1. Introduction
Staphylococcus aureus is a universal Gram positive bacteria that lives harmlessly on the skin and mucous membranes. It can cause superficial skin lesions and wound infections as well as more serious debilitating invasive diseases such as sepsis [
1]. The current principles of sepsis management are to optimise organ perfusion with intravenous fluids, support dysfunctional organ systems (e.g., ventilation) and mitigate the immediate threat of uncontrolled infection with antibiotic therapy and source control. With significant advances in sepsis recognition and supportive care, the in-hospital mortality rate has improved over time and this has resulted in a significant increase in the number of sepsis survivors. The greatest concern for those who survive sepsis is increased risk of recurrent infections. Current evidence suggests that between 30 and 42% of sepsis survivors relapse within the first 90 days after being discharged from hospital [
2,
3,
4]. Treatment failure is defined as persistence of infection following 10 days after therapy started, recurrence of infection within 60 days of discontinuation of therapy or death in a 30-day period post therapy.
The vascular endothelium is a major target of sepsis-induced events and a number of pathogens, including
S. aureus, have been shown to bind to and internalize into human vascular endothelial cells [
5,
6]. The process of internalization is mediated by the major
S. aureus cell wall proteins, fibronectin binding proteins A/B (FnbpA/B) which bind plasma fibronectin and cross link to the integrin α5β1 [
7]. Integrin engagement triggers bacterial internalization in a process driven by endothelial cell actin remodelling, focal adhesion kinase and Src family kinases [
8]. Once internalized, neither immune cells nor antibiotics can reach the bacteria, rendering them safe from host immunity and circulating anti-staphylococcal antibiotics, thus encouraging its survival and persistence. Infection recurrence occurs as a result of the re-emergence of these internalized bacteria back into the bloodstream at a later time point. Although sepsis is often treated with antibiotic cocktails composed of anti-staphylococcal compounds including flucloxacillin, teicoplanin, clindamycin and linezolid, research has shown they are unable to kill internalised bacteria since these antibiotics have poor cell penetration [
9,
10]. Therefore, relapse may occur due to the lack of access of the antibiotics to the intracellular bacteria, suggesting that bacterial invasion of endothelial cells by
S. aureus is a pathogenic mechanism used to evade antibiotic treatment and prolong infection.
The growing concern around the lack of treatment for sepsis coupled with the antimicrobial resistance crisis places an immense pressure on alleviating the mortality and morbidity associated with this pathogen. Traditional antimicrobials exhibit limitations in clinical settings, displaying minimal permeation to the infection nidus [
11,
12,
13]. However, the delivery of drugs using nanoparticles has profoundly altered the pharmaceutical landscape and can circumvent the issues associated with traditional antibiotic delivery. By fine-tuning its physiochemical properties such as size, surface charge, material composition, and drug release, nanoparticles can be optimised for specific purposes and significantly improve the efficacy of antibiotic treatment. Largely attractive aspects of nanoparticles are its small diameter (10–500 nm), high surface-to-volume ratio, capacity to encapsulate various drug compounds, sustained drug release, and controlled delivery [
14]. Whilst conventional antibiotics display poor host membrane permeability, nanoparticles can transport drugs via endocytosis which facilitates intracellular targeting. This enhances bacteria eradication and improves efficacy of the antibacterial drug. Nanoparticles can be even further customised by using various coatings including polymers, lipids, liposomes, inorganic metals, or silica which alters their biocompatibility and clearance by the reticuloendothelial system. Additionally, these surfaces can be designed with ligands to increase affinity to host cells or bacterial surfaces to encourage active targeting [
11]. Loading antibiotics into nanoparticles have displayed significant promise towards improving drug delivery and in vitro antimicrobial activity amongst notably harmful microbes, including
S. aureus,
Escherichia coli, and
Pseudomonas aeruginosa [
15,
16,
17]. Overall, coupling traditional antibiotics with drug-encapsulated nanoparticles offer clear potential for a delivery system that could be exceedingly useful in addressing the limitations with conventional antibiotic treatments.
Using an ex vivo dynamic model of human endothelial cells, we demonstrated that S. aureus was capable of binding to and re-emerging from human vascular endothelial cells via fibronectin binding proteins, thus evading immune and antibiotic attack. We also demonstrated the internalisation of S. aureus from host cells and constructed a new formulation of vancomycin-loaded nanoparticles which targeted the internalized bacterium to prevent recurrent infection. These results have profound implications for the treatment and management of recurrent infection in sepsis patients.
3. Discussion
Morbidity and mortality associated with bloodstream infections remain significantly high, particularly in patients who concomitantly develop sepsis or acute infective endocarditis. A combination therapy of vancomycin and gentamicin is appropriate for severe cases of infective endocarditis involving persistent bacteria, due to its ability to clear extracellularly bound microbes [
18]. Yet, treatment of intracellular bacteria using common antimicrobials are often ineffective. Gentamicin is unable to penetrate eukaryotic host cells. Rifampicin has demonstrated efficacy against intracellular microbes, but antibacterial resistance may render it soon unusable. Vancomycin, although remaining the initial choice for highly resistant bacteria, is poorly absorbed when administered orally or through the intestinal tract and therefore must be administered intravenously [
19]. Therefore, intracellular bacteria clearly represent a significant threat during bloodstream infections and the ability of
S. aureus to persist within host cells and later re-emerge can drastically contribute to recurrent infections experienced by some patients [
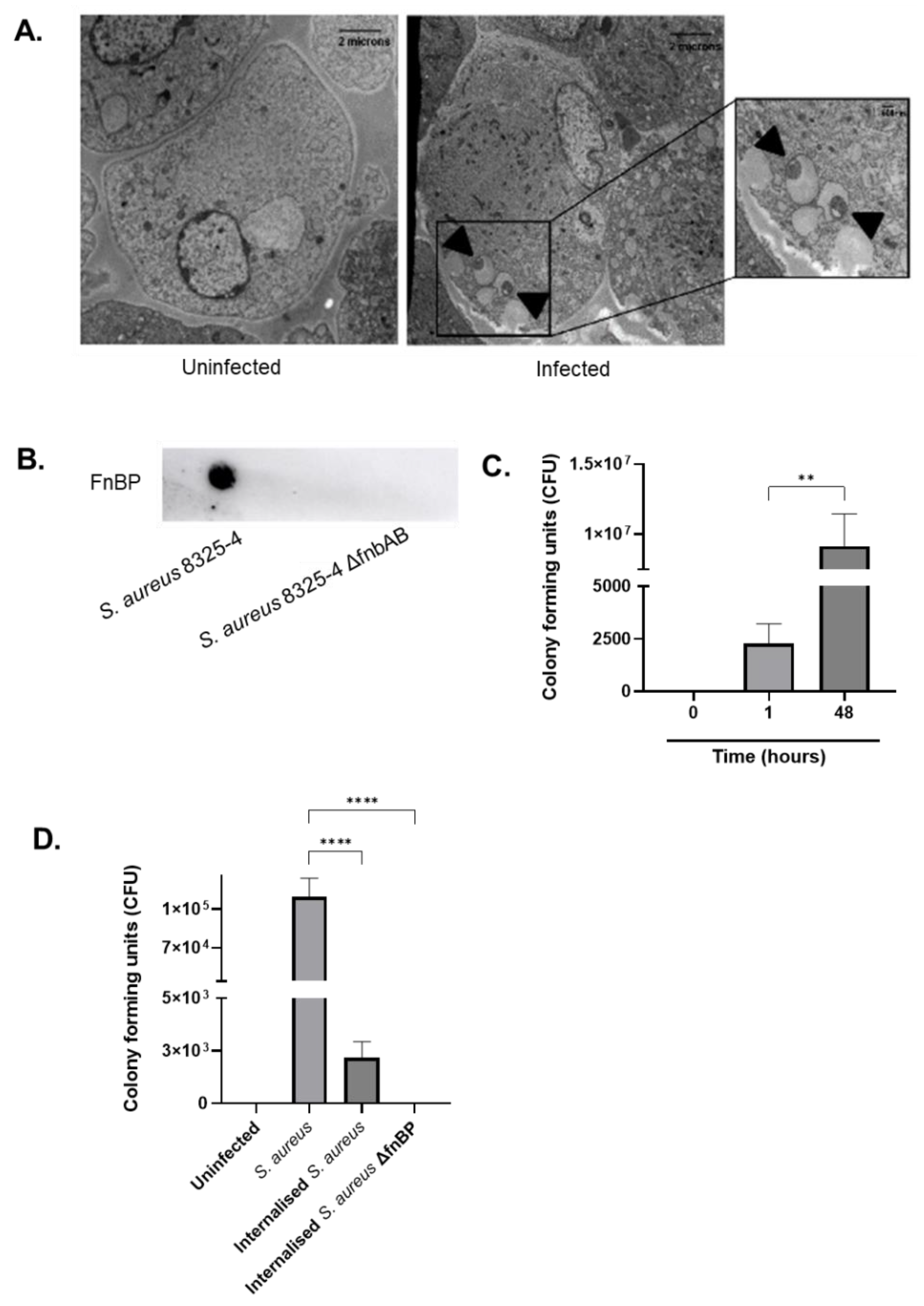
20]. Within the first hour of infection, we showed
S. aureus was capable of entering human endothelial cells through the major surface cell wall protein, FnbpA/B. This virulence factor has been demonstrated to aid in its adhesion and subsequent internalisation to host cells by binding to α5β1 integrins widely expressed across a variety of tissues and cells [
7]. This contributes to internal persistence within the host cell for over 48 h, followed by re-emergence. As demonstrated by microscopy,
S. aureus relocated to vacuoles within the endothelial cell. Rollin et al. demonstrated that
S. aureus was able to remain within endothelial cells even after 10 days post-infection, where most infected cells were incapable of restricting bacterial growth and were lysed after 48 h. However, in other cells, bacteria resided for at least 7 days without proliferating and later escaped using an unknown pathway [
21]. Synonymous with this result, we demonstrated that, in the absence of antibiotics, internalised bacteria were capable of re-emerging and remained viable, therefore rendering them infectious and capable of targeting other neighbouring cells within the monolayer. This suggests that the population of
S. aureus that was able to survive within the host cell resulted in the formation of small colony variants (SCV) [
22]. These are a slow-growing subpopulation of highly pathogenic bacteria capable of growing in externally stressful environments and contribute significantly to relapsing infections [
23]. Although the mechanism by which such bacteria are capable of evading cells remains unknown, our data suggest they could be responsible for high relapse rates observed during infective endocarditis and sepsis [
4].
Additionally, traditional antibiotics pose numerous challenges in modern medicine, including low bioavailability and lack of permeation to the infection niche, which is a significant limitation for the treatment of biofilms or intracellular microbes. Recently, new strategies have been employed to adjust to these complications, where the development of nanomedicine for targeted therapy against bacterial infections has been highlighted as a promising solution [
24]. In the present study, we proposed a new formulation of PLGA RG 503H nanoparticles loaded with vancomycin HCl to target internalised
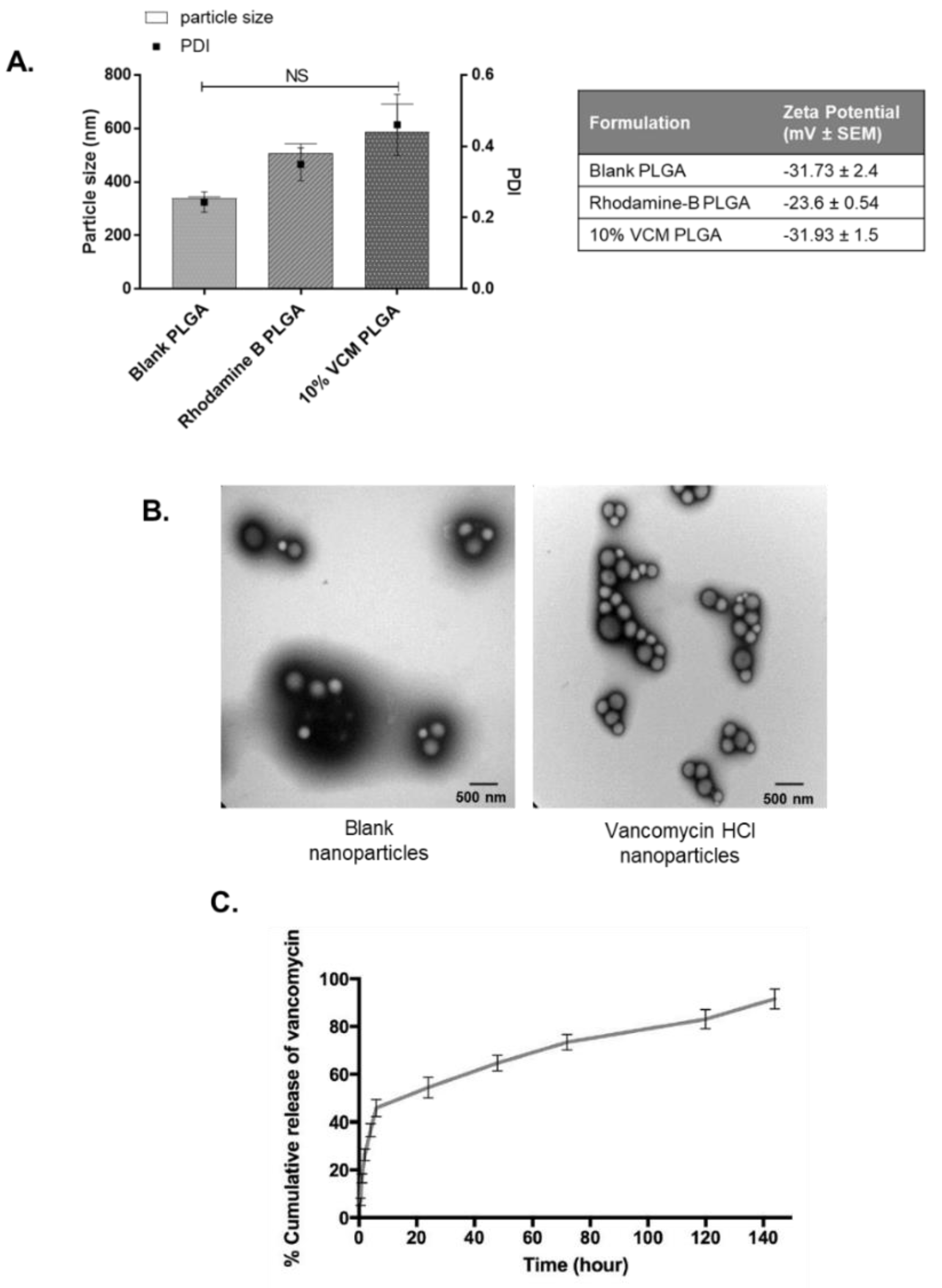
S. aureus and potentially eliminate the threat of recurrent infections. Although several delivery systems for vancomycin have been developed, only a select few have sought to identify the optimal formulation for the successful encapsulation into PLGA nanoparticles. This FDA-approved polymer is highly biocompatible and biodegradable, making it a good candidate for pharmaceutical applications such as drug delivery [
25]. We aimed to select key physio-chemical properties to construct an ideal nanoparticle in terms of drug release characteristics, cytotoxicity, and stability. The main characteristics we investigated were particle size, encapsulation efficiency, zeta potential, surface charge, and polydispersity index. These nanoparticles were fabricated using a modified double emulsion w1/o/w2 solvent evaporation method using ethyl alcohol as an organic solvent [
26]. Data have shown that using ethyl alcohol produces stable nanoparticles, whereas other solvents including acetone, chloromethane, and dichloromethane failed to do so [
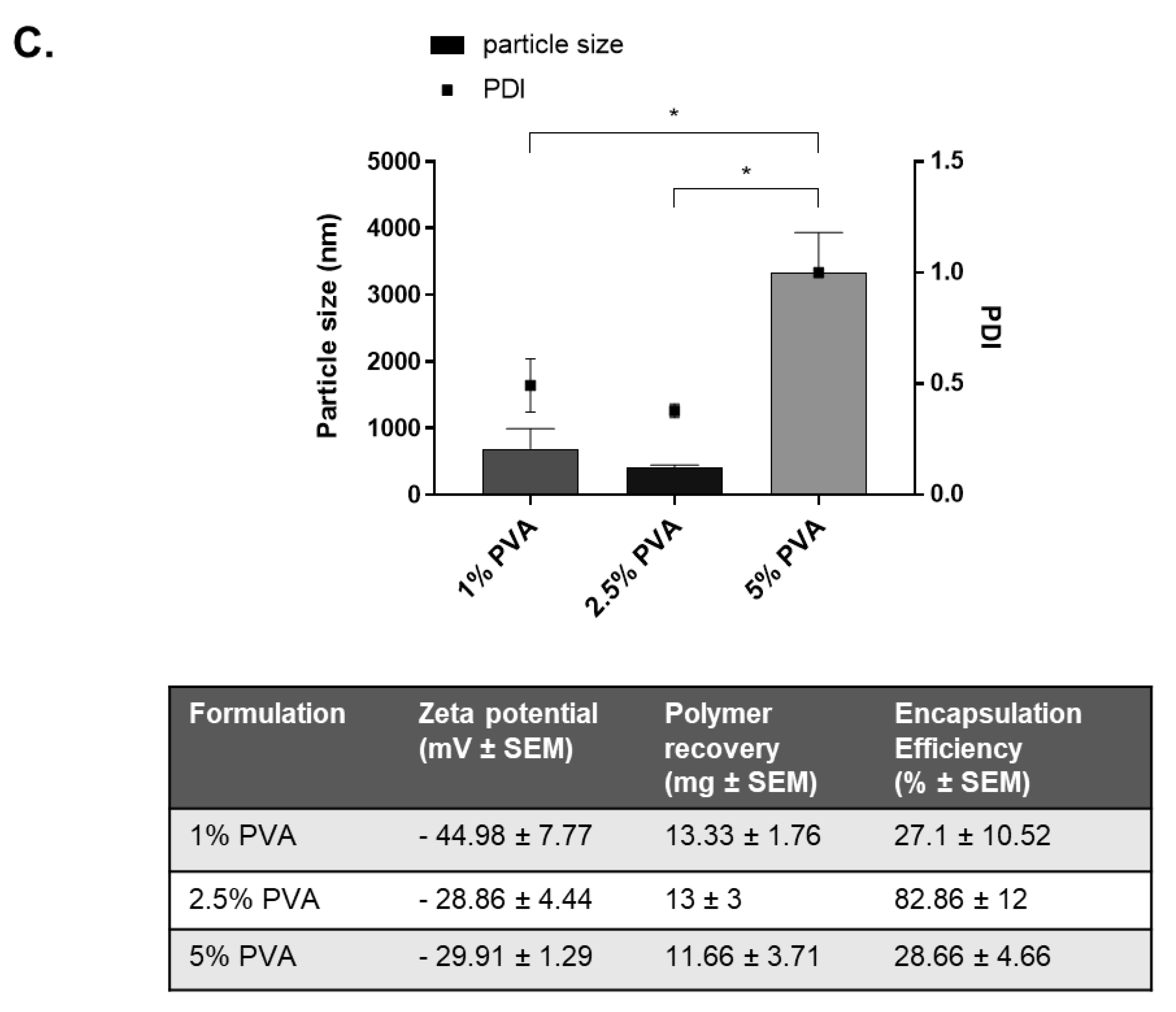
27]. Polyvinyl alcohol was chosen as a stabilising agent for the second aqueous phase due to its ability to control the size of the formulation and produce particles with high encapsulation [
28]. PVA biomaterials display high in vivo biocompatibility, low toxicity, and low biodegradability which is advantageous if the nanoparticles were administered intravenously. Particle size also is an important parameter to consider for cellular uptake, permeability, and avoiding rapid clearance. Some size limitations are known; very small nanoparticles of 10 nm are subject to renal clearance and phagocytosis, and therefore significantly reduce the accumulation of such particles at the target site [
29]. Therefore, taking these into consideration, we constructed our nanoparticles to be within the recommended range of 10–1000 nm.
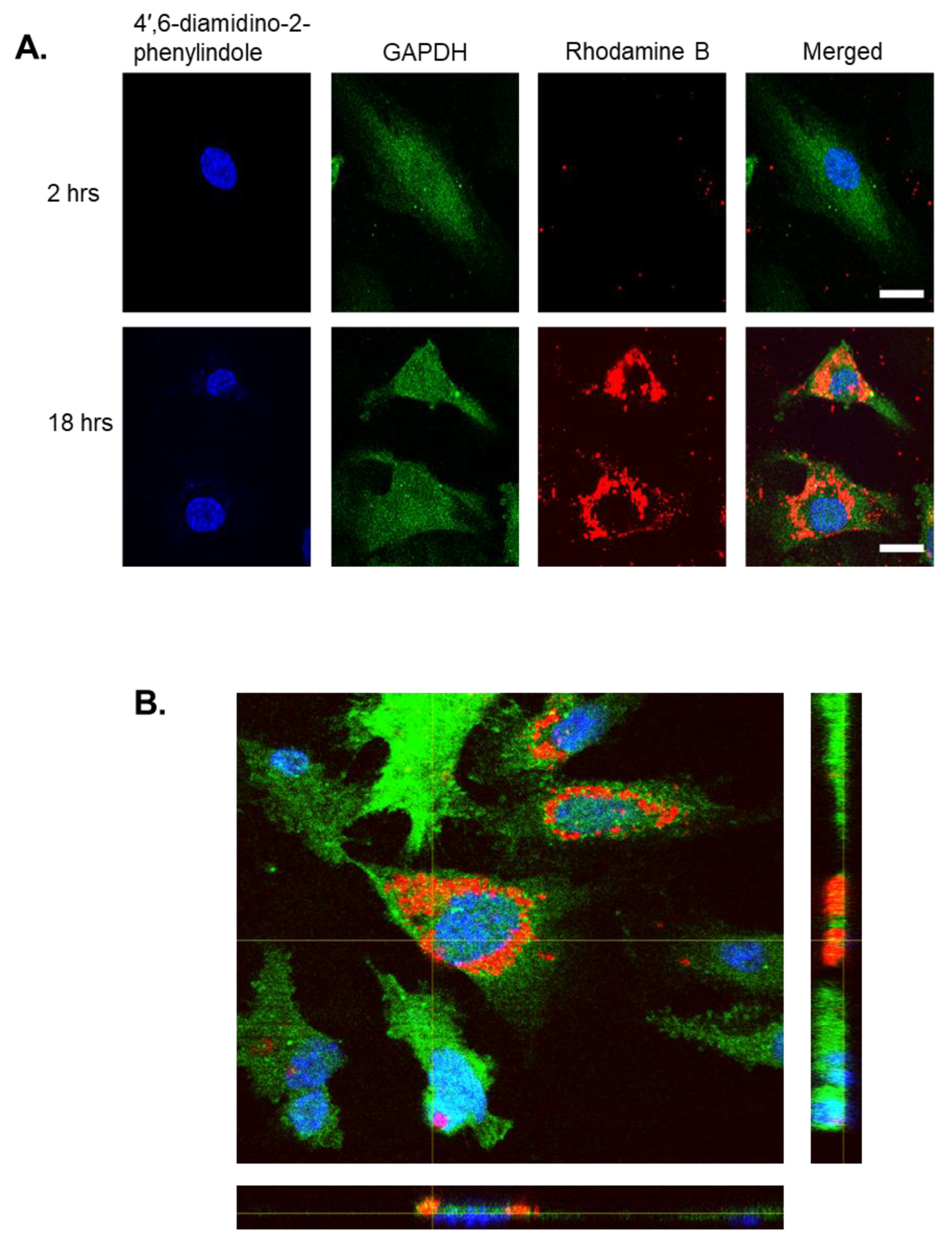
Rhodamine B, a fluorescent hydrophilic dye, was selected as a drug model since it was unable to penetrate the membrane of HEK293 cells when un-encapsulated yet, when loaded into nanoparticles, it could enter easily [
30]. After 18 h, Rhodamine B-loaded PLGA nanoparticles were able to internalise into human endothelial cells and were visualised by TEM within the cystol. In comparison to other research, it appears that the rate at which PLGA nanoparticles are internalised is dependent upon incubation time, concentration of nanoparticles, and cell line. For instance, renal proximal epithelial cells were capable of internalising nanoparticles within 30 min, whereas colon Caco-2 epithelial cells only began uptake after 24 h [
31]. The encapsulation efficiency of our vancomycin-loaded PLGA nanoparticles was optimised by altering the volume of inner aqueous phase, drug:polymer ratios, and stabiliser concentrations. Our final nanoparticles were capable of 82%
w/
w ± 12 vancomycin encapsulation, comparable to the highest value reported in the literature of 94.76% [
32].
The in vitro efficacy model evaluated the ability of the formulated vancomycin nanoparticles to treat S. aureus-infected endothelial cells. As anticipated, cells that were treated with concentrated vancomycin solution showed reduced S. aureus growth, confirming the antibiotics’ ability to target extracellularly-bound bacterium. However, cells that were additionally supplemented with the vancomycin nanoparticles displayed significantly less S. aureus growth compared to cells only treated with vancomycin solution. This demonstrates the remarkable antimicrobial capability of the 10% vancomycin PLGA RG 503H nanoparticles, able to successfully target the internalised S. aureus compared to the application of free vancomycin alone.
The prevalence of persistent S. aureus infections is an enormous concern for survivors of sepsis or bacteraemia. We have found that S. aureus is capable of internalising into human endothelial cells through its surface expressed fibronectin binding proteins. This suggests that invasion of endothelial cells may be a virulence mechanism employed by S. aureus to evade circulating antibiotic treatment and cause recurrent infections. Anti-staphylococcal compounds like vancomycin are unable to internalise into cells, and therefore cannot target intracellular bacteria. Limitations surrounding current sepsis treatments can be overcome by coupling traditional antibiotics with drug-encapsulated nanoparticles to offer a significantly improved delivery system to the infection nidus. We have formulated 10% vancomycin HCl PLGA RG 503H nanoparticles that display 91% drug release in vitro, which were capable of significantly reducing bacterial growth within infected endothelial cells. These nanoparticles are highly efficient to counteract the virulent internalisation mechanisms employed by the bacterium and display future potential to target bacterial infections caused by S. aureus.
4. Materials and Methods
4.1. Bacterial Isolates and Cell Culture Conditions
The bacterial strains used in this study are S. aureus NCTC 8325-4 and isogenic mutant lacking the fibronectin binding proteins, S. aureus 8325-4 ΔfnbpAB. Both isolates were grown in Brain Heart Isolation (BHI) broth to mid-exponential growth phase at 37 °C (optical density 600 nm = 1). Human Aortic Endothelial Cells (HAoEC) (PromoCell) were maintained in Endothelial Cell Media supplemented with basal medium, penicillin (200 U/mL) and streptomycin (200 µg/mL).
4.2. Dot Blot Test
S. aureus isolates were cultured overnight as previously described and centrifuged at 3800 rpm for 7 min. After re-suspending the pellet in PBS, a 5 µL drop was placed onto a nitrocellulose membrane and allowed to dry at room temperature. The membrane was blocked for 20 min in 10% TBST (tris-buffered saline tween) followed by addition of a S. aureus FnBP primary antibody at 1:1000. Following washing, an HRB-donkey polyclonal secondary antibody was added at 1:10,000. The membrane was analysed for protein expression using an ECL detection kit.
4.3. Internalisation and Visualisation of S. aureus in Human Endothelial Cells
Human endothelial cells were seeded at a density of 5 × 104 cells per well and infected with S. aureus 8325-4 at a Multiplicity of Infection of 1000. After 1 h, cells were washed with PBS and lysed using 0.1% Triton X-100 to measure total bacteria. To measure intracellular bacteria, cells were washed and supplemented with 100 µg/mL gentamicin HCl for 1 h. Cells were lysed as described. Total bacterial counts were determined using colony forming units (CFU/mL). Internalised bacteria were visualised using Transmission Electron Microscopy (TEM). Briefly, infected endothelial cells were fixed in 2.5% glutaraldehyde, mounted on Pioloform copper coated grids, and imaged using a Hitachi H-7650 electron microscope. To confirm the re-emergence of internalised S. aureus after 0, 1, and 48 h of infection, cells were treated with gentamicin as described. The infected cells were lysed as described with 0.1% Triton X-100, and a 50 µL cell lysate sample was plated onto BHI agar to determine and quantify bacterial growth.
4.4. Preparation of Rhodamine B Nanoparticles
To confirm that the proposed formulation method (double-emulsion (water1-in-oil-in-water2) solvent evaporation) would encapsulate a hydrophilic molecule and result in nanoparticles suitable for cellular uptake in terms of size and physicochemical characteristics, the model hydrophilic drug Rhodamine B was first encapsulated. Rhodamine B is also fluorescent and therefore its location can be tracked intracellularly. Briefly, 0.5 mg Rhodamine B was dissolved in deionised water (1.0 mL) and emulsified dropwise with sonication (70%) into the PLGA-RG-503H polymer (99.5 mg dissolved in 4 mL ethyl acetate) in an ice bath. This primary emulsion was then emulsified by sonication (70%) into 10 mL PVA 2.5% w/v in an ice-bath. This mixture was poured into 35 mL PVA 1% (w/v) under magnetic stirring at 200 rpm for 16 h at room temperature. The nanoparticles were collected by ultracentrifugation at 11,000 rpm for 15 min at 20 °C. The pellet was washed three times in 20 mL deionized water and re-centrifuged to remove any residual PVA. Samples were resuspended in a cryoprotectant, 5% glucose (w/v) prior to long term storage by freeze drying. Nanoparticles intended for TEM imaging were resuspended in deionised water.
4.5. Preparation of Vancomycin HCl-Loaded Nanoparticles
Vancomycin HCl nanoparticles were first formulated using a base formulation. Briefly, 90mg of Vancomycin HCl was dissolved in deionised water (1.0 mL) and emulsified dropwise with sonication (70%) into the PLGA -RG-503H polymer (90 mg dissolved in 4 mL ethyl acetate) in an ice bath. This primary emulsion was then emulsified by sonication (70%) into 10 mL PVA 2.5% w/v in an ice-bath. This mixture was poured into 35 mL PVA 1% (w/v) under magnetic stirring at 200 rpm for 16 h at room temperature. The nanoparticles were collected by ultracentrifugation at 11,000 rpm for 15 min at 20 °C. The pellet was washed three times 20 mL deionized water and re-centrifuged. Samples were resuspended in a cryoprotectant, 5% glucose (w/v) prior to long term storage by freeze drying. Nanoparticles intended for TEM imaging were resuspended in deionised water. Subsequently, formulation parameters (drug:polymer ratio; volume of inner aqueous phase; PVA concentration) were varied to optimise the vancomycin HCl nanoparticle properties in terms of nanoparticle size, zeta potential and encapsulation efficiency. The optimal formulation parameters included a drug:polymer ratio of 1:9, an inner aqueous phase of 0.3 mL and PVA concentration of 2.5% w/v. Blank nanoparticles were formulated as per the base formulation, with the exception of the addition of Vancomycin HCl.
4.6. Immunofluorescence
Human endothelial cells were seeded onto glass coverslips at 3 × 105 cells then treated with 1 mg/mL Rhodamine B-loaded nanoparticles for 18 h. Cells were fixed using 4% formaldehyde and permeabilized with 0.1% Triton X-100. After blocking using 1% Bovine Serum Albumin (BSA), GAPDH 14C10 Rabbit mAb 1:50 was added followed by Alexa Fluor 647 goat anti-rabbit IgG 1:200. Coverslips were transferred to a glass slide with ProLong Diamond Antifade Mountant with DAPI then visualised using a Zeiss Axioscope for fluorescent imaging.
4.7. Characterising PLGA Nanoparticles
The average particle size, zeta potential, and polydispersity index (PDI) of either blank or loaded nanoparticles were analysed by photon correlation spectroscopy at 25 °C, using a Zetasizer Nano Series. The shape and surface morphology of the nanoparticles was observed by TEM on a silicon monoxide/formvar coated copper grid.
4.8. Determination of Encapsulation Efficiency
The encapsulation efficiency of nanoparticles was determined on freeze-dried samples. A weighted amount of sample was dissolved in acetonitrile and sonicated for 40 min. An equal quantity of water was added to precipitate the PLGA. The pellet was ultracentrifuged at 24,000 rpm for 20 min to pellet the precipitated PLGA. The supernatant was analysed using UV spectrophotometry at 280 nm to calculate drug loading per nanoparticle. The encapsulation efficiency (% w/w) was determined by the following equation: (amount of drug loaded/theoretical amount of drug loaded) × 100.
4.9. Nanoparticle Drug Release
To determine the amount of released vancomycin HCl from nanoparticles, in vitro release studies were performed using the PUR-A-LYZER mini 6000 dialysis kit. A dialysis bag containing a weighted amount of vancomycin PLGA nanoparticles in PBS at pH 7.4 was immersed in a shaking water bath at 100 rpm at 37 °C. At fixed time intervals, withdrawn samples were measured using UV spectrophotometry at 280 nm to quantify released vancomycin HCl.
4.10. Cytotoxic Evaluation of Formulation Nanoparticles
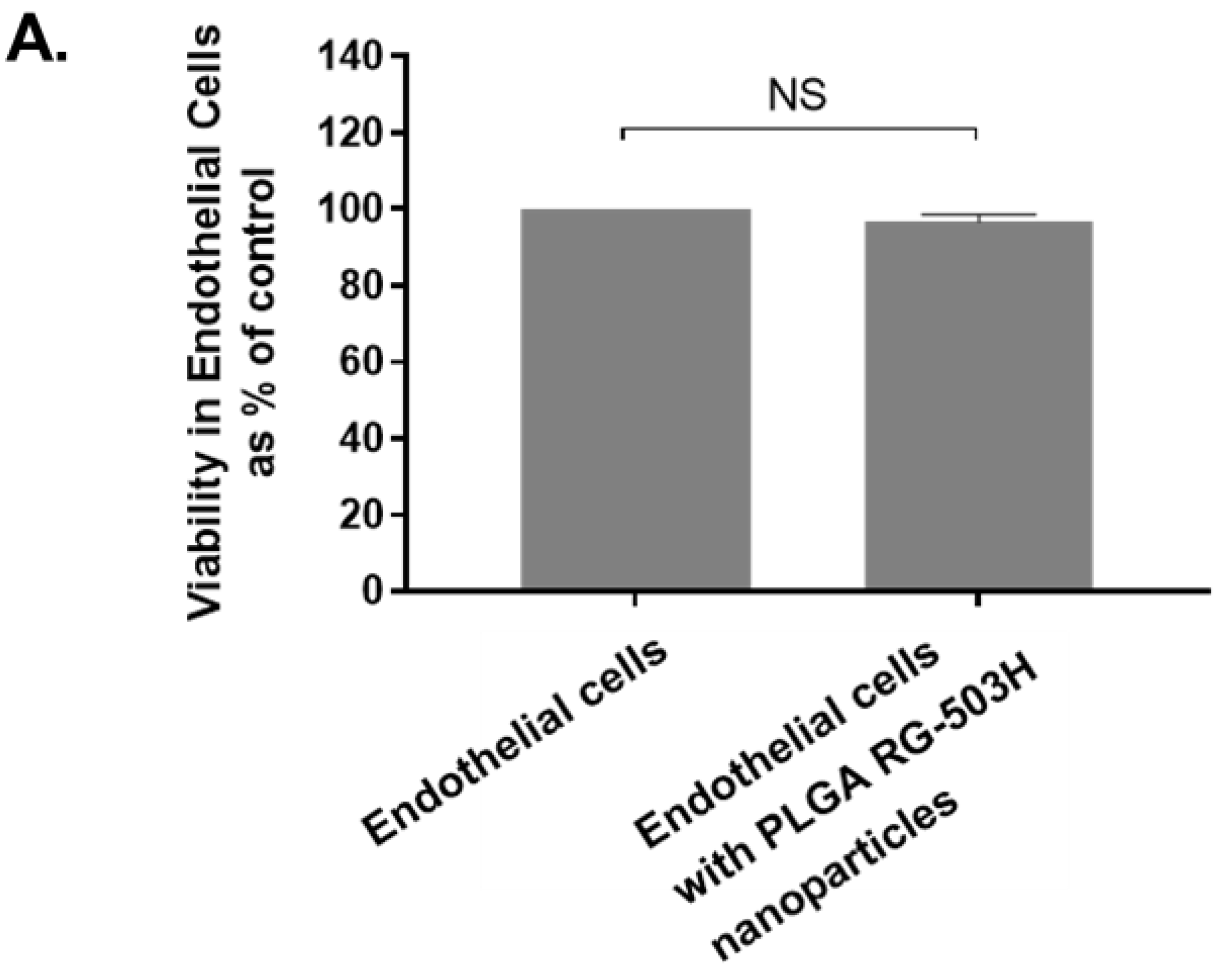
The MTT assay ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was used to examine cellular toxicity of the nanoparticles. Human endothelial cells were cultured at 2 × 105 cells/mL then treated for 24 h with 1 mg/mL PLGA nanoparticles. The viability of human endothelial cells was measured according to the MTT protocol followed by examination by UV spectrophotometry at 570 nm.
4.11. Pre-Clinical Evaluation of Formulated Nanoparticles
To examine whether vancomycin HCl PLGA nanoparticles were capable of treating human endothelial cells, they were infected with S. aureus 8325-4 as previously described and treated with 0.5 mg/mL vancomycin (10% w/w)-loaded nanoparticles for 18 h. After being lysed, the cell supernatant was plated onto BHI agar to calculate CFU/mL.
4.12. Statistical Analysis
Experiments were performed in independent triplicates, and statistically significant differences were analysed using one-way ANOVA followed by post hoc tests or t-tests using GraphPad Prism. Results were considered significant at p ≤ 0.05, and data was represented as mean ± standard error of mean (SEM).


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






