M.; Wang, S.; Butcher, S.J.; Dokland, T. Structure and Size Determination of Bacteriophage P2 and P4 Procapsids: Function of Size Responsiveness Mutations. J. Struct. Biol. 2012, 178, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Sandmeier, H.; Iida, S.; Arber, W. DNA Inversion Regions Min of Plasmid P15B and Cin of Bacteriophage P1: Evolution of Bacteriophage Tail Fiber Genes. J. Bacteriol. 1992, 174, 3936–3944. [Google Scholar] [CrossRef] [Green Version]
- Camacho, A.; Jiménez, F.; de la Torre, J.; Carrascosa, J.L.; Mellado, R.P.; Viñuela, E.; Salas, M.; Vásquez, C. Assembly of Bacillus Subtilis Phage Φ29: 1. Mutants in the Cistrons Coding for the Structural Proteins. Eur. J. Biochem. 1977, 73, 39–55. [Google Scholar] [CrossRef]
- Harshey, R.M. The Mu Story: How a Maverick Phage Moved the Field Forward. Mob. DNA 2012, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Gorzelnik, K.V.; Cui, Z.; Reed, C.A.; Jakana, J.; Young, R.; Zhang, J. Asymmetric Cryo-EM Structure of the Canonical Allolevivirus Qβ Reveals a Single Maturation Protein and the Genomic SsRNA in Situ. Proc. Natl. Acad. Sci. USA 2016, 113, 11519–11524. [Google Scholar] [CrossRef] [Green Version]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, J.C.; Hutchison, C.A.; Slocombe, P.M.; Smith, M. Nucleotide Sequence of Bacteriophage Φx174 DNA. Nature 1977, 265, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Fiers, W.; Contreras, R.; Duerinck, F.; Haegeman, G.; Iserentant, D.; Merregaert, J.; Min Jou, W.; Molemans, F.; Raeymaekers, A.; Van den Berghe, A.; et al. Complete Nucleotide Sequence of Bacteriophage MS2 RNA: Primary and Secondary Structure of the Replicase Gene. Nature 1976, 260, 500–507. [Google Scholar] [CrossRef]
- Yuan, Y.; Gao, M. Jumbo Bacteriophages: An Overview. Front. Microbiol. 2017, 8, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza, S.D.; Nieweglowska, E.S.; Govindarajan, S.; Leon, L.M.; Berry, J.D.; Tiwari, A.; Chaikeeratisak, V.; Pogliano, J.; Agard, D.A.; Bondy-Denomy, J. A Bacteriophage Nucleus-like Compartment Shields DNA from CRISPR Nucleases. Nature 2020, 577, 244–248. [Google Scholar] [CrossRef]
- Pausch, P.; Al-Shayeb, B.; Bisom-Rapp, E.; Tsuchida, C.A.; Li, Z.; Cress, B.F.; Knott, G.J.; Jacobsen, S.E.; Banfield, J.F.; Doudna, J.A. Crispr-Casf from Huge Phages Is a Hypercompact Genome Editor. Science 2020, 369, 333–337. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, S.; Ross, R.P.; Coffey, A. Bacteriophage and Their Lysins for Elimination of Infectious Bacteria: Review Article. FEMS Microbiol. Rev. 2009, 80, 801–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Obstacle in Phage Therapy | Conventional Approach | Synthetic Approach | |
|---|---|---|---|
| 1 | Narrow host range | Application of phage cocktail [17,18]. | Genetic manipulation of receptor-binding protein [19,20]. |
| 2 | Emergence of phage-resistant bacteria | Phage cocktail; combination of antibiotic and phage [9,21]. | Genetic manipulation of receptor-binding protein [22]; incorporation of small RNAs or CRISPR-Cas system to silence antibiotic resistance determinant [23,24] or delivery of genes encoding proteins capable of increasing bacteria susceptibility to antibiotics [25]. |
| 3 | Low stability of phage in blood circulation due to rapid clearance by reticuloendothelial system (RES) | Multiple doses of phage administration [26]. | Introduction of mutation in phage capsid protein [27]; introduction of PEG into phage particle (PEGylation) [28]. |
| 4 | Safety concern due to difficulty of standardization and the presence of many unknown genes in phage genome | Application of phage-derived endolysin [29]. | Development of well-characterized, non-propagating phages [30]; development of antimicrobial payload using a phagemid and phage-inducible chromosomal islands (PICIs) [24,31]. |
| 5 | Presence of potential hazardous genes (toxin, virulence, and antibiotic resistance genes) in phage genome | Only strictly virulent phage is recommended for therapy [9], and whole-genome analysis should be done in the first place. | Custom-made phage can be generated easily using current techniques [19,20,32]. |
| 6 | Safety concern due to low purity of phage preparation and potential toxin contamination from bacterial propagation cell | Removal of toxins by CsCl purification and ion exchange column [9] or affinity chromatography [33]. | Phage production using cell-free system, such as cell-free transcription–translation (TXTL) [32]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azam, A.H.; Tan, X.-E.; Veeranarayanan, S.; Kiga, K.; Cui, L. Bacteriophage Technology and Modern Medicine. Antibiotics 2021, 10, 999. https://doi.org/10.3390/antibiotics10080999
Azam AH, Tan X-E, Veeranarayanan S, Kiga K, Cui L. Bacteriophage Technology and Modern Medicine. Antibiotics. 2021; 10(8):999. https://doi.org/10.3390/antibiotics10080999
Chicago/Turabian StyleAzam, Aa Haeruman, Xin-Ee Tan, Srivani Veeranarayanan, Kotaro Kiga, and Longzhu Cui. 2021. "Bacteriophage Technology and Modern Medicine" Antibiotics 10, no. 8: 999. https://doi.org/10.3390/antibiotics10080999
APA StyleAzam, A. H., Tan, X. -E., Veeranarayanan, S., Kiga, K., & Cui, L. (2021). Bacteriophage Technology and Modern Medicine. Antibiotics, 10(8), 999. https://doi.org/10.3390/antibiotics10080999