
Metagenomics and Other Omics Approaches to Bacterial Communities and Antimicrobial Resistance Assessment in Aquacultures



Abstract
:1. Introduction
2. Microbial Communities and Antimicrobial Resistance in Aquacultures
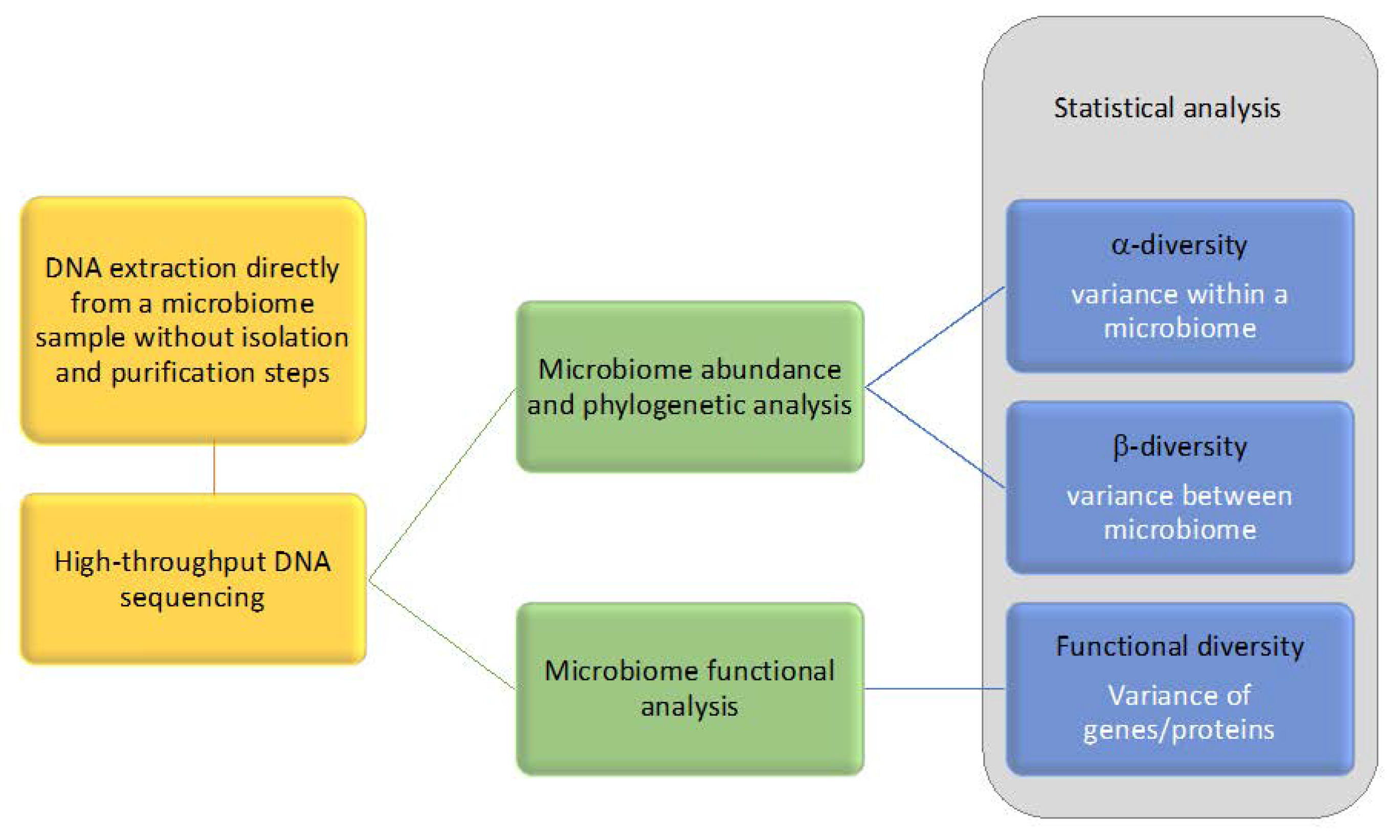
3. Omics Technologies to Address Microbial Communities
4. Omics Technologies to Characterize Aquatic Resistomes
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Population Day: Influencing Our Future Well-Being. Available online: https://www.compassion.com/world-days/world-population-day.htm (accessed on 30 November 2020).
- Santos, L.; Ramos, F. Antimicrobial Resistance in Aquaculture: Current Knowledge and Alternatives to Tackle the Problem. Int. J. Antimicrob. Agents 2018, 52, 135–143. [Google Scholar] [CrossRef]
- Naylor, R.L.; Hardy, R.W.; Buschmann, A.H.; Bush, S.R.; Cao, L.; Klinger, D.H.; Little, D.C.; Lubchenco, J.; Shumway, S.E.; Troell, M. A 20-Year Retrospective Review of Global Aquaculture. Nature 2021, 591, 551–563. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control (ECDC); European Food Safety Authority (EFSA); European Medicines Agency (EMA). ECDC/EFSA/EMA Second Joint Report on the Integrated Analysis of the Consumption of Antimicrobial Agents and Occurrence of Antimicrobial Resistance in Bacteria from Humans and Food-producing Animals. EFS2 2017, 15, e04872. [Google Scholar] [CrossRef]
- Rico, A.; Satapornvanit, K.; Haque, M.M.; Min, J.; Nguyen, P.T.; Telfer, T.C.; van den Brink, P.J. Use of Chemicals and Biological Products in Asian Aquaculture and Their Potential Environmental Risks: A Critical Review. Rev. Aquac. 2012, 4, 75–93. [Google Scholar] [CrossRef]
- Seyfried, E.E.; Newton, R.J.; Rubert, K.F.; Pedersen, J.A.; McMahon, K.D. Occurrence of Tetracycline Resistance Genes in Aquaculture Facilities with Varying Use of Oxytetracycline. Microb. Ecol. 2010, 59, 799–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, C.D. Antimicrobial Resistance Associated with Salmonid Farming. In Antimicrobial Resistance in the Environment; Keen, P.L., Montforts, M.H.M.M., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 423–451. ISBN 978-1-118-15624-7. [Google Scholar]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilbäck, C.; Sandegren, L.; Hughes, D.; Andersson, D.I. Selection of Resistant Bacteria at Very Low Antibiotic Concentrations. PLoS Pathog. 2011, 7, e1002158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabello, F.C. Heavy Use of Prophylactic Antibiotics in Aquaculture: A Growing Problem for Human and Animal Health and for the Environment. Environ. Microbiol. 2006, 8, 1137–1144. [Google Scholar] [CrossRef]
- Wall, B.A.; Mateus, A.; Marshall, L.; Pfeiffer, D.; Lubroth, J.; Ormel, H.J.; Otto, P.; Patriarchi, A. Drivers, Dynamics and Epidemiology of Antimicrobial Resistance in Animal Production; Food and Agriculture Organization of the United Nations: Rome, Italy, 2016; ISBN 978-92-5-109441-9. [Google Scholar]
- Perry, J.; Waglechner, N.; Wright, G. The Prehistory of Antibiotic Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025197. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.L.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic Resistance Is Ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef]
- Brouchkov, A.; Kabilov, M.; Filippova, S.; Baturina, O.; Rogov, V.; Galchenko, V.; Mulyukin, A.; Fursova, O.; Pogorelko, G. Bacterial Community in Ancient Permafrost Alluvium at the Mammoth Mountain (Eastern Siberia). Gene 2017, 636, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Mindlin, S.Z.; Petrova, M.A. On the Origin and Distribution of Antibiotic Resistance: Permafrost Bacteria Studies. Mol. Genet. Microbiol. Virol. 2017, 32, 169–179. [Google Scholar] [CrossRef]
- Sommer, M.O.A.; Church, G.M.; Dantas, G. The Human Microbiome Harbors a Diverse Reservoir of Antibiotic Resistance Genes. Virulence 2010, 1, 299–303. [Google Scholar] [CrossRef] [Green Version]
- Olivares, J.; Bernardini, A.; Garcia-Leon, G.; Corona, F.B.; Sanchez, M.; Martinez, J.L. The Intrinsic Resistome of Bacterial Pathogens. Front. Microbiol. 2013, 4, 103. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Edwards, R. Insights into Antibiotic Resistance through Metagenomic Approaches. Future Microbiol. 2011, 7, 73–89. [Google Scholar] [CrossRef] [Green Version]
- Martínez, J.L. Antibiotics and Antibiotic Resistance Genes in Natural Environments. Science 2008, 321, 365–367. [Google Scholar] [CrossRef]
- Sultan, I.; Rahman, S.; Jan, A.T.; Siddiqui, M.T.; Mondal, A.H.; Haq, Q.M.R. Antibiotics, Resistome and Resistance Mechanisms: A Bacterial Perspective. Front. Microbiol. 2018, 9, 2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raja, A.; Prabakaran, P. Actinomycetes and Drug-An Overview. Am. J. Drug Discov. Dev. 2011, 1, 75–84. [Google Scholar] [CrossRef]
- Genilloud, O. Actinomycetes: Still a Source of Novel Antibiotics. Nat. Prod. Rep. 2017, 34, 1203–1232. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.H.; Moore, L.S.P.; Sundsfjord, A.; Steinbakk, M.; Regmi, S.; Karkey, A.; Guerin, P.J.; Piddock, L.J.V. Understanding the Mechanisms and Drivers of Antimicrobial Resistance. Lancet 2016, 387, 176–187. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Czekalski, N.; Sigdel, R.; Birtel, J.; Matthews, B.; Bürgmann, H. Does Human Activity Impact the Natural Antibiotic Resistance Background? Abundance of Antibiotic Resistance Genes in 21 Swiss Lakes. Environ. Int. 2015, 81, 45–55. [Google Scholar] [CrossRef]
- Teglia, C.M.; Perez, F.A.; Michlig, N.; Repetti, M.R.; Goicoechea, H.C.; Culzoni, M.J. Occurrence, Distribution and Ecological Risk of Fluoroquinolones in Rivers and Wastewaters. Environ. Toxicol. Chem. 2019, 38, 2305–2313. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D. Microbiological Effects of Sublethal Levels of Antibiotics. Nat. Rev. Microbiol. 2014, 12, 465–478. [Google Scholar] [CrossRef]
- Barr, V.; Barr, K.; Millar, M.R.; Lacey, R.W. Beta-Lactam Antibiotics Increase the Frequency of Plasmid Transfer in Staphylococcus Aureus. J. Antimicrob. Chemother. 1986, 17, 409–413. [Google Scholar] [CrossRef]
- Lopez, E.; Blazquez, J. Effect of Subinhibitory Concentrations of Antibiotics on Intrachromosomal Homologous Recombination in Escherichia Coli. Antimicrob. Agents Chemother. 2009, 53, 3411–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, A.; Laureti, L.; Crussard, S.; Abida, H.; Rodríguez-Rojas, A.; Blázquez, J.; Baharoglu, Z.; Mazel, D.; Darfeuille, F.; Vogel, J.; et al. β-Lactam Antibiotics Promote Bacterial Mutagenesis via an RpoS-Mediated Reduction in Replication Fidelity. Nat. Commun. 2013, 4, 1610. [Google Scholar] [CrossRef] [Green Version]
- Baharoglu, Z.; Krin, E.; Mazel, D. Connecting Environment and Genome Plasticity in the Characterization of Transformation-Induced SOS Regulation and Carbon Catabolite Control of the Vibrio Cholerae Integron Integrase. J. Bacteriol. 2012, 194, 1659–1667. [Google Scholar] [CrossRef] [Green Version]
- Campoy, S.; Hervàs, A.; Busquets, N.; Erill, I.; Teixidó, L.; Barbé, J. Induction of the SOS Response by Bacteriophage Lytic Development in Salmonella Enterica. Virology 2006, 351, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, M.O.A.; Dantas, G.; Church, G.M. Functional Characterization of the Antibiotic Resistance Reservoir in the Human Microflora. Science 2009, 325, 1128–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San Millan, A.; MacLean, R.C. Fitness Costs of Plasmids: A Limit to Plasmid Transmission. Microbiol. Spectr. 2017, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G.J. Environmental Factors Influencing the Development and Spread of Antibiotic Resistance. FEMS Microbiol. Rev. 2018, 42, fux053. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Resistance from Environmental Pollution among Biggest Emerging Health Threats, Says UN Environment. Available online: http://www.unep.org/news-and-stories/press-release/antimicrobial-resistance-environmental-pollution-among-biggest (accessed on 10 April 2021).
- Sargenti, M.; Bartolacci, S.; Luciani, A.; Di Biagio, K.; Baldini, M.; Galarini, R.; Giusepponi, D.; Capuccella, M. Investigation of the Correlation between the Use of Antibiotics in Aquaculture Systems and Their Detection in Aquatic Environments: A Case Study of the Nera River Aquafarms in Italy. Sustainability 2020, 12, 5176. [Google Scholar] [CrossRef]
- Tiedje, J.M.; Wang, F.; Manaia, C.M.; Virta, M.; Sheng, H.; Ma, L.; Zhang, T.; Topp, E. Antibiotic Resistance Genes in the Human-Impacted Environment: A One Health Perspective. Pedosphere 2019, 29, 273–282. [Google Scholar] [CrossRef]
- Pulling-Together-to-Beat-Superbugs-Knowledge-and-Implementation-Gaps-in-Addressing-Antimicrobial-Resistance.Pdf. Available online: https://www.worldbank.org/en/topic/agriculture/publication/pulling-together-to-beat-superbugs-knowledge-and-implementation-gaps-in-addressing-antimicrobial-resistance (accessed on 15 April 2021).
- Ryu, S.-H.; Park, S.-G.; Choi, S.-M.; Hwang, Y.-O.; Ham, H.-J.; Kim, S.-U.; Lee, Y.-K.; Kim, M.-S.; Park, G.-Y.; Kim, K.-S.; et al. Antimicrobial Resistance and Resistance Genes in Escherichia Coli Strains Isolated from Commercial Fish and Seafood. Int. J. Food Microbiol. 2012, 152, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Ozaktas, T.; Taskin, B.; Gozen, A.G. High Level Multiple Antibiotic Resistance among Fish Surface Associated Bacterial Populations in Non-Aquaculture Freshwater Environment. Water Res. 2012, 46, 6382–6390. [Google Scholar] [CrossRef] [PubMed]
- Cabello, F.C.; Godfrey, H.P.; Tomova, A.; Ivanova, L.; Dölz, H.; Millanao, A.; Buschmann, A.H. Antimicrobial Use in Aquaculture Re-Examined: Its Relevance to Antimicrobial Resistance and to Animal and Human Health: Aquacultural Antimicrobial Use and Antimicrobial Resistance. Env. Microbiol. 2013, 15, 1917–1942. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.Q.A.; Cabello, F.C.; L’Abée-Lund, T.M.; Tomova, A.; Godfrey, H.P.; Buschmann, A.H.; Sørum, H. Antimicrobial Resistance and Antimicrobial Resistance Genes in Marine Bacteria from Salmon Aquaculture and Non-Aquaculture Sites. Environ. Microbiol. 2014, 16, 1310–1320. [Google Scholar] [CrossRef]
- Aich, N.; Ahmed, N.; Paul, A. Issues of Antibiotic Resistance in Aquaculture Industry and Its Way Forward. Int. J. Curr. Microbiol. Appl. Sci. 2018, 7, 26–41. [Google Scholar] [CrossRef]
- Cabello, F.C.; Godfrey, H.P.; Buschmann, A.H.; Dölz, H.J. Aquaculture as yet Another Environmental Gateway to the Development and Globalisation of Antimicrobial Resistance. Lancet Infect. Dis. 2016, 16, e127–e133. [Google Scholar] [CrossRef]
- O’Neill, J. Antimicrobials in Agriculture and the Environment: Reducing Unnecessary Use and Waste. Available online: https://wellcomecollection.org/works/x88ast2u/items (accessed on 19 April 2021).
- Baquero, F.; Martínez, J.-L.; Cantón, R. Antibiotics and Antibiotic Resistance in Water Environments. Curr. Opin. Biotechnol. 2008, 19, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Tacon, A.G.J.; Metian, M. Global Overview on the Use of Fish Meal and Fish Oil in Industrially Compounded Aquafeeds: Trends and Future Prospects. Aquaculture 2008, 285, 146–158. [Google Scholar] [CrossRef]
- Allen, H.K.; Looft, T.; Bayles, D.O.; Humphrey, S.; Levine, U.Y.; Alt, D.; Stanton, T.B. Antibiotics in Feed Induce Prophages in Swine Fecal Microbiomes. mBio 2011, 2, e00260-11. [Google Scholar] [CrossRef] [Green Version]
- Udikovic-Kolic, N.; Wichmann, F.; Broderick, N.A.; Handelsman, J. Bloom of Resident Antibiotic-Resistant Bacteria in Soil Following Manure Fertilization. Proc. Natl. Acad. Sci. USA 2014, 111, 15202–15207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Su, J. Vulnerability of China’s Nearshore Ecosystems under Intensive Mariculture Development. Environ. Sci Pollut. Res. 2017, 24, 8957–8966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.; Huang, K.; Yu, J.; Ding, C.; Wang, Z.; Zhao, C.; Yuan, H.; Wang, Z.; Wang, S.; Hu, J.; et al. Metagenomic Analysis of Bacterial Communities and Antibiotic Resistance Genes in the Eriocheir Sinensis Freshwater Aquaculture Environment. Chemosphere 2019, 224, 202–211. [Google Scholar] [CrossRef]
- Xiong, W.; Sun, Y.; Zhang, T.; Ding, X.; Li, Y.; Wang, M.; Zeng, Z. Antibiotics, Antibiotic Resistance Genes, and Bacterial Community Composition in Fresh Water Aquaculture Environment in China. Microb. Ecol. 2015, 70, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Marti, E.; Huerta, B.; Rodríguez-Mozaz, S.; Barceló, D.; Marcé, R.; Balcázar, J.L. Abundance of Antibiotic Resistance Genes and Bacterial Community Composition in Wild Freshwater Fish Species. Chemosphere 2018, 196, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Muziasari, W.I.; Pitkänen, L.K.; Sørum, H.; Stedtfeld, R.D.; Tiedje, J.M.; Virta, M. The Resistome of Farmed Fish Feces Contributes to the Enrichment of Antibiotic Resistance Genes in Sediments below Baltic Sea Fish Farms. Front. Microbiol. 2017, 7, 2137. [Google Scholar] [CrossRef]
- Mog, M.; Ngasotter, S.; Tesia, S.; Waikhom, D.; Panda, P.; Sharma, S.; Varshney, S. Problems of Antibiotic Resistance Associated with Oxytetracycline Use in Aquaculture: A Review. J. Entomol. Zool. Stud. 2020, 8, 1075–1082. [Google Scholar]
- Elmahdi, S.; DaSilva, L.V.; Parveen, S. Antibiotic Resistance of Vibrio Parahaemolyticus and Vibrio Vulnificus in Various Countries: A Review. Food Microbiol. 2016, 57, 128–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, B.; Ba, Y.; Niu, L.; Lou, F.; Zhang, Z.; Liu, H.; Pan, Y.; Zhao, Y. A Comprehensive Research on Antibiotic Resistance Genes in Microbiota of Aquatic Animals. Front. Microbiol. 2018, 9, 1617. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Su, Y.; Deng, Y.; Guo, Z.; Mao, C.; Liu, G.; Xu, L.; Cheng, C.; Bei, L.; Feng, J. Prevalence and Distribution of Antibiotic Resistance in Marine Fish Farming Areas in Hainan, China. Sci. Total Environ. 2019, 653, 605–611. [Google Scholar] [CrossRef]
- Harnisz, M.; Korzeniewska, E.; Gołaś, I. The Impact of a Freshwater Fish Farm on the Community of Tetracycline-Resistant Bacteria and the Structure of Tetracycline Resistance Genes in River Water. Chemosphere 2015, 128, 134–141. [Google Scholar] [CrossRef]
- Watts, J.E.M.; Schreier, H.J.; Lanska, L.; Hale, M.S. The Rising Tide of Antimicrobial Resistance in Aquaculture: Sources, Sinks and Solutions. Mar. Drugs 2017, 15, 158. [Google Scholar] [CrossRef] [Green Version]
- Done, H.Y.; Venkatesan, A.K.; Halden, R.U. Does the Recent Growth of Aquaculture Create Antibiotic Resistance Threats Different from Those Associated with Land Animal Production in Agriculture? AAPS J. 2015, 17, 513–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FAO (Ed.) Contributing to Food Security and Nutrition for All. In The State of World Fisheries and Aquaculture; FAO: Rome, Italy, 2016; ISBN 978-92-5-109185-2. [Google Scholar]
- Henriksson, P.J.G.; Rico, A.; Troell, M.; Klinger, D.H.; Buschmann, A.H.; Saksida, S.; Chadag, M.V.; Zhang, W. Unpacking Factors Influencing Antimicrobial Use in Global Aquaculture and Their Implication for Management: A Review from a Systems Perspective. Sustain. Sci. 2018, 13, 1105–1120. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.Q.A.; Colquhoun, D.J.; Nikuli, H.L.; Sørum, H. Prevalence of Antibiotic Resistance Genes in the Bacterial Flora of Integrated Fish Farming Environments of Pakistan and Tanzania. Environ. Sci. Technol. 2012, 46, 8672–8679. [Google Scholar] [CrossRef]
- Marshall, B.M.; Levy, S.B. Food Animals and Antimicrobials: Impacts on Human Health. Clin. Microbiol. Rev. 2011, 24, 718–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, R.H.S.D.F.; Carvalho, E.M.R.; Carvalho, F.C.T.; Silva, C.M.; Sousa, O.V.; Rodrigues, D.P. Antimicrobial Susceptibility of Escherichia Coli Isolated from Shrimp (Litopenaeus Vannamei) and Pond Environment in Northeastern Brazil. J. Environ. Sci. Health Part B 2010, 45, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Capita, R.; Alonso-Calleja, C. Antibiotic-Resistant Bacteria: A Challenge for the Food Industry. Crit. Rev. Food Sci. Nutr. 2013, 53, 11–48. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Use in Aquaculture and Antimicrobial Resistance. In Report of a Joint FAO/OIE/WHO Expert Consultation on Antimicrobial Use in Aquaculture and Antimicrobial Resistance; 13–16 June 2006; WHO: Seoul, Korea, 2006; p. 107.
- Gauthier, D.T. Bacterial Zoonoses of Fishes: A Review and Appraisal of Evidence for Linkages between Fish and Human Infections. Vet. J. 2015, 203, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Annual_Report_AMR_3.Pdf. Available online: https://www.oie.int/fileadmin/Home/eng/Our_scientific_expertise/docs/pdf/AMR/Annual_Report_AMR_3.pdf (accessed on 15 April 2021).
- Miranda, C.D.; Tello, A.; Keen, P.L. Mechanisms of Antimicrobial Resistance in Finfish Aquaculture Environments. Front. Microbiol. 2013, 4, 233. [Google Scholar] [CrossRef] [Green Version]
- Lulijwa, R.; Rupia, E.J.; Alfaro, A.C. Antibiotic Use in Aquaculture, Policies and Regulation, Health and Environmental Risks: A Review of the Top 15 Major Producers. Rev. Aquacult 2020, 12, 640–663. [Google Scholar] [CrossRef]
- Hamza, D.; Dorgham, S.; Ismael, E.; El-Moez, S.I.A.; Elhariri, M.; Elhelw, R.; Hamza, E. Emergence of β-Lactamase- and Carbapenemase-Producing Enterobacteriaceae at Integrated Fish Farms. Antimicrob. Resist. Infect. Control 2020, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.J.; Rubin, J.E. Carbapenemase Producing Bacteria in the Food Supply Escaping Detection. PLoS ONE 2015, 10, e0126717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, V.; Nunes, J.; Gomes, A.; Capita, R.; Alonso-Calleja, C.; Pereira, J.E.; Torres, C.; Igrejas, G.; Poeta, P. Detection of Antibiotic Resistance in Escherichia Coli Strains: Can Fish Commonly Used in Raw Preparations Such as Sushi and Sashimi Constitute a Public Health Problem? J. Food Prot. 2019, 82, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Grevskott, D.H.; Svanevik, C.S.; Sunde, M.; Wester, A.L.; Lunestad, B.T. Marine Bivalve Mollusks As Possible Indicators of Multidrug-Resistant Escherichia Coli and Other Species of the Enterobacteriaceae Family. Front. Microbiol. 2017, 8, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, A.R.; Mok, J.S.; Lee, D.E.; Kwon, J.Y.; Park, K. Occurrence, Virulence, and Antimicrobial Resistance of Vibrio Parahaemolyticus Isolated from Bivalve Shellfish Farms along the Southern Coast of Korea. Environ. Sci. Pollut. Res. 2019, 26, 21034–21043. [Google Scholar] [CrossRef]
- Agnoletti, F.; Arcangeli, G.; Barbanti, F.; Barco, L.; Brunetta, R.; Cocchi, M.; Conedera, G.; D’Este, L.; Drigo, I.; Spigaglia, P.; et al. Survey, Characterization and Antimicrobial Susceptibility of Clostridium Difficile from Marine Bivalve Shellfish of North Adriatic Sea. Int. J. Food Microbiol. 2019, 298, 74–80. [Google Scholar] [CrossRef]
- Moura, A.; Henriques, I.; Smalla, K.; Correia, A. Wastewater Bacterial Communities Bring Together Broad-Host Range Plasmids, Integrons and a Wide Diversity of Uncharacterized Gene Cassettes. Res. Microbiol. 2010, 161, 58–66. [Google Scholar] [CrossRef]
- Microarray-Based Detection of 90 Antibiotic Resistance Genes of Gram-Positive Bacteria. J. Clin. Microbiol. 2005, 43, 2291–2302. Available online: https://jcm.asm.org/content/43/5/2291 (accessed on 19 April 2021). [CrossRef] [Green Version]
- Bragg, L.; Tyson, G.W. Metagenomics Using Next-Generation Sequencing. In Environmental Microbiology; Paulsen, I.T., Holmes, A.J., Eds.; Humana Press: Totowa, NJ, USA, 2014; Volume 1096, pp. 183–201. ISBN 978-1-62703-711-2. [Google Scholar]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and Assembly of Genomes and Genetic Elements in Complex Metagenomic Samples without Using Reference Genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Locey, K.J.; Lennon, J.T. Scaling Laws Predict Global Microbial Diversity. Proc. Natl. Acad. Sci. USA 2016, 113, 5970–5975. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, T.; David, P.H.C.; Pothier, J. Antibiotics as Both Friends and Foes of the Human Gut Microbiome: The Microbial Community Approach. Drug Dev. Res. 2019, 80, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Ponomarova, O.; Patil, K.R. Metabolic Interactions in Microbial Communities: Untangling the Gordian Knot. Curr. Opin. Microbiol. 2015, 27, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, A.Z.; Matson, E.G.; Eldar, A.; Leadbetter, J.R. RNA-Seq Reveals Cooperative Metabolic Interactions between Two Termite-Gut Spirochete Species in Co-Culture. ISME J. 2011, 5, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Freilich, S.; Zarecki, R.; Eilam, O.; Segal, E.S.; Henry, C.S.; Kupiec, M.; Gophna, U.; Sharan, R.; Ruppin, E. Competitive and Cooperative Metabolic Interactions in Bacterial Communities. Nat. Commun. 2011, 2, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horgan, R.P.; Kenny, L.C. ‘Omic’ Technologies: Genomics, Transcriptomics, Proteomics and Metabolomics: The Obstetrician & Gynaecologist. Obstet. Gynaecol. 2011, 13, 189–195. [Google Scholar] [CrossRef]
- Westerhoff, H.V.; Palsson, B.O. The Evolution of Molecular Biology into Systems Biology. Nat. Biotechnol. 2004, 22, 1249–1252. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, J. Metagenetics: Spending Our Inheritance on the Future. Microb. Biotechnol. 2009, 2, 138–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woese, C.R.; Fox, G.E. Phylogenetic Structure of the Prokaryotic Domain: The Primary Kingdoms. Proc. Natl. Acad. Sci.USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [Green Version]
- Patané, J.S.L.; Martins, J.; Setubal, J.C. Phylogenomics. In Comparative Genomics; Methods in Molecular Biology; Setubal, J.C., Stoye, J., Stadler, P.F., Eds.; Springer: New York, NY, USA, 2018; Volume 1704, pp. 103–187. ISBN 978-1-4939-7461-0. [Google Scholar]
- Jansson, J.K.; Hofmockel, K.S. The Soil Microbiome—From Metagenomics to Metaphenomics. Curr. Opin. Microbiol. 2018, 43, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Alpha and Beta Diversity—Metagenomics. Available online: http://www.metagenomics.wiki/pdf/definition/alpha-beta-diversity (accessed on 21 August 2019).
- Calle, M.L. Statistical Analysis of Metagenomics Data. Genom. Inf. 2019, 17, e6. [Google Scholar] [CrossRef]
- Angly, F.E.; Dennis, P.G.; Skarshewski, A.; Vanwonterghem, I.; Hugenholtz, P.; Tyson, G.W. CopyRighter: A Rapid Tool for Improving the Accuracy of Microbial Community Profiles through Lineage-Specific Gene Copy Number Correction. Microbiome 2014, 2, 11. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S RRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D. Reintroducing Mothur: 10 Years Later. Appl. Environ. Microbiol. 2019, 86, e02343-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Ren, J.; Michail, S.; Sun, F. MicroPro: Using Metagenomic Unmapped Reads to Provide Insights into Human Microbiota and Disease Associations. Genome Biol. 2019, 20, 154. [Google Scholar] [CrossRef]
- Bowman, J.S.; Ducklow, H.W. Microbial Communities Can Be Described by Metabolic Structure: A General Framework and Application to a Seasonally Variable, Depth-Stratified Microbial Community from the Coastal West Antarctic Peninsula. PLoS ONE 2015, 10, e0135868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpton, T.J.; Riesenfeld, S.J.; Kembel, S.W.; Ladau, J.; O’Dwyer, J.P.; Green, J.L.; Eisen, J.A.; Pollard, K.S. PhylOTU: A High-Throughput Procedure Quantifies Microbial Community Diversity and Resolves Novel Taxa from Metagenomic Data. PLoS Comput. Biol. 2011, 7, e1001061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2: An Improved and Extensible Approach for Metagenome Inference. bioRxiv 2019, 672295. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and Tools for High Throughput RRNA Analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoddard, S.F.; Smith, B.J.; Hein, R.; Roller, B.R.K.; Schmidt, T.M. RrnDB: Improved Tools for Interpreting RRNA Gene Abundance in Bacteria and Archaea and a New Foundation for Future Development. Nucleic Acids Res. 2015, 43, D593–D598. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting Functional Profiles from Metagenomic 16S RRNA Data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Mori, H.; Maruyama, T.; Yano, M.; Yamada, T.; Kurokawa, K. VITCOMIC2: Visualization Tool for the Phylogenetic Composition of Microbial Communities Based on 16S RRNA Gene Amplicons and Metagenomic Shotgun Sequencing. BMC Syst. Biol. 2018, 12, 30. [Google Scholar] [CrossRef]
- Escudeiro, P.; Pothier, J.; Dionisio, F.; Nogueira, T. Antibiotic Resistance Gene Diversity and Virulence Gene Diversity Are Correlated in Human Gut and Environmental Microbiomes. Msphere 2019, 4, e00135-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, B.P.; Mohanty, S.; Mitra, T.; Mahanty, A.; Ganguly, S.; Singh, S. Omics Technology in Fisheries and Aquaculture. Adv. Fish Res. 2019, 8, 1–30. [Google Scholar]
- Liu, Z.; Ma, H.; Goryanin, I. A Semi-Automated Genome Annotation Comparison and Integration Scheme. BMC Bioinform. 2013, 14, 172. [Google Scholar] [CrossRef] [Green Version]
- Sharpton, T.J. An Introduction to the Analysis of Shotgun Metagenomic Data. Front. Plant Sci. 2014, 5, 209. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, D.; Walsh, F. Antibiotic Resistance Genes across a Wide Variety of Metagenomes. FEMS Microbiol. Ecol. 2016, 92, fiv168. [Google Scholar] [CrossRef]
- Xavier, B.B.; Das, A.J.; Cochrane, G.; Ganck, S.D.; Kumar-Singh, S.; Aarestrup, F.M.; Goossens, H.; Malhotra-Kumar, S. Consolidating and Exploring Antibiotic Resistance Gene Data Resources. J. Clin. Microbiol. 2016, 54, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Gibson, M.K.; Forsberg, K.J.; Dantas, G. Improved Annotation of Antibiotic Resistance Determinants Reveals Microbial Resistomes Cluster by Ecology. ISME J. 2015, 9, 207–216. [Google Scholar] [CrossRef]
- Yin, X.; Jiang, X.-T.; Chai, B.; Li, L.; Yang, Y.; Cole, J.R.; Tiedje, J.M.; Zhang, T. ARGs-OAP v2.0 with an Expanded SARG Database and Hidden Markov Models for Enhancement Characterization and Quantification of Antibiotic Resistance Genes in Environmental Metagenomes. Bioinformatics 2018, 34, 2263–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.-M. ARG-ANNOT, a New Bioinformatic Tool To Discover Antibiotic Resistance Genes in Bacterial Genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.-H.; McDermott, P.F.; et al. Validating the AMRFinder Tool and Resistance Gene Database by Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of Isolates. Antimicrob. Agents Chemother. 2019, 63, e00483-19. [Google Scholar] [CrossRef] [Green Version]
- Ruppé, E.; Ghozlane, A.; Tap, J.; Pons, N.; Alvarez, A.-S.; Maziers, N.; Cuesta, T.; Hernando-Amado, S.; Clares, I.; Martínez, J.L.; et al. Prediction of the Intestinal Resistome by a Three-Dimensional Structure-Based Method. Nat. Microbiol. 2019, 4, 112. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Zhou, R.; Zeng, S.; Hou, D.; Liu, J.; Weng, S.; He, J.; Huang, Z. Occurrence of Human Pathogenic Bacteria Carrying Antibiotic Resistance Genes Revealed by Metagenomic Approach: A Case Study from an Aquatic Environment. J. Environ. Sci. 2019, 80, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Nordahl Petersen, T.; Rasmussen, S.; Hasman, H.; Carøe, C.; Bælum, J.; Charlotte Schultz, A.; Bergmark, L.; Svendsen, C.A.; Lund, O.; Sicheritz-Pontén, T.; et al. Meta-Genomic Analysis of Toilet Waste from Long Distance Flights; a Step towards Global Surveillance of Infectious Diseases and Antimicrobial Resistance. Sci. Rep. 2015, 5, 11444. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, T.; Rankin, D.J.; Touchon, M.; Taddei, F.; Brown, S.P.; Rocha, E.P.C. Horizontal Gene Transfer of the Secretome Drives the Evolution of Bacterial Cooperation and Virulence. Curr. Biol. 2009, 19, 1683–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, T.; Touchon, M.; Rocha, E.P.C. Rapid Evolution of the Sequences and Gene Repertoires of Secreted Proteins in Bacteria. PLoS ONE 2012, 7, e49403. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Mao, D.; Luo, Y.; Wang, L.; Xu, B.; Xu, L. Occurrence of Sulfonamide and Tetracycline-Resistant Bacteria and Resistance Genes in Aquaculture Environment. Water Res. 2012, 46, 2355–2364. [Google Scholar] [CrossRef]
- Huang, L.; Xu, Y.-B.; Xu, J.-X.; Ling, J.-Y.; Chen, J.-L.; Zhou, J.-L.; Zheng, L.; Du, Q.-P. Antibiotic Resistance Genes (ARGs) in Duck and Fish Production Ponds with Integrated or Non-Integrated Mode. Chemosphere 2017, 168, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Kobiyama, A.; Ikeo, K.; Reza, M.S.; Rashid, J.; Yamada, Y.; Ikeda, Y.; Ikeda, D.; Mizusawa, N.; Sato, S.; Ogata, T.; et al. Metagenome-Based Diversity Analyses Suggest a Strong Locality Signal for Bacterial Communities Associated with Oyster Aquaculture Farms in Ofunato Bay. Gene 2018, 665, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Reza, M.S.; Kobiyama, A.; Yamada, Y.; Ikeda, Y.; Ikeda, D.; Mizusawa, N.; Ikeo, K.; Sato, S.; Ogata, T.; Jimbo, M.; et al. Taxonomic Profiles in Metagenomic Analyses of Free-Living Microbial Communities in the Ofunato Bay. Gene 2018, 665, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Li, Y.; Qi, Z.; Yue, Y.; Min, M.; Peng, S.; Shi, Z.; Gao, Y. Diverse and Abundant Antibiotic Resistance Genes from Mariculture Sites of China’s Coastline. Sci. Total Environ. 2018, 630, 117–125. [Google Scholar] [CrossRef]
- Ng, C.; Tay, M.; Tan, B.; Le, T.-H.; Haller, L.; Chen, H.; Koh, T.H.; Barkham, T.M.S.; Thompson, J.R.; Gin, K.Y.-H. Characterization of Metagenomes in Urban Aquatic Compartments Reveals High Prevalence of Clinically Relevant Antibiotic Resistance Genes in Wastewaters. Front. Microbiol. 2017, 8, 2200. [Google Scholar] [CrossRef] [Green Version]
- The Global Sewage Surveillance Project Consortium; Hendriksen, R.S.; Munk, P.; Njage, P.; van Bunnik, B.; McNally, L.; Lukjancenko, O.; Röder, T.; Nieuwenhuijse, D.; Pedersen, S.K.; et al. Global Monitoring of Antimicrobial Resistance Based on Metagenomics Analyses of Urban Sewage. Nat. Commun. 2019, 10, 1124. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, R.; Zhang, M.; Cheng, Z.; Li, J.; Zhang, G.; Chen, B.; Zou, S.; Yang, Y. Exploring the Differences of Antibiotic Resistance Genes Profiles between River Surface Water and Sediments Using Metagenomic Approach. Ecotoxicol. Environ. Saf. 2018, 161, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, X.; Zhao, Z.; Duan, C.; Chen, H.; Wang, M.; Ren, H.; Yin, Y.; Ye, L. Metagenomic Analysis Revealed the Prevalence of Antibiotic Resistance Genes in the Gut and Living Environment of Freshwater Shrimp. J. Hazard. Mater. 2018, 350, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Brunton, L.A.; Desbois, A.P.; Garza, M.; Wieland, B.; Mohan, C.V.; Häsler, B.; Tam, C.C.; Le, P.N.T.; Phuong, N.T.; Van, P.T.; et al. Identifying Hotspots for Antibiotic Resistance Emergence and Selection, and Elucidating Pathways to Human Exposure: Application of a Systems-Thinking Approach to Aquaculture Systems. Sci. Total Environ. 2019, 687, 1344–1356. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Khelaifia, S.; Alou, M.T.; Ndongo, S.; Dione, N.; Hugon, P.; Caputo, A.; Cadoret, F.; Traore, S.I.; Seck, E.H.; et al. Culture of Previously Uncultured Members of the Human Gut Microbiota by Culturomics. Nat. Microbiol. 2016, 1, 16203. [Google Scholar] [CrossRef] [PubMed]
- Seng, P.; Drancourt, M.; Gouriet, F.; La Scola, B.; Fournier, P.-E.; Rolain, J.M.; Raoult, D. Ongoing Revolution in Bacteriology: Routine Identification of Bacteria by Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry. Clin. Infect. Dis. 2009, 49, 543–551. [Google Scholar] [CrossRef]
- Croxatto, A.; Prod’hom, G.; Greub, G. Applications of MALDI-TOF Mass Spectrometry in Clinical Diagnostic Microbiology. FEMS Microbiol. Rev. 2012, 36, 380–407. [Google Scholar] [CrossRef]
- Bilen, M.; Dufour, J.-C.; Lagier, J.-C.; Cadoret, F.; Daoud, Z.; Dubourg, G.; Raoult, D. The Contribution of Culturomics to the Repertoire of Isolated Human Bacterial and Archaeal Species. Microbiome 2018, 6, 94. [Google Scholar] [CrossRef] [Green Version]
- Nowrotek, M.; Jałowiecki, Ł.; Harnisz, M.; Płaza, G.A. Culturomics and Metagenomics: In Understanding of Environmental Resistome. Front. Environ. Sci. Eng. 2019, 13, 40. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An Improved Greengenes Taxonomy with Explicit Ranks for Ecological and Evolutionary Analyses of Bacteria and Archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive Functional Profiling of Microbial Communities Using 16S RRNA Marker Gene Sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The Ribosomal Database Project: Improved Alignments and New Tools for RRNA Analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classifier for Rapid Assignment of RRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-Species Living Tree Project (LTP)” Taxonomic Frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- López-García, A.; Pineda-Quiroga, C.; Atxaerandio, R.; Pérez, A.; Hernández, I.; García-Rodríguez, A.; González-Recio, O. Comparison of Mothur and QIIME for the Analysis of Rumen Microbiota Composition Based on 16S RRNA Amplicon Sequences. Front. Microbiol. 2018, 9, 3010. [Google Scholar] [CrossRef]
- Mothur. Available online: https://mothur.org/ (accessed on 10 September 2019).
- Balvočiūtė, M.; Huson, D.H. SILVA, RDP, Greengenes, NCBI and OTT—How Do These Taxonomies Compare? BMC Genomics 2017, 18, 114. [Google Scholar] [CrossRef] [Green Version]
- Federhen, S. The NCBI Taxonomy Database. Nucleic Acids Res. 2012, 40, D136–D143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staley, C.; Gould, T.J.; Wang, P.; Phillips, J.; Cotner, J.B.; Sadowsky, M.J. Sediments and Soils Act as Reservoirs for Taxonomic and Functional Bacterial Diversity in the Upper Mississippi River. Microb. Ecol. 2016, 71, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Roller, B.R.K.; Stoddard, S.F.; Schmidt, T.M. Exploiting RRNA Operon Copy Number to Investigate Bacterial Reproductive Strategies. Nat. Microbiol. 2016, 1, 16160. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Name of the Tool | Ref. | Link (Accessed on 24 June 2021) |
|---|---|---|
| AMRFinderPlus | [122] | https://github.com/ncbi/amr/wiki |
| ARG-ANNOT | [121] | https://github.com/tseemann/abricate/pull/82 |
| ARGs-OAP | [119] | https://galaxyproject.org/use/args-oap/ |
| CARD 2020 | [120] | https://card.mcmaster.ca |
| MUSTARD | [123] | http://mgps.eu/Mustard/ |
| Resfams | [118] | https://github.com/dantaslab/resfams |
| ResFinder 4.0 | [124] | https://cge.cbs.dtu.dk/services/ResFinder/ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nogueira, T.; Botelho, A. Metagenomics and Other Omics Approaches to Bacterial Communities and Antimicrobial Resistance Assessment in Aquacultures. Antibiotics 2021, 10, 787. https://doi.org/10.3390/antibiotics10070787
Nogueira T, Botelho A. Metagenomics and Other Omics Approaches to Bacterial Communities and Antimicrobial Resistance Assessment in Aquacultures. Antibiotics. 2021; 10(7):787. https://doi.org/10.3390/antibiotics10070787
Chicago/Turabian StyleNogueira, Teresa, and Ana Botelho. 2021. "Metagenomics and Other Omics Approaches to Bacterial Communities and Antimicrobial Resistance Assessment in Aquacultures" Antibiotics 10, no. 7: 787. https://doi.org/10.3390/antibiotics10070787
APA StyleNogueira, T., & Botelho, A. (2021). Metagenomics and Other Omics Approaches to Bacterial Communities and Antimicrobial Resistance Assessment in Aquacultures. Antibiotics, 10(7), 787. https://doi.org/10.3390/antibiotics10070787