2.1. Benzimidazoles Substituted in the “2” Position with Pyrazole Moiety
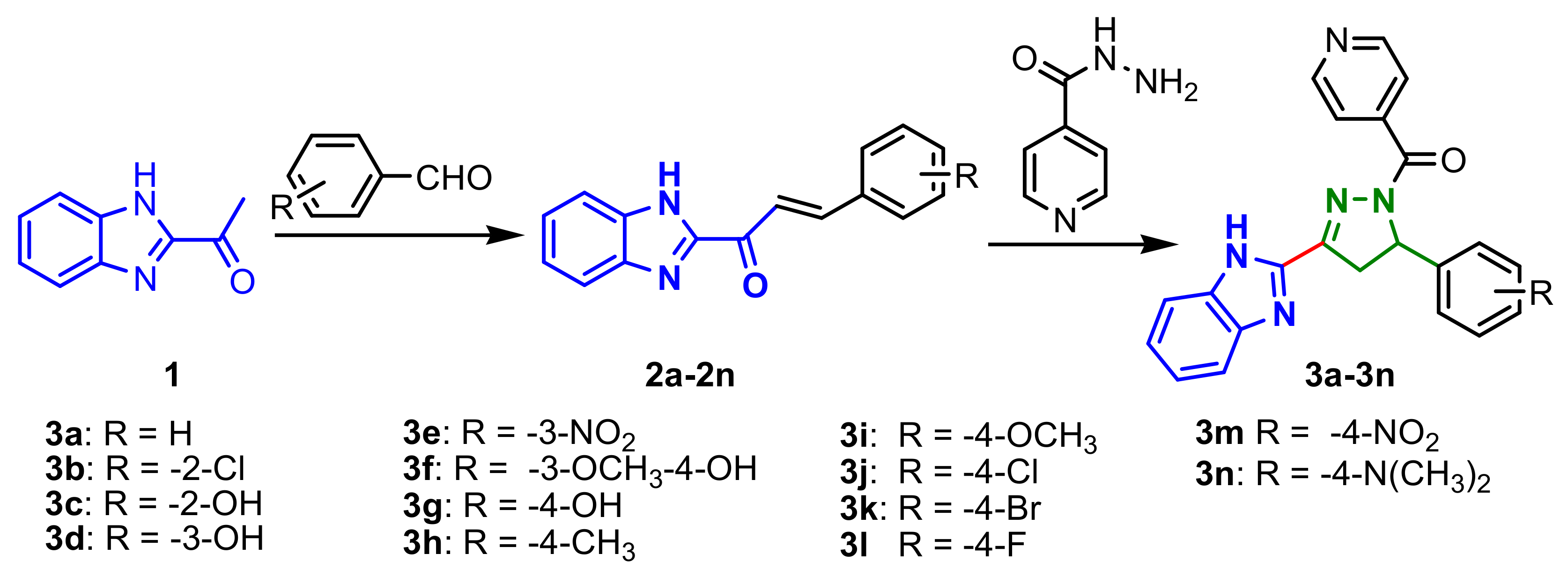
Benzimidazole chalcones
2a–
2n, synthesized from 2-acetylbenzimidazole
1 and aldehydes in ethanolic KOH by a Claisen–Schmidt condensation, were cyclocondensated with izoniazide, to give (3-(1
H-benzo[d]imidazol-2-yl)-5-(aryl)-4,5-dihydro-1
H-pyrazol-1-yl)(pyridin-4-yl)methanones
3a–
3n in good yields (
Scheme 1). All compounds showed antimicrobial activity against bacterial strains
E. coli,
P. aeruginosa,
S. aureus,
S. pyogenes and fungi
C. albicans,
A. niger and
A. clavatus. The compounds
3d,
3g,
3h, were found to be the best antibacterials, with MIC of 25 μg mL
−1 against
P. aeuginosa (
3d) and
E. coli (
3g,
3h) and compound
3n the best antifungal, with MIC 25 μg mL
−1 against
A. niger [
55].
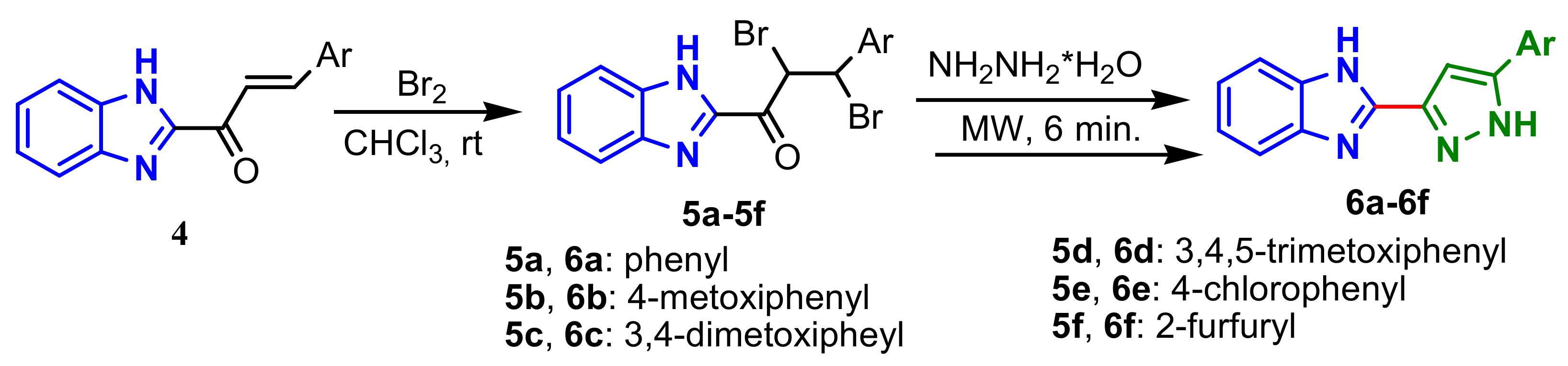
Rajora and Srivastava reported the synthesis of some 2-(1
H-pyrazol-3-yl)-1
H-benzo[d]imidazoles by bromination of benzimidazolyl chalcone
4, with the formation of dibrominated intermediates
5a–
5f, followed by cyclization in the presence of hydrazine hydrate and dehydrobromination, with the formation of compounds
6a–
6f (
Scheme 2).
Compounds
6a–
6f showed good antimicrobial activity on four bacterial strains,
E. coli,
P. aeruginusa,
B. subtilis,
K. pneumoniae and two fungi,
Candida albicons and
Aspergillus niger, considering ciprofoxacin and fluconazole as standard drugs [
56].
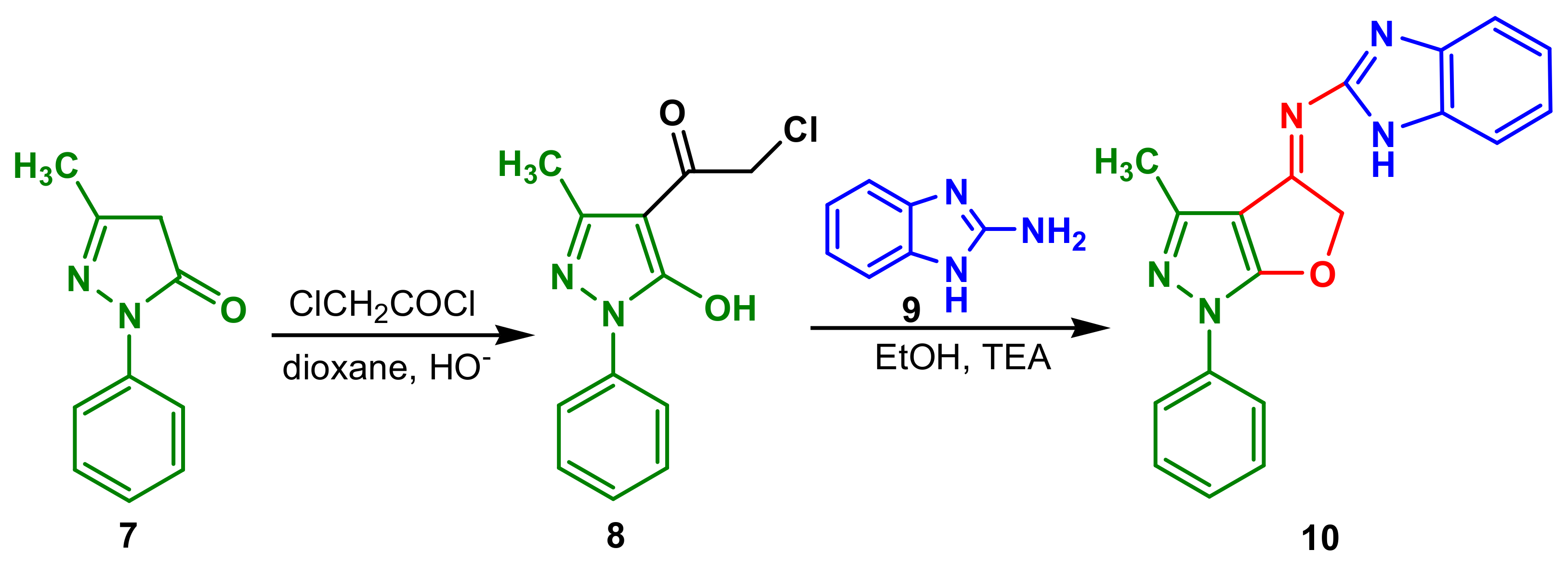
2-Chloro-1-(5-hydroxy-3-methyl-1-phenyl-1
H-pyrazol-4-yl)ethanone
8 synthesized by refluxing 5-pyrazolone
7 with chloroacetyl chloride in a basic dioxane solution reacted with 2-aminobenzimidazole
9 to give N-(3-methyl-1-phenyl-1
H-furo [2,3-c]pyrazol-4(5
H)-ylidene)-1
H-benzimidazol-2-amine
10 (
Scheme 3) [
57]. Compound
10 showed a very good anti-Gram-positive profile, being equivalent to chloramphenicol against
B. subtilis (MIC 3.125 μg mL
−1), significant activity against
B. thuringiensis (MIC 6.25 μg mL
−1) and also good antibacterial activities against Gram-positive bacteria,
E. coli (MIC 50 μg mL
−1) and
P. aeruginosa (MIC 50 μg mL
−1). Antifungal activity of the compound
10 was 50% lower than cycloheximide in inhibitory the growth of
B. fabae and
F. oxysporum (MIC 6.25 μg mL
−1).
A similar condensation of chalcones
11a–
11k with intermediate hydrazide
12 in acetic acid, at 130 °C afforded new benzimidazole bearing pyrazoline derivatives
13a–
13k in excellent yields (
Scheme 4). All compounds showed antimicrobial activity against bacteria
E. coli MTCC443,
P. aeruginosa MTCC1688,
S. aureus MTCC 96,
S. pyogenes MTCC 442 and fungi
C. albicans MTCC 227,
A. niger MTCC 282 and
A. clavatus MTCC 1323. Compounds
13a–
13d showed the highest inhibition against almost all bacteria tested with values of minimum inhibitory concentrations of 25–50 mg mL
−1, while derivatives
13e–
13k had antifungal activity against almost all strains tested, with similar CMI values [
58]. Structure–activity relationship studies have shown that the presence of electron-withdrawing groups in the aromatic ring, like F, Cl, Br and NO
2, are responsible for increasing antimicrobial activity for most microorganisms tested.
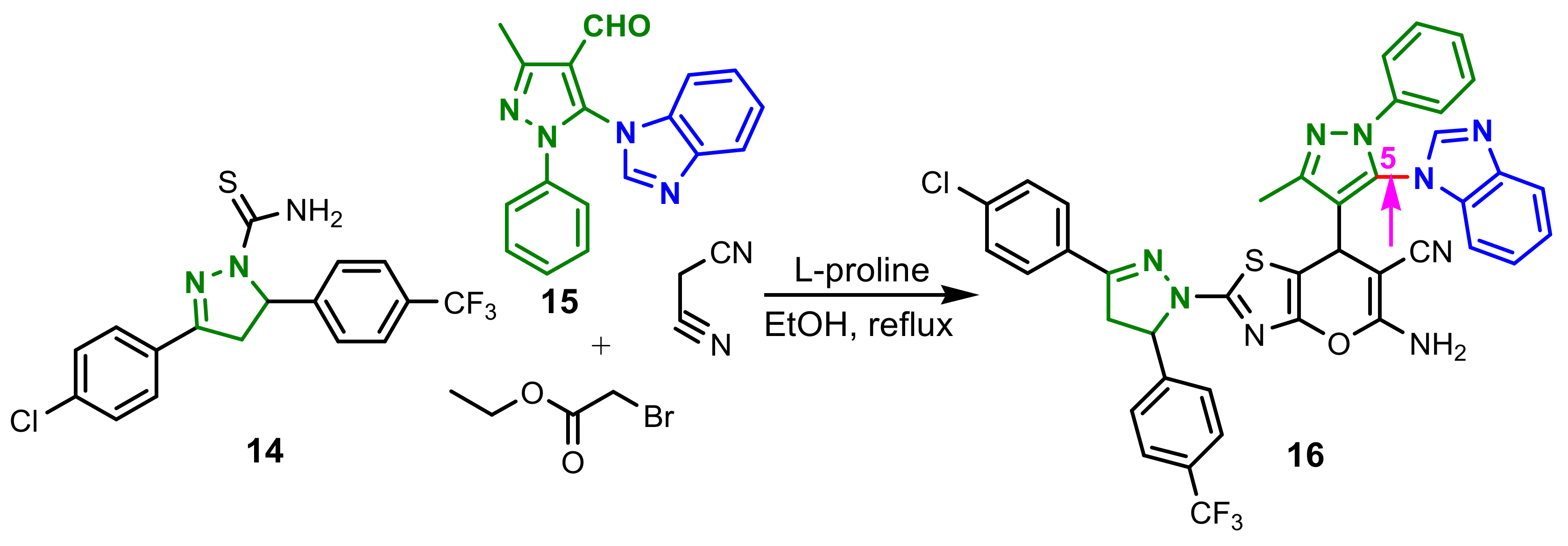
Kalaria et al., reported a L-proline promoted one-pot four-component tandem reaction for synthesis of compound
16, starting from carbothioamide
14, pyrazolyl aldehyde
15, α-bromoethylacetate and malononitrile (
Scheme 5) [
59]. Antibacterial activity of the compounds
16 was screened against three Gram-positive bacteria (
Streptococcus pneumoniae MTCC 1936,
Bacillus subtilis MTCC 441 and
Clostridium tetani MTCC 449) and three Gram-negative bacteria (
Escherichia coli MTCC 443,
Salmonella typhi MTCC 98,
Vibrio cholerae MTCC 3906) using ampicillin, norfloxacin and ciprofloxacin as the standard antibacterial drugs. Compound
16 illustrated an excellent activity against Gram-positive bacteria
B. subtilis (62.5 μg mL
−1), being more potent than ampicillin (250 μg mL
−1) and norfloxacin (100 μg mL
−1) and also against
C. tetani, with a CMI of 200 μg mL
−1 compared with 250 μg mL
−1 for ampicilin. Additionally, the structure–activity relationship (SAR) showed that the presence of benzimidazole in the fifth position in the pyrazole ring is responsible for its biological activity.
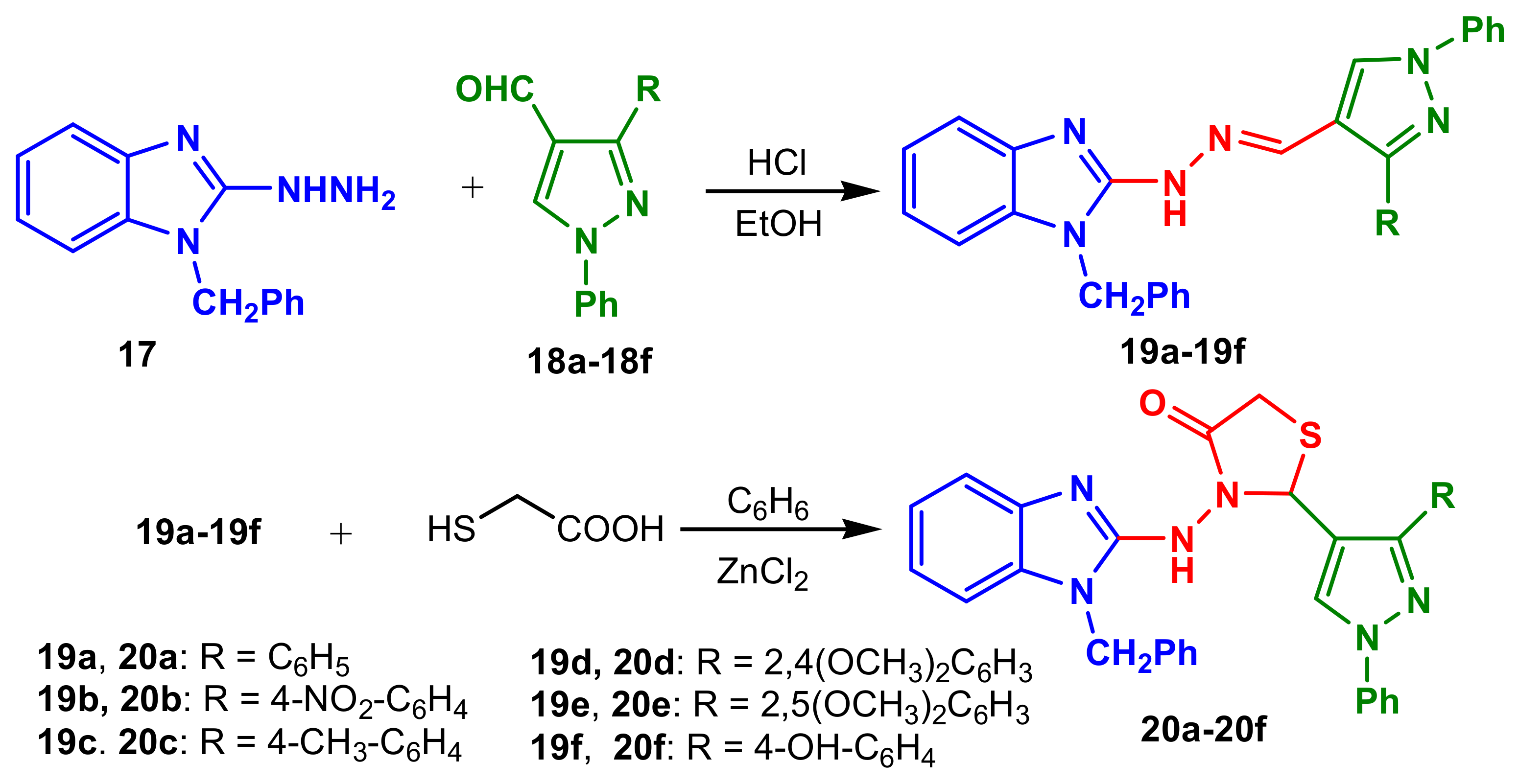
Patil et al., reported two series o benzimidazole–pyrazole compounds
19a–
19f and
20a–
20f in two steps: a condensation between 2-benzimidazolehydrazine
17 and pyrazole
18a–
18f, followed by cyclization with thioglicolic acid (
Scheme 6) [
60]. The compounds
19b,
19d,
20a and
20f show good activity against bacteria
P. aeruginosa,
S. aureus and
P. vulgaris, while the others show moderate to poor activity against all pathogens. The compounds
19a and
19c exhibited good activity against fungal strains
A. niger and
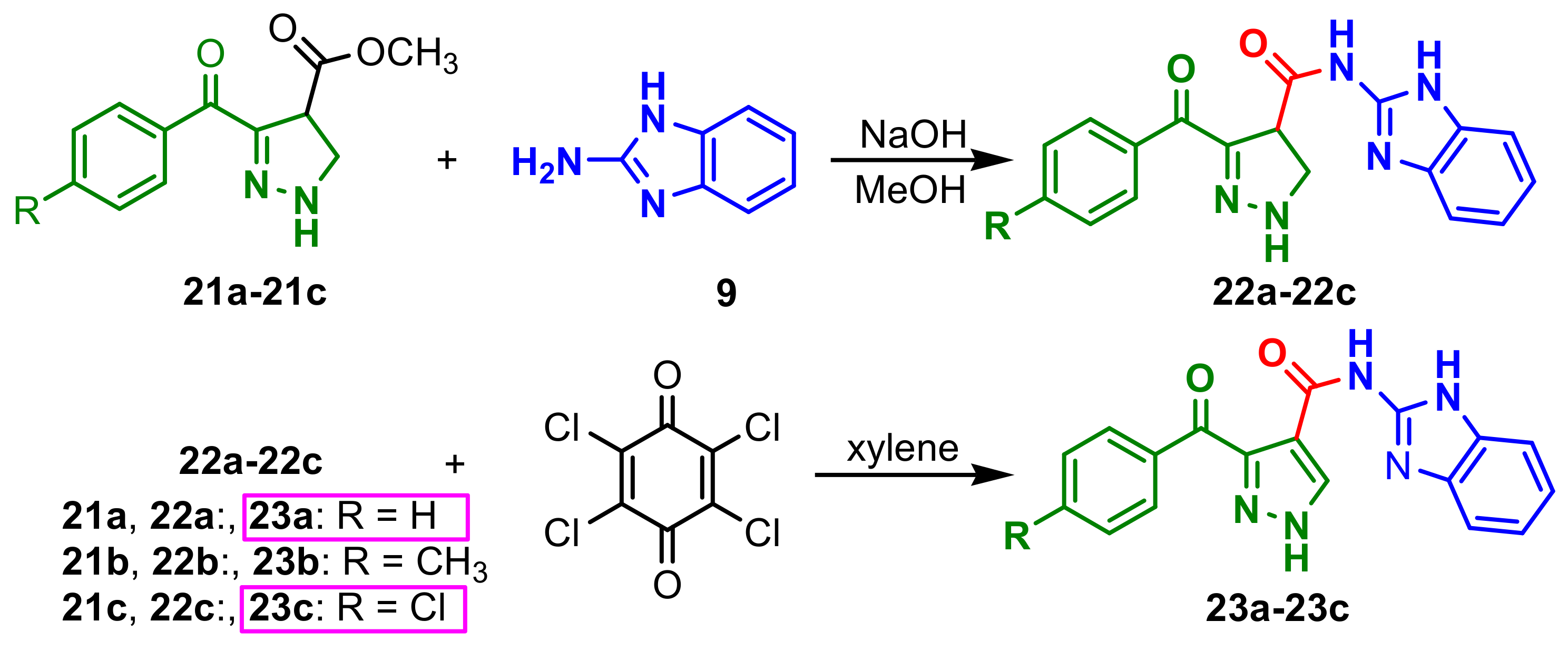
A. flavus.Reddy et al., reported the synthesis of a new class of pyrazolyl–benzimidazoles
23a–
23c possessing an amide group by reaction between pyrazolones
21a–
21c with 1
H-benzo[d]imidazol-2-amine
9, and the oxidation of the intermediate compounds
22a–
22c with chloranil (
Scheme 7) [
61]. It was found that the presence of electron-withdrawing substituent “Cl” on the aromatic ring increases the antimicrobial activity, compound
23c being a potent antifungal agent against
A. niger considering ketoconazole as standard. Additionally, compounds
23a and
23c possess antimicrobial activity against
B. subtilis and
P. aeruginosa (chloramphenicol standard).
Padalkar et al., synthesized a new class of antimicrobial agents, by reaction of phenyl hydrazine with substituted acetophenones
24 to give the corresponding hydrazones
25, which on Vilsmeier–Haack reaction with POCl
3–DMF gave substituted 3-aryl-4-formyl pyrazoles
26. Compounds
25a–
25b were condensed with o-substituted aromatic amines
27 in the presence of PCl
3 in ethanol to obtain corresponding 2-[substituted-1
H-pyrazol-4-yl]-1
H-benzimidazoles
28b–
28i (
Scheme 8) [
62]. The compound
28g showed good antibacterial activity against
Escherichia coli and
Staphylococcus aureus, and compounds
28d,
28e,
28h exhibited weak to moderate growth inhibitory activity against both
E. coli and
S. aureus as revealed from their MIC values (
Table 1). Compounds
28f and
28h show good inhibitory growth in the case of
Candida albicans (MIC = 62.5 μg mL
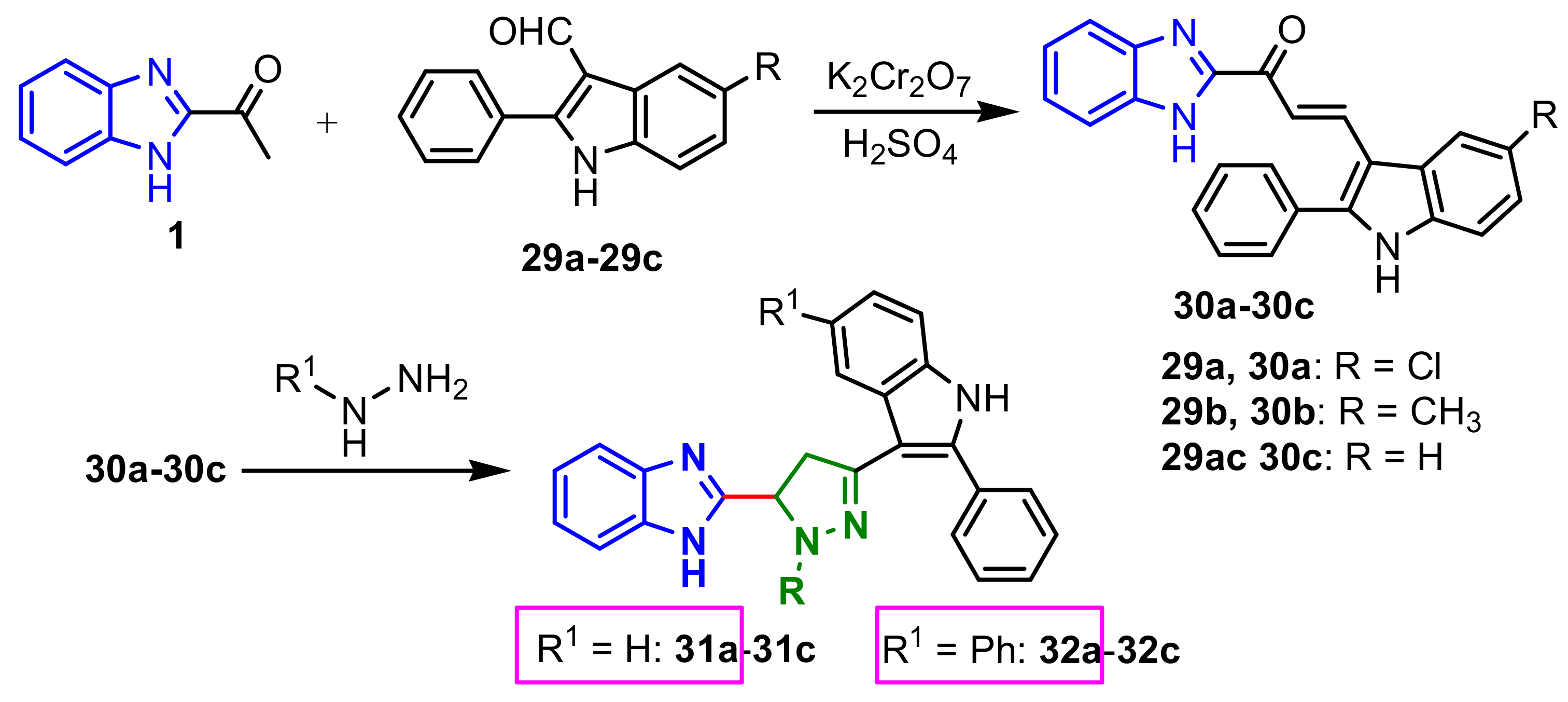
−1). Saundane et al., reported the synthesis of a series of benzimidazole–pyrazole compounds using a two-step strategy (
Scheme 9): synthesis of intermediate chalcones
25a–
25b by a condensation reaction, followed by a cyclization reaction with hydazine (compounds
31) or phenylhydrazine (compounds
32) [
63]. All compounds were assessed for their in vitro antibacterial activity against four representative bacterial species
E. coli (MTCC-723),
S. aureus (ATCC-29513),
K. pneumonia (NCTC-13368) and
P. aeruginosa (MTCC-1688) using gentamycin as a reference and for their antifungal activity against
A. oryzae (MTCC-3567T),
A. niger (MTCC-281),
A. flavus (MTCC-1973),
A. terreus (MTCC-1782). Compounds
31a and
32a possess good antibacterial and antifungal activity (
Table 2), against
E. coli,
S. aureus (MIC = 8 μg mL
−1) and
A. niger (MIC = 8 μg mL
−1 for
31a). Additionally, all compounds possess antioxidant activity.
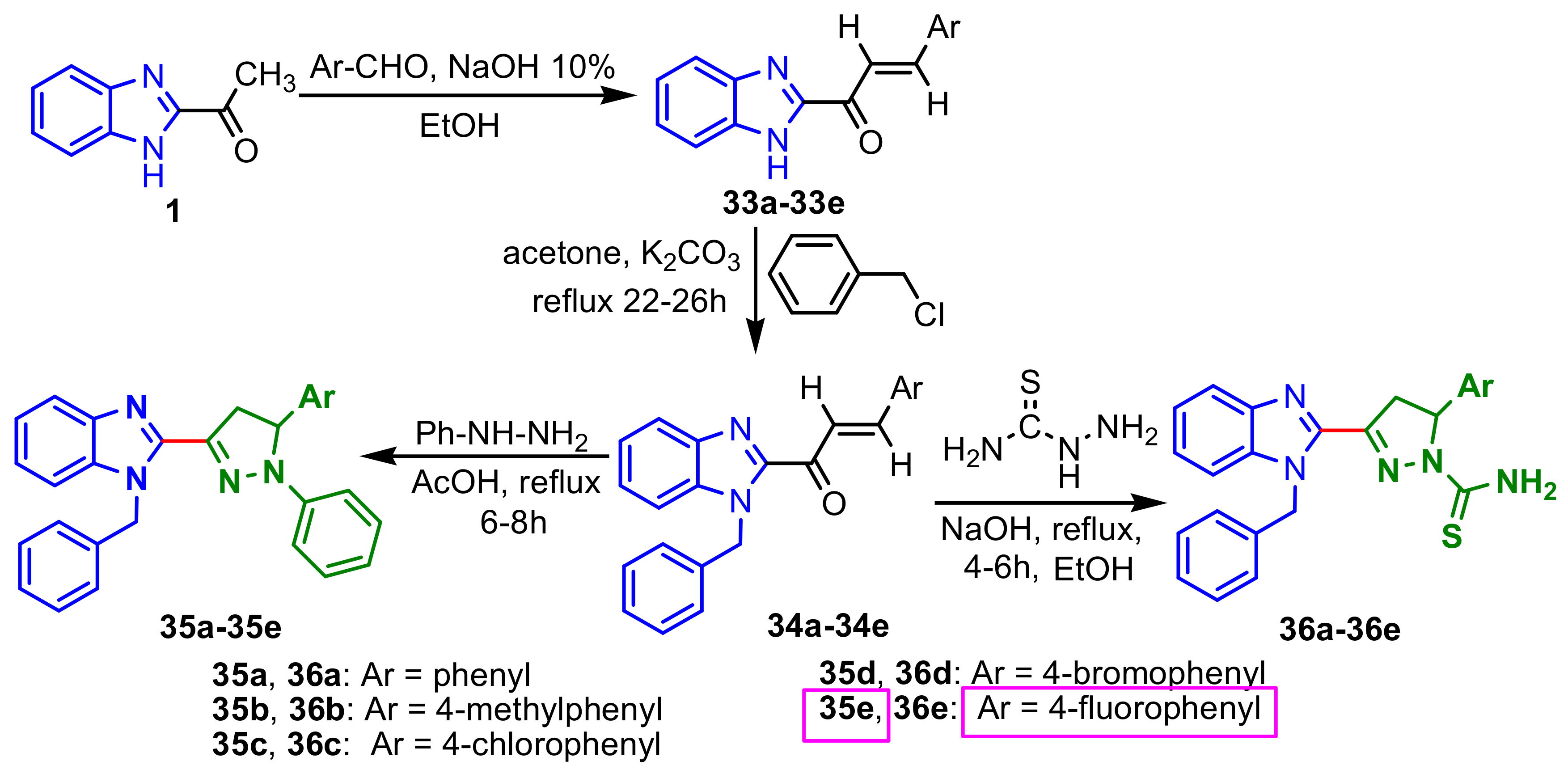
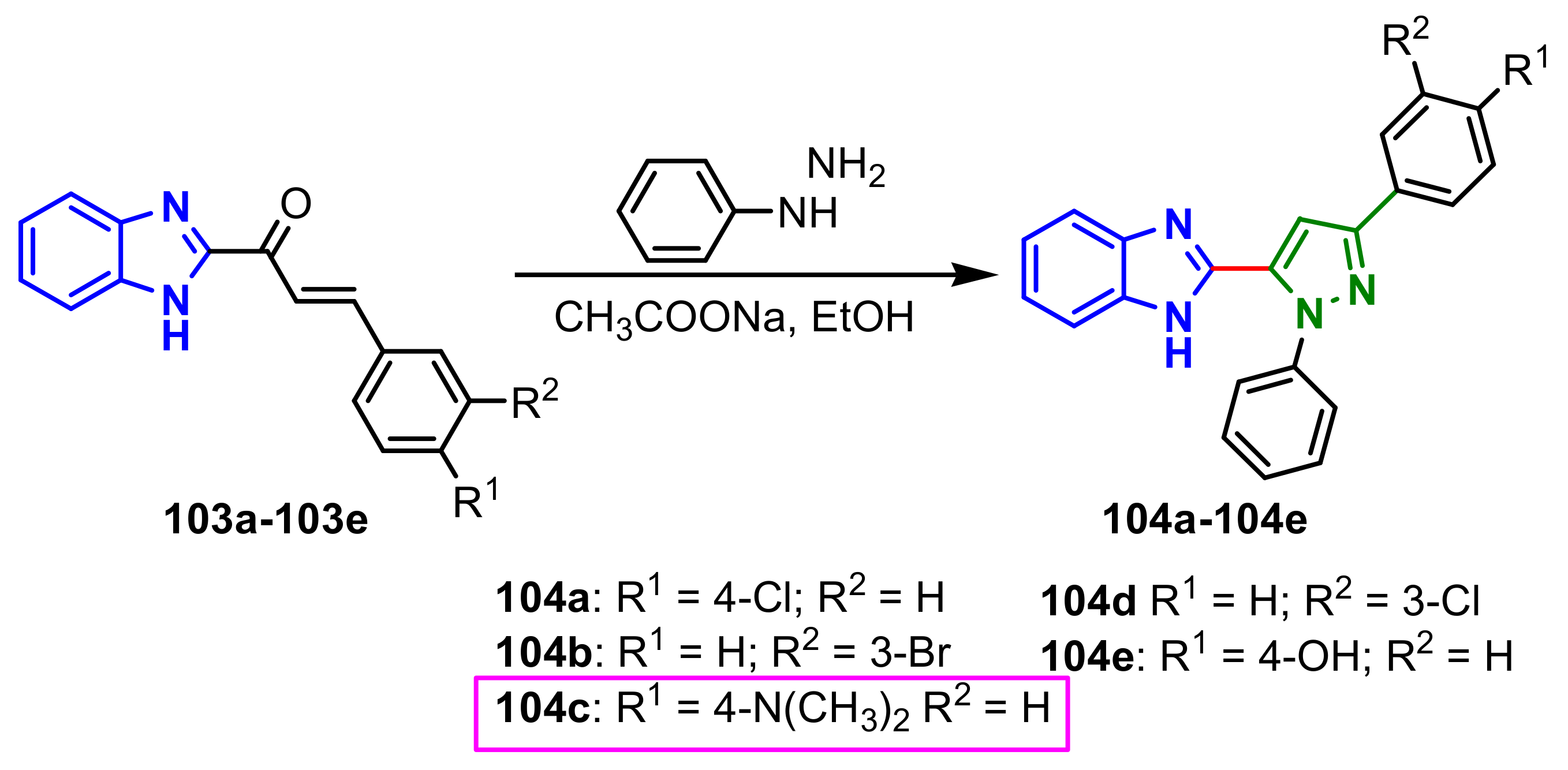
Padhy et al., synthesized two series of benzimidazole–pyrazole compounds in three steps: (i) Claisen–Schmidt condensation of 2-acetylbezimidazole
1 with substituted aromatic aldehydes in presence of NaOH, to give the intermediates chalcones
33a–
33e; (ii) condensation of the chalcones
33 with benzyl chloride gave the corresponding 1-benzyl substituted compounds
34a–
34e; (iii) the reaction of compounds
34 with phenylhydrazine in the presence of acetic acid afforded 1-benzyl-2-(5-aryl-1-phenyl-4,5-dihydro-1
H-pyrazol-3-yl)-1
H-benzimidazoles
35a–
35e, while (iv) condensation with thiosemicarbazide in presence of NaOH, give 5-aryl-3-(1-benzyl-1
H-benzimidazol-2-yl)-4,5-dihydro-1
H-pyrazole-1-carbothioamides
36a–
36e in good yields (
Scheme 10). The in vitro antimicrobial activity of compounds
35–
36 was tested against four bacterial strains,
S. aureus,
B. subtilis,
E. coli,
P. aeruginosa, and one fungus,
C. albicans. The compounds exhibited weaker antimicrobial activities compared to those of the control drugs (Ciprofloxacin and Fluconazole), the MIC values of the compounds ranged between 64–1024 μg mL
–1 for the 1-phenylpyrazolines
35a–
35e and between 128–512 μg mL
–1 for the pyrazoline-1-carbothioamides
36a–
36e. Compound
35e showed good activity (64 μg mL
–1) against all tested bacterial strains [
64].
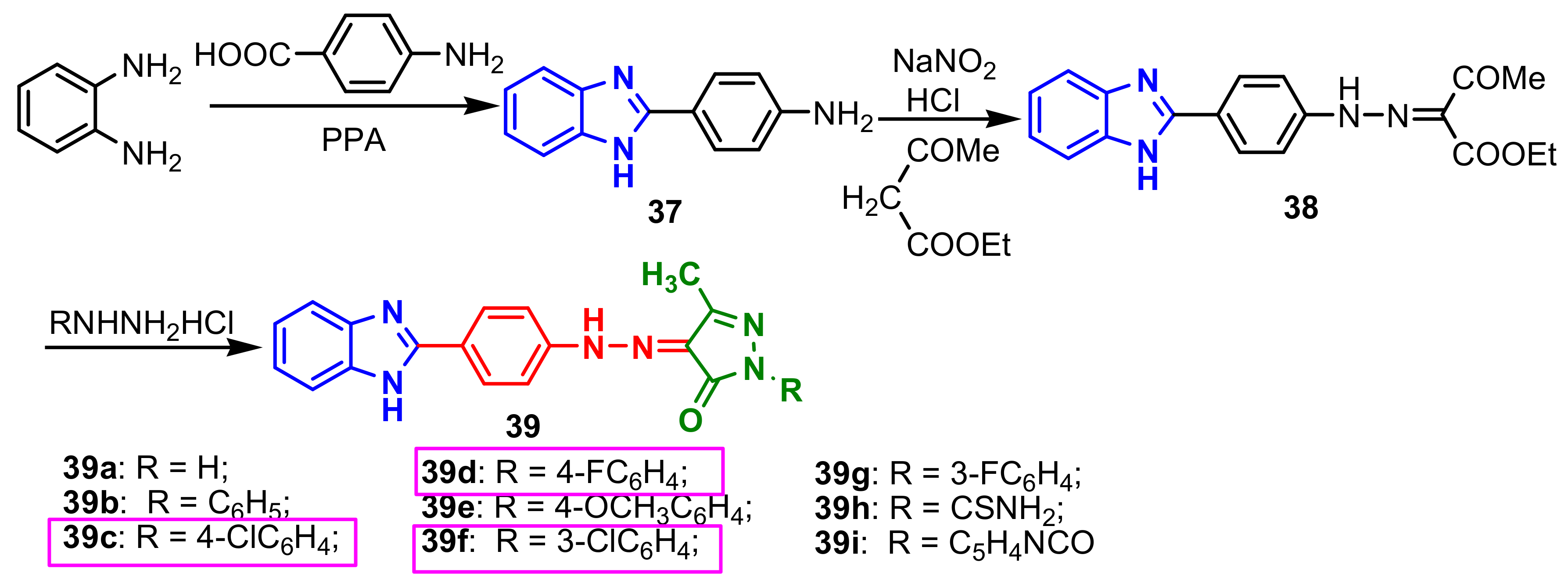
4-(1
H-benzimidazol-2-yl)benzenamine
37, obtained by cyclization reaction of 1,2-phenylenediamine with 4-amino benzoic acid, was diazotized and treated with ethylacetoacetate to produce ethyl 2-(2-(4-(1
H-benzimidazol-2-yl)phenyl) hydrazono)-3-oxobutanoate
38 through intramolecular rearrangement reaction. Dehydrative cyclisation of
38 in the with different hydrazine hydrochlorides produce corresponding benzimidazole–pyrazole
39a–
39i (
Scheme 11) [
65]. The antitubercular and antimicrobial activity of compounds
39 was determined on four Gram-positive strains, three Gram-negative strains and 2 fungi. In
Table 3 we marked in green the very good antimicrobial activities of compounds
39c and
39f, as well as of the compounds
3d and
3g, for the labeled strains, and the very good antibacterial activities of all compounds against
Staphylococcus aureus. The values of the minimum inhibitory concentrations (MIC) in
Table 3 showed that compounds,
39c and
39f, possess almost all MICs as good as the standards used for antitubercular and antimicrobial activities, and their antifungal activities are twice as high, compared to Ketoconazole, ie 3.9 μg mL
–1 against
Aspergillus niger ATCC 9029 and 1.95 μg mL
–1 against
Aspergillus fumigatus ATCC 46645. With the exception of compounds
39f,
39h and
39i, all compounds showed better antibacterial activity (MIC = 125 μg mL
–1) than standard, Ciprofloxacin, compounds
39c and
39d being 16 times more active(MIC = 7.81 μg mL
–1) than standard.
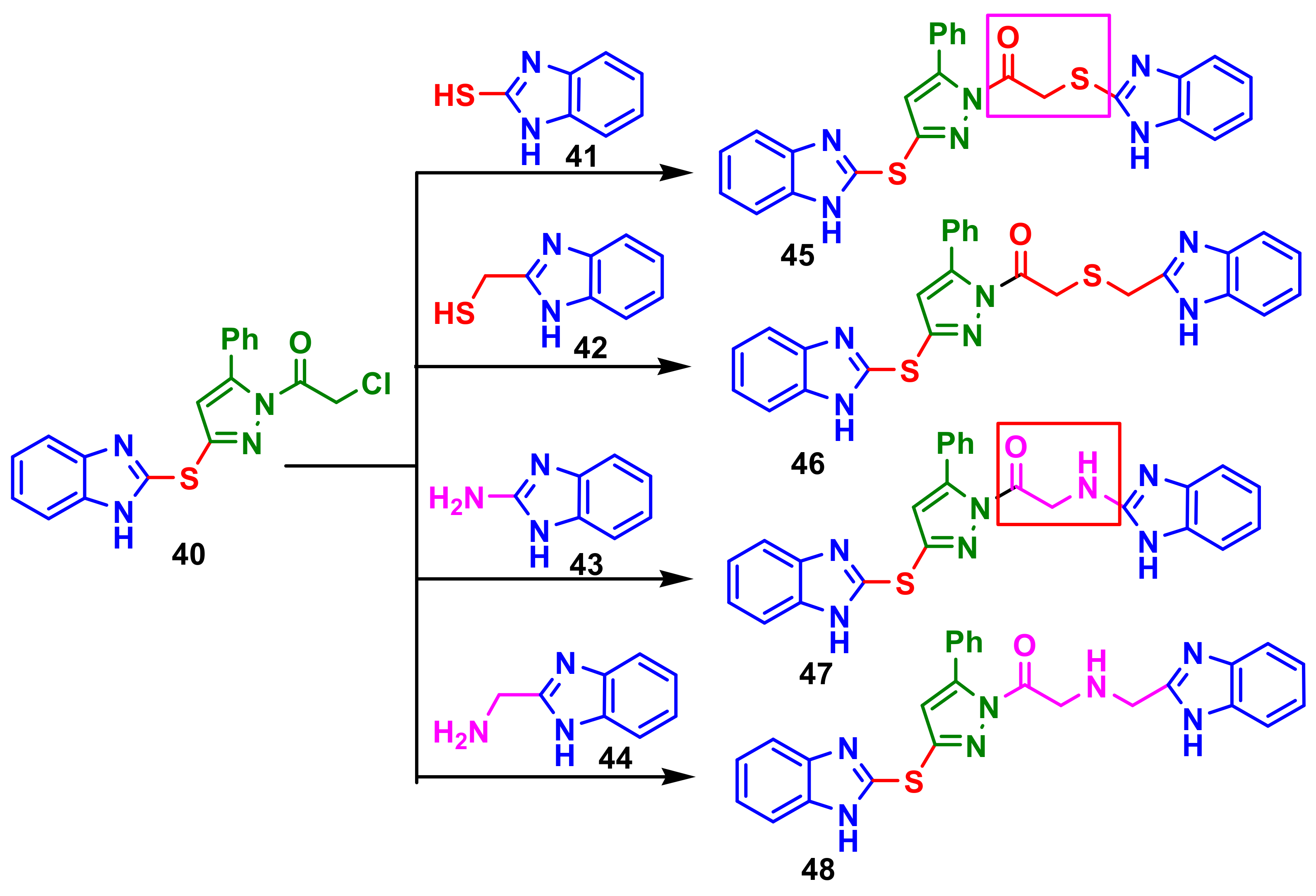
Suram et al., reported the synthesis of a series of bis(benzimidazolyl)pyrazole compounds from chlororacetylpyrazole-benzimidazole
40 and benzimidazoles
41–
44, to obtain compounds
45–
48 (
Scheme 12) [
66]. It was observed that the compound with
thio ethanone linkage
45 and
amino ethanone linkage
47 displayed slightly higher activity than that with methyl thio ethanone
46 and methyl amino ethanone linkage
48 on the microbial tested strains,
S. aureus,
B. subtilis,
P. aeruginosa,
K. pneumoniae,
A. niger and
P. chrysogenum, when compared with the standard drugs chloramphenicol and ketoconazole.
A new class of benzimidazole–pyrazoles was prepared using a Claisen–Schmidt reaction [
67]. From all synthesized compounds, derivative
51, obtained by cyclocondensation reaction of thioamide
49 with 4-fluorophenacyl bromide
50 (
Scheme 13), having nitro substituent on the aromatic ring showed greater antimicrobial activity particularly against
Pseudomonas aeruginosa, with an inhibition zone of 34 mm at 100 μg per well, and
Penicillium chrysogenum, with an inhibition zone of 41 mm at 100 μg per well.
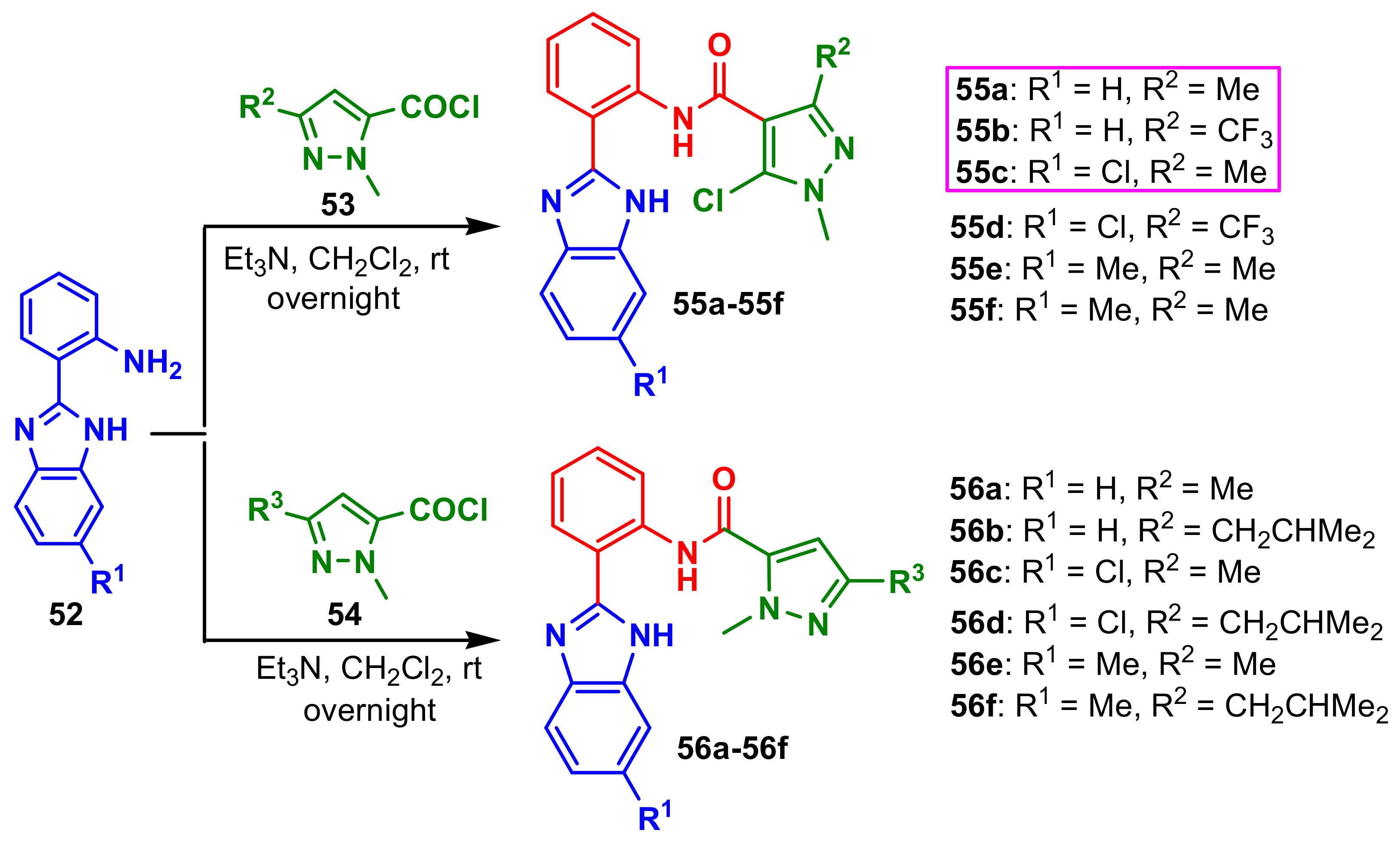
Si et al., synthesized two series of benzimidazole–pyrazoles by reaction of benzimidazole
52 with pyrazole-5-carbonyl chlorides
53 and
54 to afford the final compounds
55a–
55f and
56a–
56f (
Scheme 14) [
68]. The authors reported the antifungal activities against four fungi,
B. cinerea,
R. solani,
F. graminearum,
A. solani, considering hymexazol as the positive control at 100 μg mL
−1 (
Table 4). All compounds showed better inhibitory activity against
B. cinerea. The inhibition rates of compounds
55a–
55f exceeded 60% against
R. solani and the inhibition rates of compounds
55a–
55f ranged from 58.28% to 68.28% against
A. solani, which were better than 55.43% of the control hymexazol. The compounds with pyrazole-4-carboxamide moiety
55a–
55f showed higher activities than the target compounds with pyrazole-5-carboxamide moiety
56a–
56f. Thus, the activities of
55a and
55b were better than those of
56a and
56b and the activities of
55c and
55d were better than those of
56c and
56d.
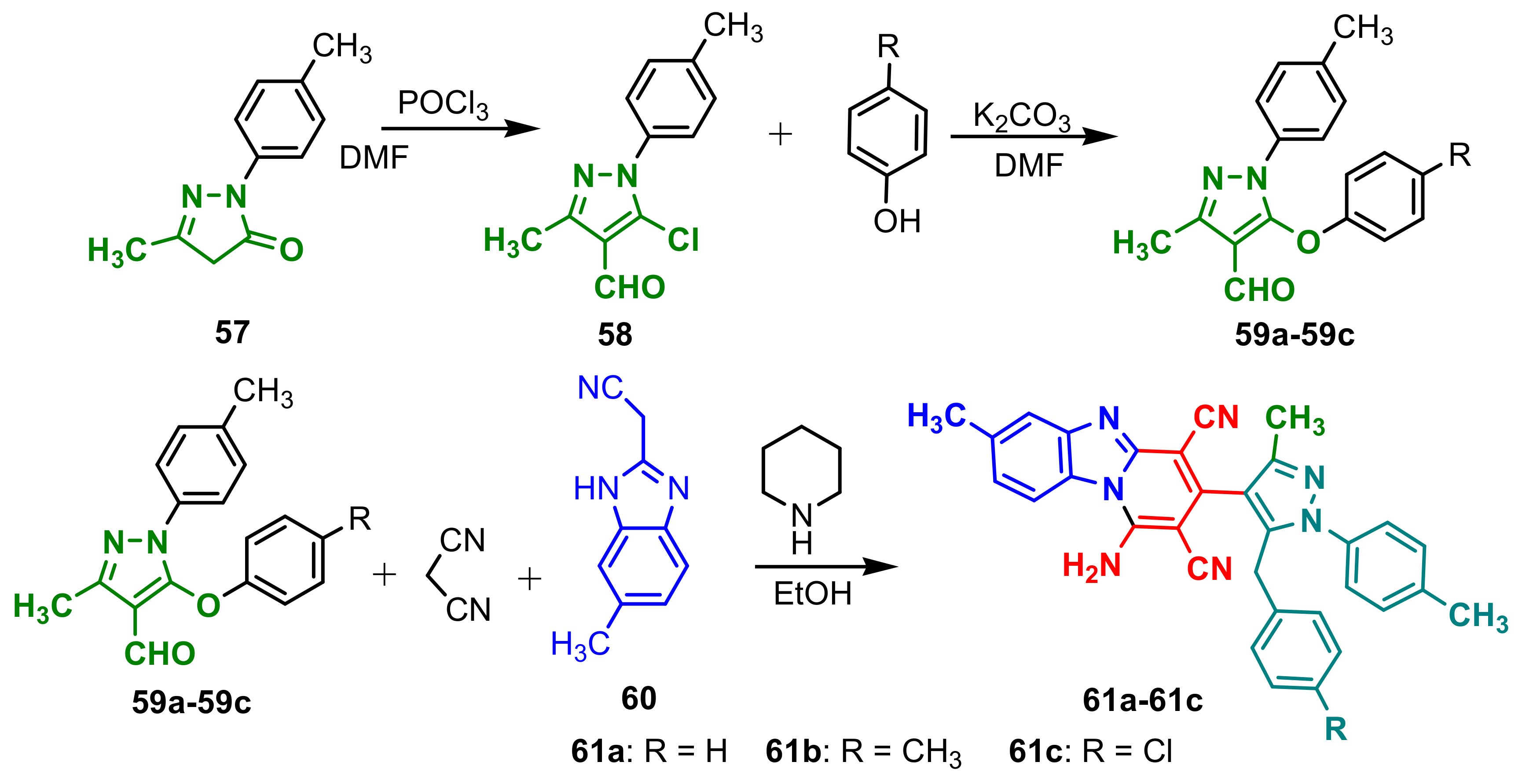
Jardosh et al., synthesized new pyrido[1,2-a]benzimidazoles starting chloroformilation and alkilation of 4-methyl-2-
p-tolylcyclopent-3-enone
57. In the next step, a one-pot three-component reaction was used to afford final compounds
61a–
61c (
Scheme 15). The in vitro antimicrobial activity of
61a–
61c against
S. typhi,
S. pneumoniae,
E. coli,
C. tetani,
V. cholera,
B. subtilis,
C. albicans and
A. fumigatus using broth microdilution technique was assessed. All compounds
61a–
61c displayed good antimicrobial activity compared to standard drugs, as can be seen in
Table 5 [
69].
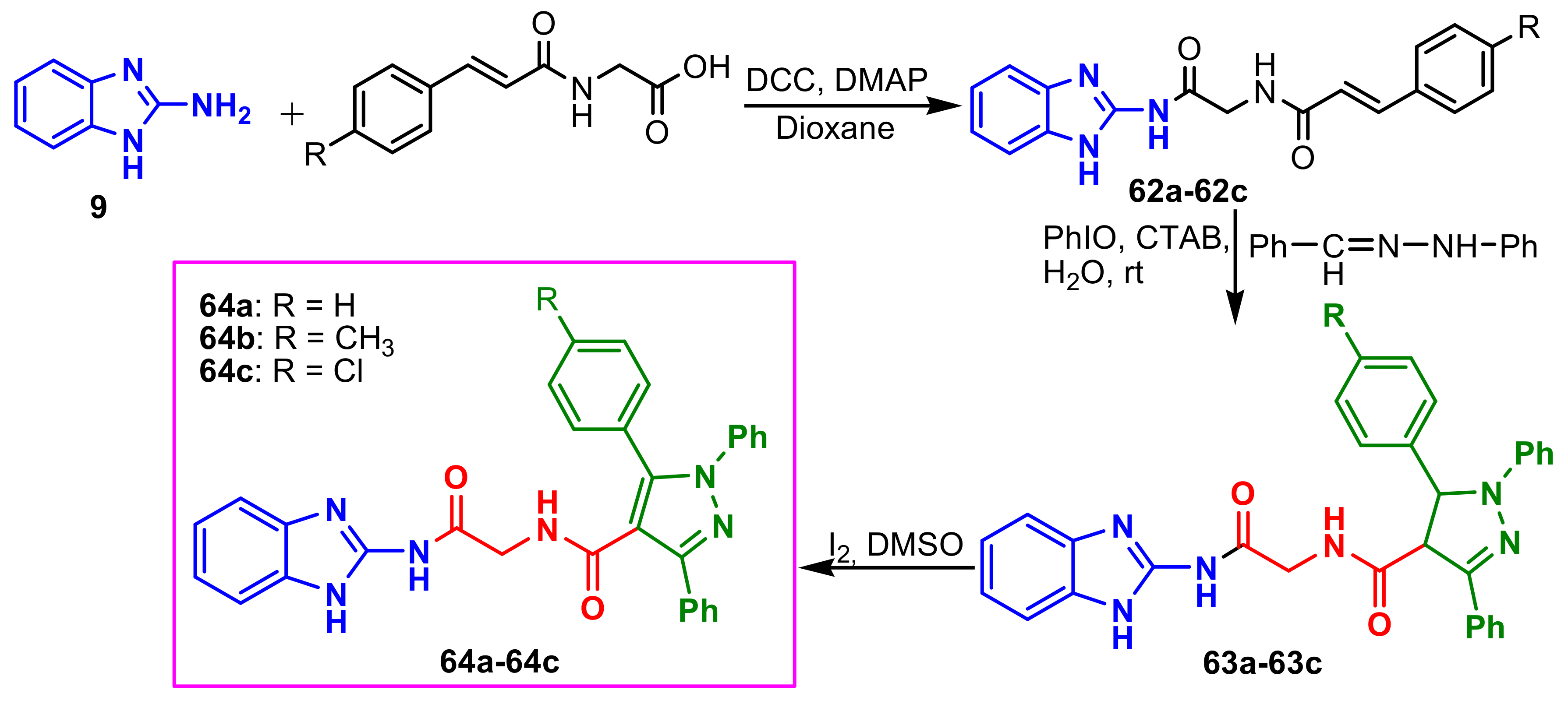
Sowdari et al., synthesized a new class of diamidomethane-linked benzazolyl–pyrazoles
64a–
64c by a green approach, using the synthesis strategy indicated in
Scheme 16 [
70]. Compounds
64a and
64c were found to be potential antifungal agents against
Aspergillus niger (MIC = 50 and 25 μg mL
−1, respectively) and
Penicillium chrysogenum (MIC = 12.5 and 12.5 μg mL
−1, respectively) compared to the standard drug, Ketoconazole.
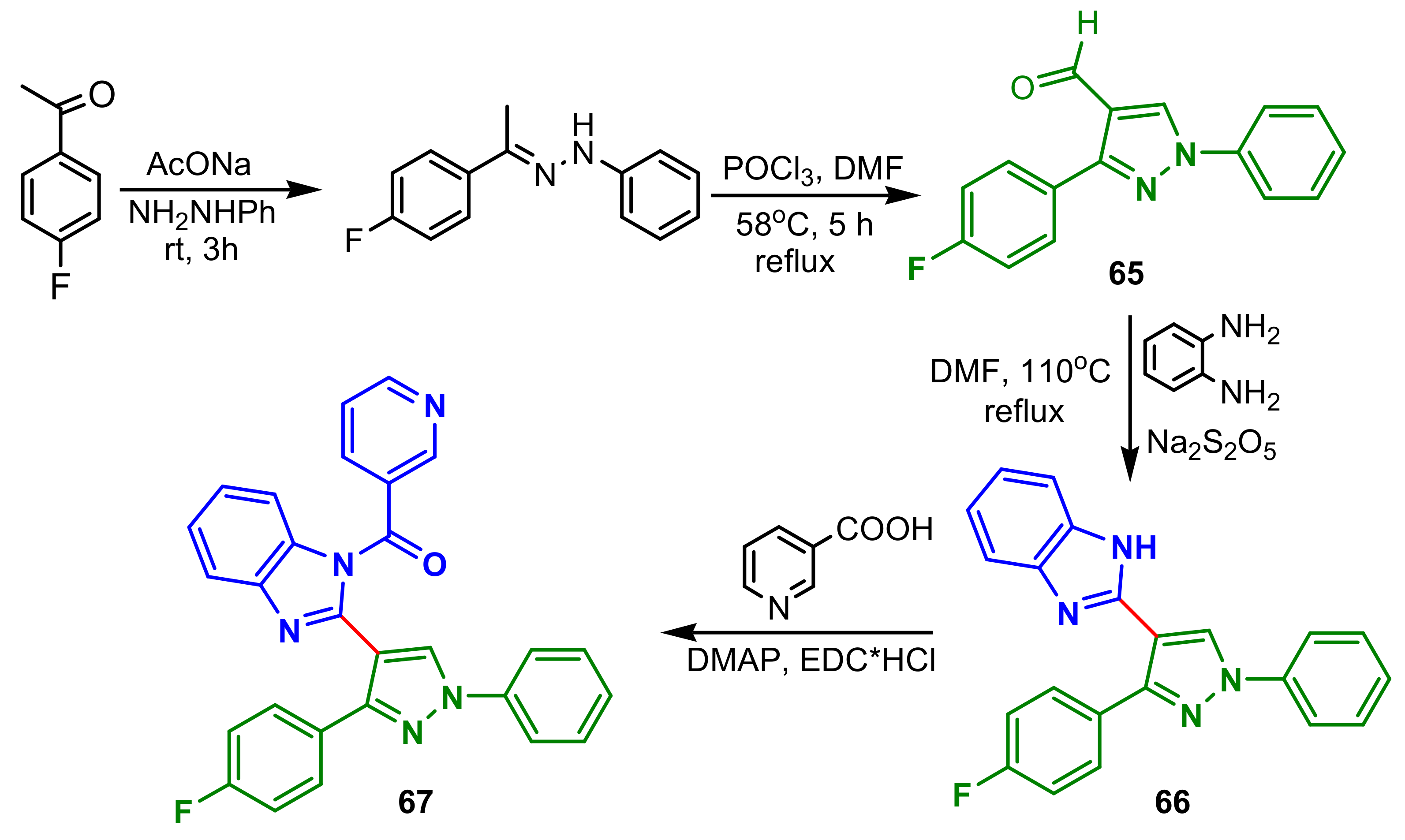
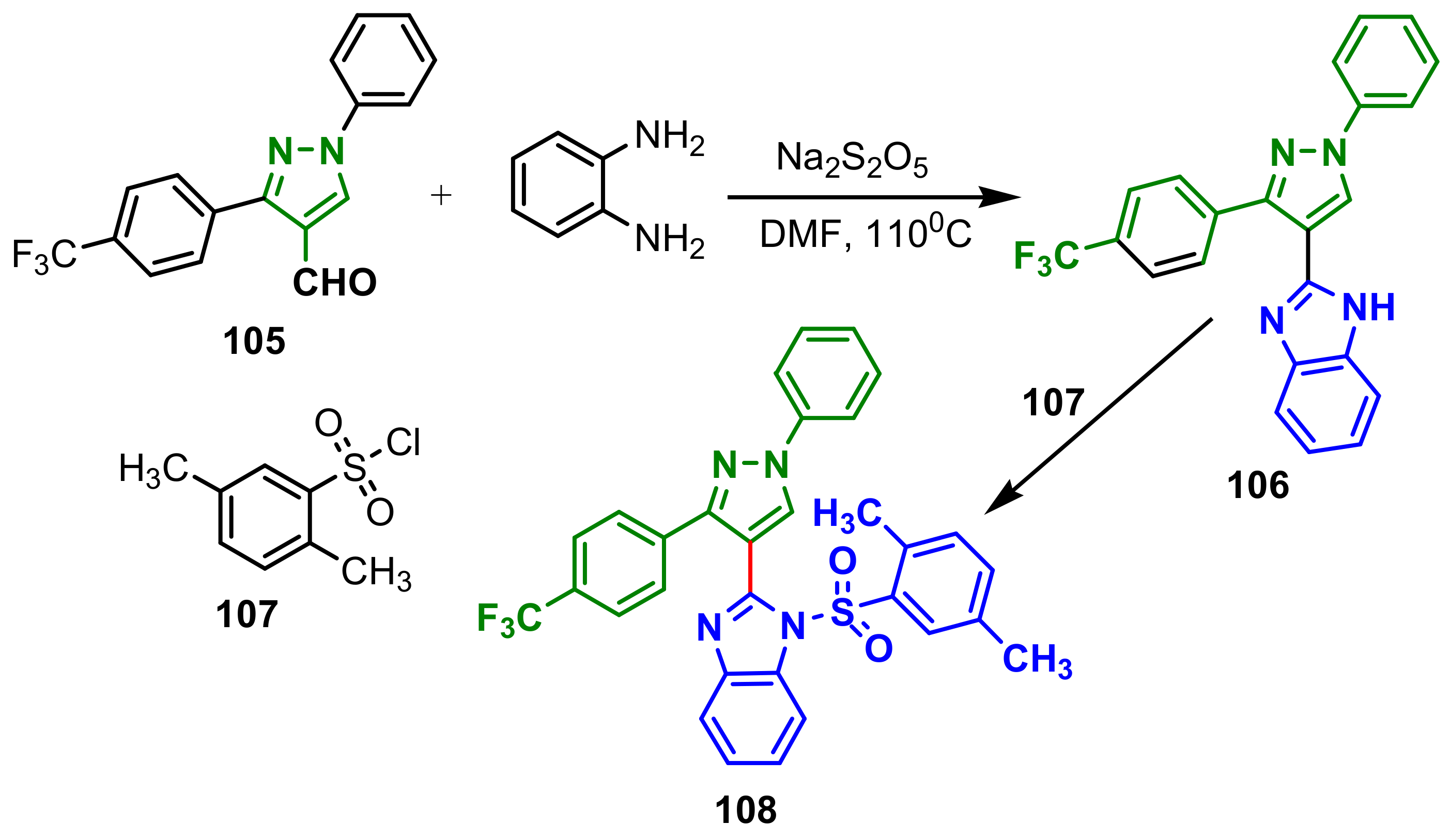
β-ketoacyl-acyl carrier protein synthase III (FabH) is an attractive target for the development of new antibacterial agents, because it catalyzes the initial step of fatty acid biosynthesis, essential for bacterial survival. Thus, Wang et al., reported the synthesis of a new series of benzimidazole–pyrazol amides with low toxicity and potent FabH inhibitory. Synthesis of compound
67 from 1-(4-fluorophenyl)ethanone is accomplished in four steps: condensation with phenylhydrazine, followed by cyclization by reflux with POCl
3 in DMF for 5 h, to obtain pyrazole
65, which with 1,2-phenylenediamine and Na
2S
2O
5 has provided benzimidazole–pyrazole intermediate
66, which by acylation with nicotinic acid, DMAP and EDC hydrochloride led to the final product
67 (
Scheme 17). Compound
67 showed the most potent inhibition activity against four bacteria strains (with MIC of 0.98, 0.49, 0.98, 0.98 μg mL
−1, respectively, against
E. coli,
P. aeruginosa,
B. subtilis and
S. aureus) and FabH (with IC
50 of 1.22 μM). Additionally, FabH mutant
Xanthomonas Campestris experiment validated that compounds binding site outcomes FabH.
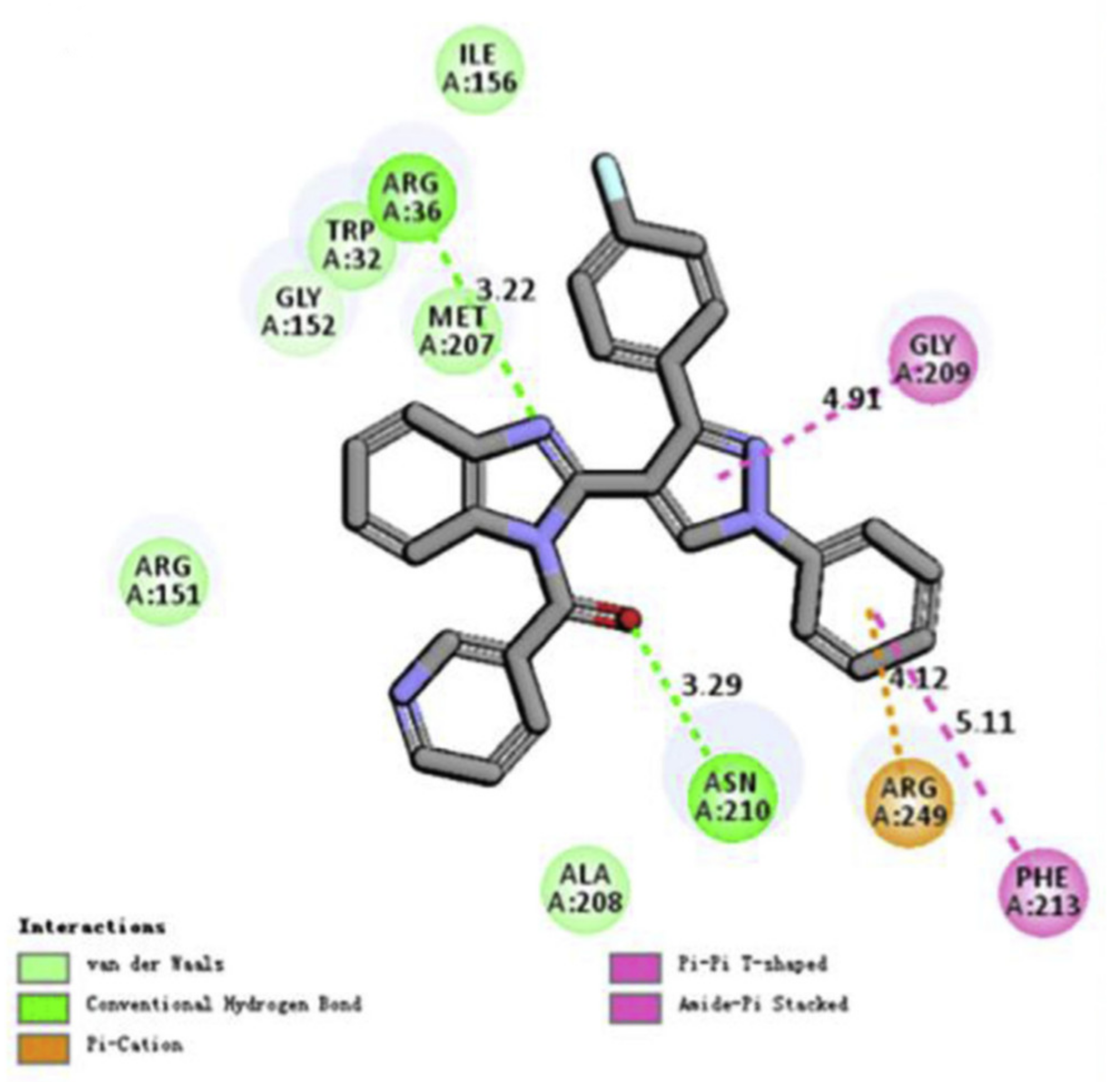
2D molecular docking modeling and surrounding residues of
E. coli FabH was also performed for compound
67 (
Figure 2) [
71].
A special method for the synthesis of a benzimidazolo–pyrazole compound was reported by Chkirate et al., [
72]. Thus, condensation of 1,2-phenylenediamine with dehydroacetic acid afford
68 which reacted with 1-bromobutane to give the alkylated 1,5-benzodiazepine
69. Compound
69 reacts with an excess of hydrazine monohydrate to afford the pyrazolyl–benzimidazole
70 (
Scheme 18). The minimum inhibitory concentration (MIC) of
70 against
S. aureus,
E. coli and
P. aeruginosa was evaluated at 12.5 μg mL
−1, 50 μg mL
−1 and 50 μg mL
−1, respectively, compared to standard drug Chloramphenicol. Additionally, Co(II) and Zn(II) complexes of
70 possess remarkable antibacterial activity.
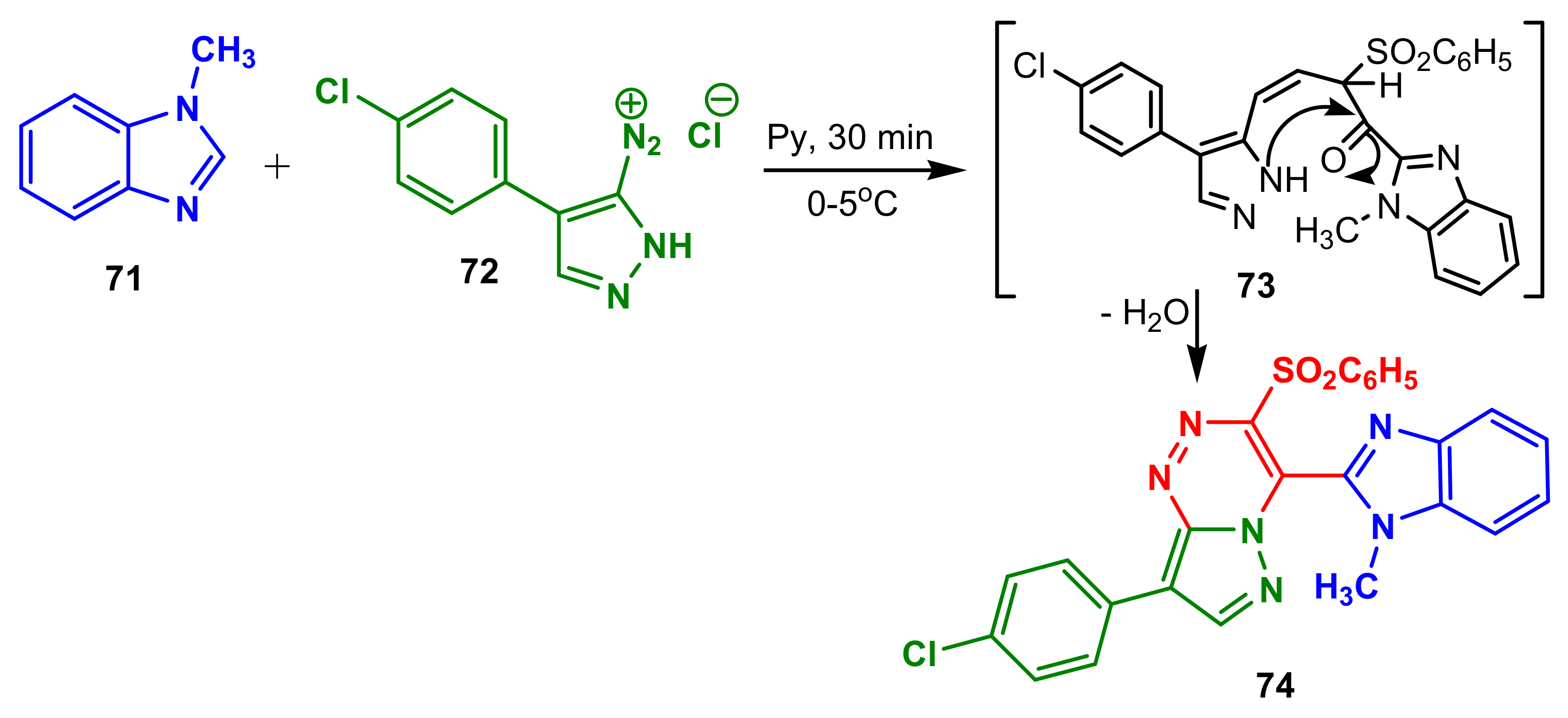
Elaziz et al., synthesized benzimidazole–pyrazole
74 from 1-methylbenzimdazole
71 and diazonium salt
72, through the intermediate
73 [73]. Compound
74 possessed better antibacterial activity than standard Cephalothin against anaerobic
E. coli (16.5 μg mL
−1 versus 24.3 μg mL
−1),
Salmonella typhimurium (13.4 μg mL
−1 versus 28.5 μg mL
−1), and better antibacterial activity than standard Chloramphenicol against
Bacillus subtilis (23.3 μg mL
−1 versus 32.4 μg mL
−1,
Scheme 19,
Table 6).
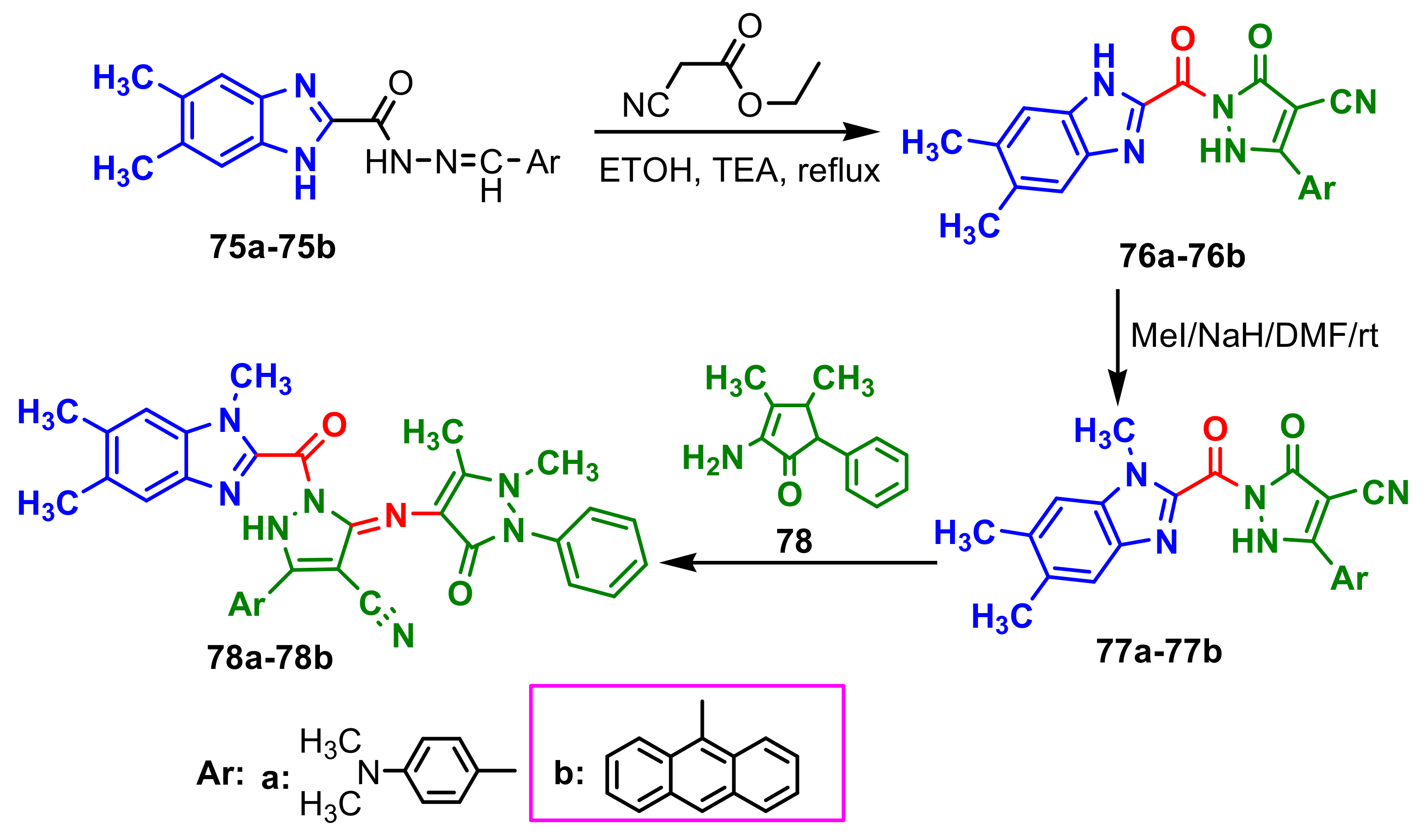
Bassyouni et al., synthesized three series of benzimidazole–pyrazoles
76–
78b [74]. Compounds
76a and
76b were synthesized by the reaction of
75a and
75b with ethyl cyanoacetate in ethanol in the presence of triethylamine, respectively (
Scheme 20). Methylation of
76a and
76b was achieved by their reaction with methyl iodide or DMC that yielded compounds
77a and
77b. Compounds
77a and
77b reacted with 4-aminoantipyrine in ethanol, in the presence of catalytic amounts of acetic acid to give
78a and
78b. The antibacterial activity of the compounds
76a,
76b,
77b,
78a and
78b was examined with Gram-positive bacteria
Bacillus subtilis,
Bacillus cereus and
Staphylococcus aureus, Gram-negative bacteria
Escherichia coli,
Pseudomonas aeruginosa and
Salmonella typhimurium. The antibacterial activity showed that compound
76a was the most active against
S. typhimurium and its activity exceeded the activity of the reference antibiotic amoxicillin. Compounds
77b and
78b exhibited high antimicrobial activity against
S. aureus (
Table 7).
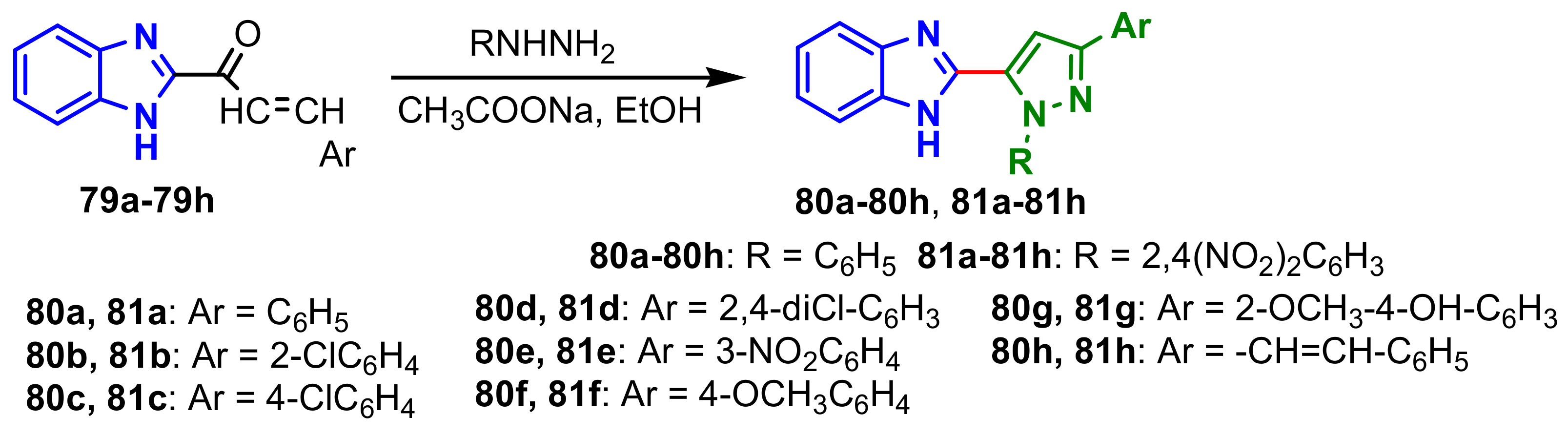
Benzimidazolo–pyrazole compounds
80a–
80h and
81a–
81h were synthesized from the reaction of chalcones
79a–
79h with phenylhydrazine and 2,4-dinitrophenylhydrazine, respectively (
Scheme 21) [
75]. All compounds were screened for their antimicrobial activities against
E. coli,
P. aeruginosa,
S. aureus,
B. subtilis,
C. albicans and
A. niger (
Table 8). The best antimicrobial activities of the compounds are marked in green in
Table 8. It is observed that compounds
80b and
80h showed a good antibacterial activity against all the strains tested, and compounds with 2,4-dinitrophenylhydrazine had better antifungal activity than the antibacterial one, e.g., compounds
81b and
81f. Only compound
80a showed significant antitubercular activity at the concentration of 100μg mL
−1 compared with the standard drug, Rifampicin.
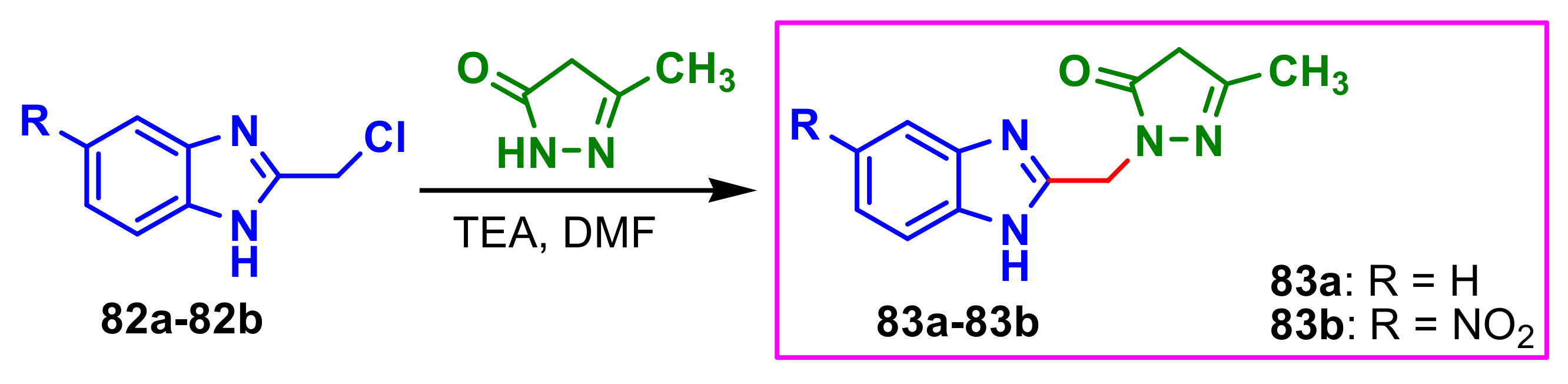
El-Gohary et al., synthesized benzimidazole–pyrazole molecules
83a–
83b with antimicrobial properties, using the reaction between benzimidazoles
82a–
82b and 3-methyl-1
H-pyrazol-5(4
H)-one in dimethylformamide (DMF), in presence of triethyl-amine (TEA) as catalyst (
Scheme 22) [
76]. The compounds
83a–
83b showed very good antimicrobial activity against two bacteria
B. cereus and
S. aureus, against two fungi,
C. albicans and
A. fumigatus, compared to the standards used, ampicillin and fluconazole (
Table 9).
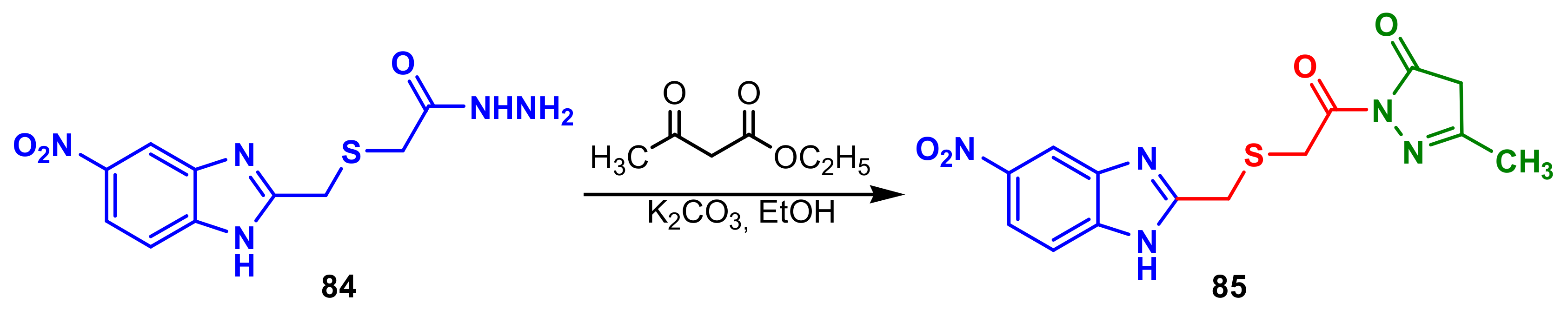
The benzimidazole–pyrazole
85 synthesized by cyclization of benzimidazole
84 in the reaction with of ethyl 3-oxobutanoate (
Scheme 23), possessed good antifungal activity, against
C. albicans (MIC = 2500 μg mL
−1) compared with standard Fluconazole (MIC = 2500 μg mL
−1) [
77].







































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
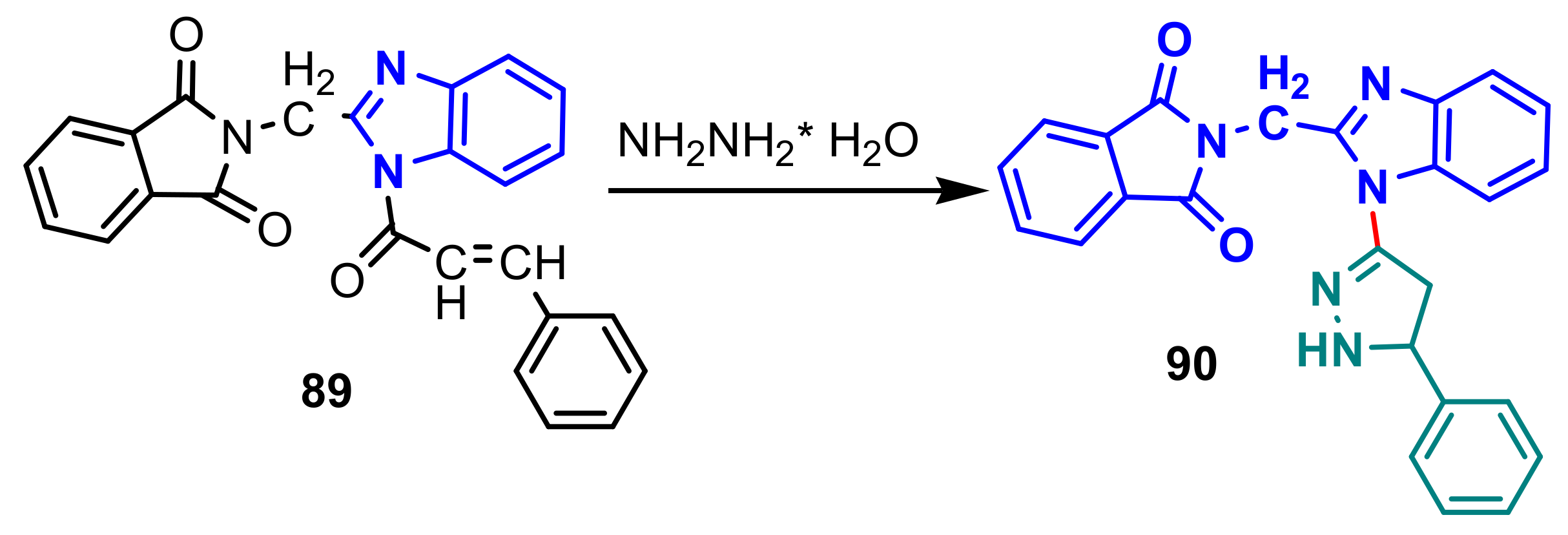{kind=link}
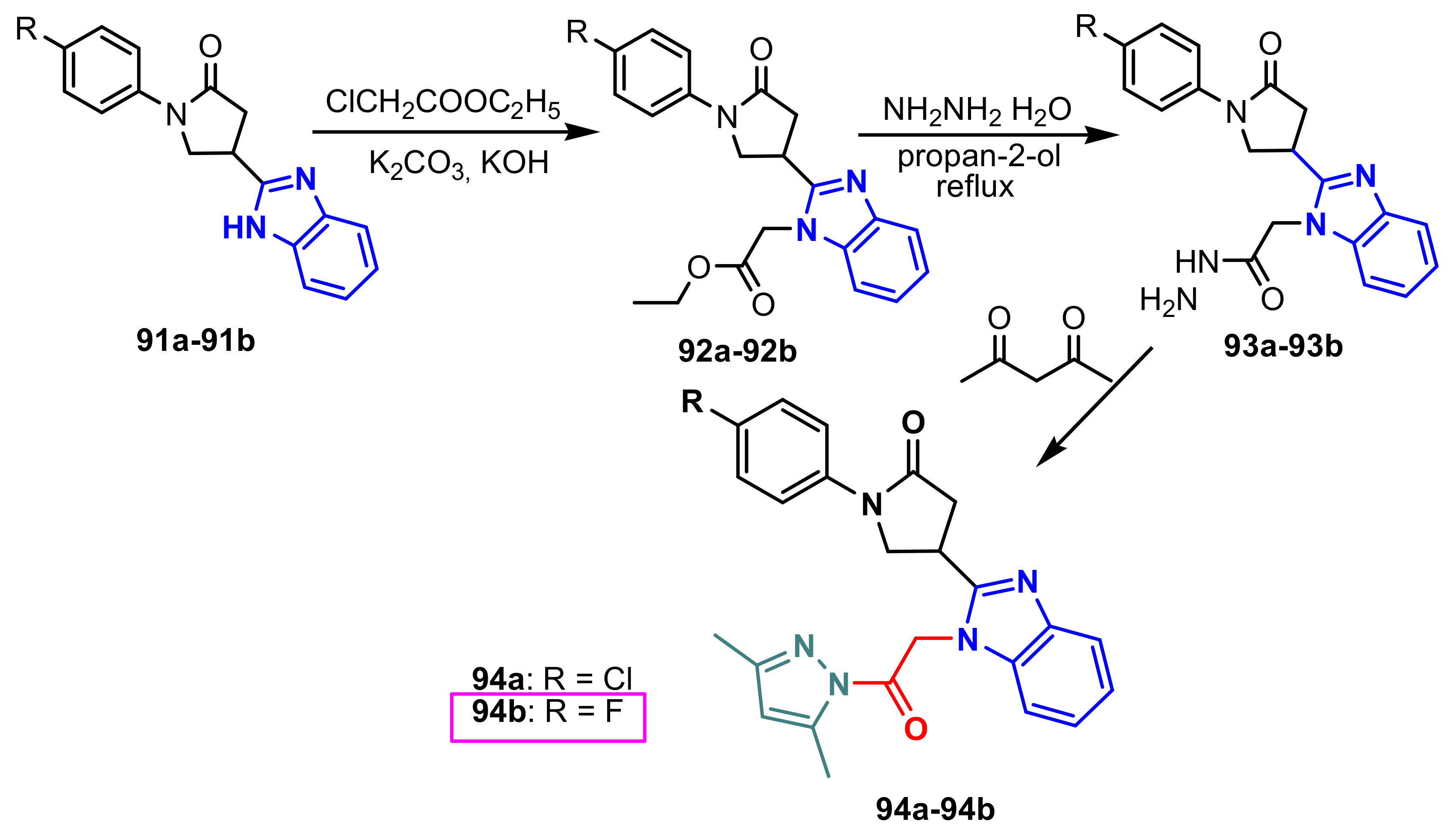{kind=link}
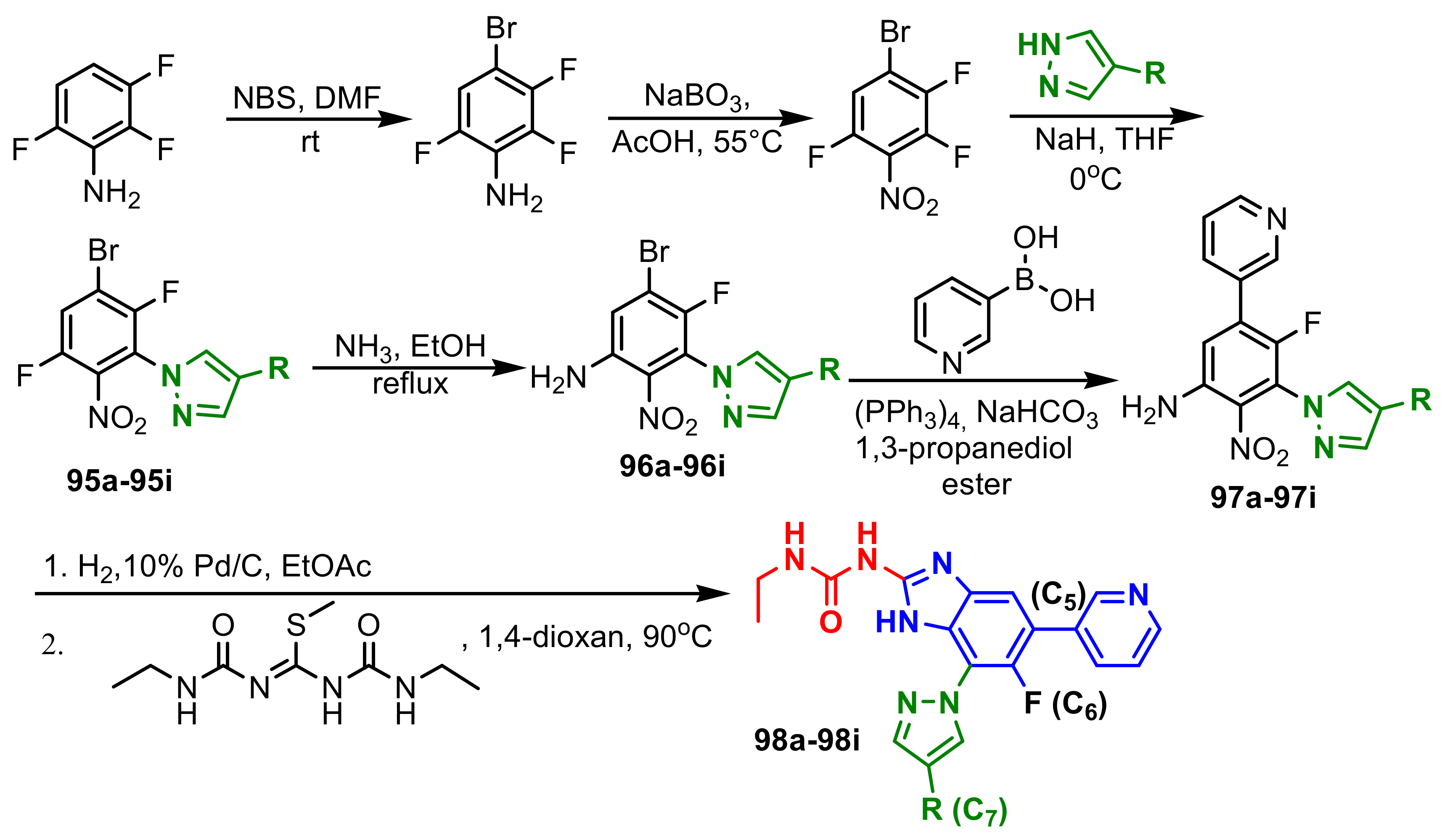{kind=link}
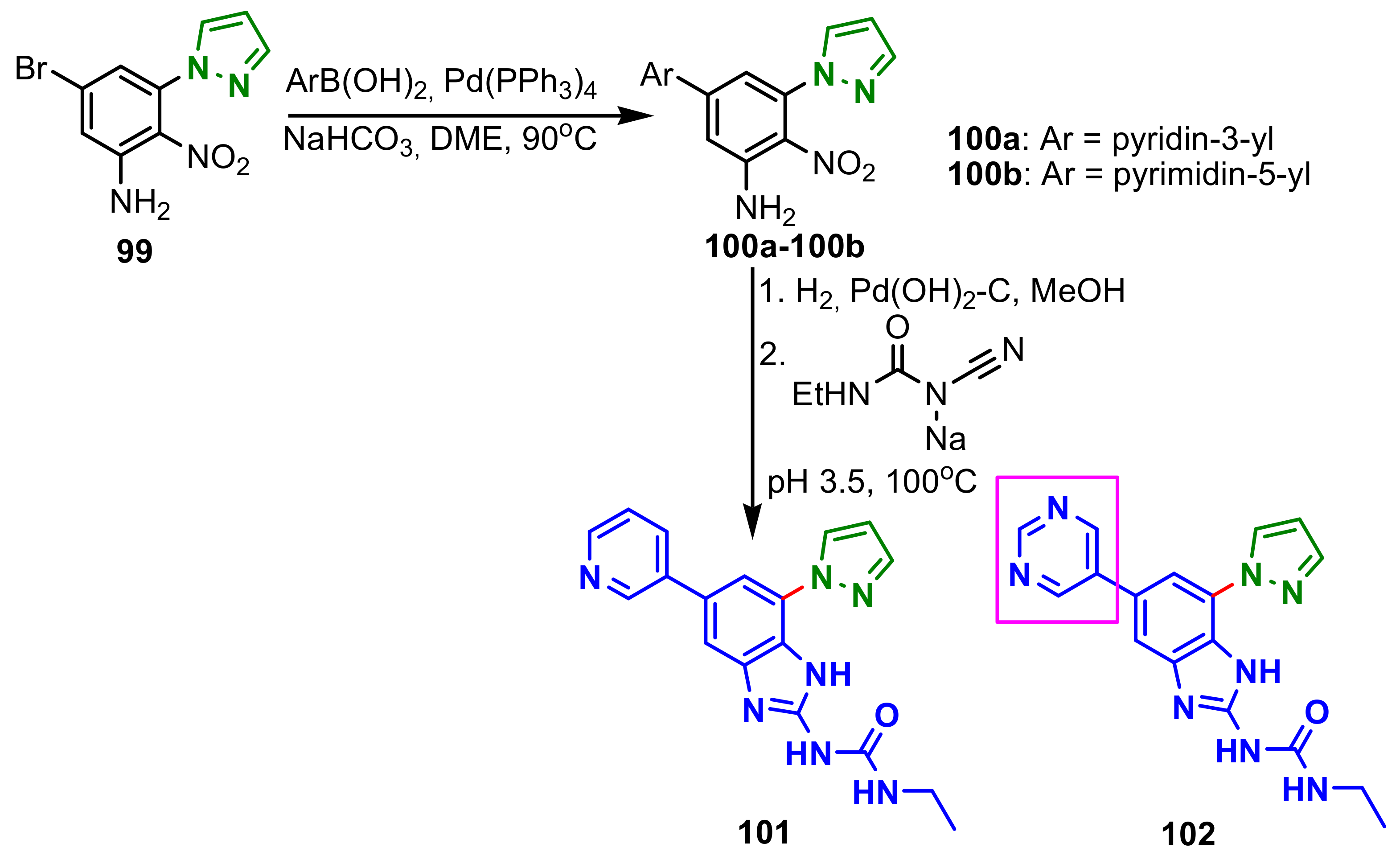{kind=link}
{kind=link}
{kind=link}
{kind=link}






