
The Biology of Colicin M and Its Orthologs

Abstract
:1. Colicins among Bacteriocins
2. Colicin M
2.1. General Characteristics
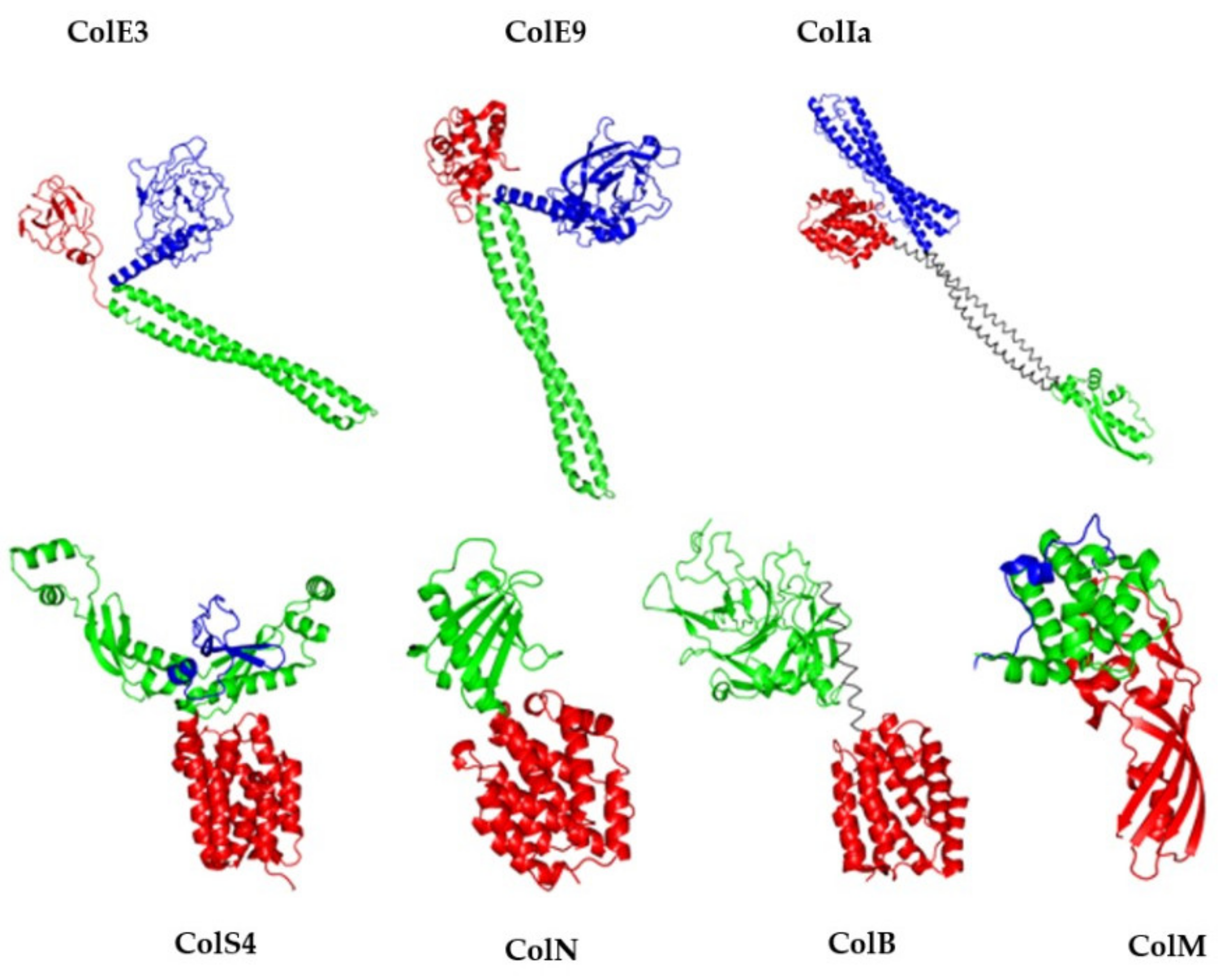
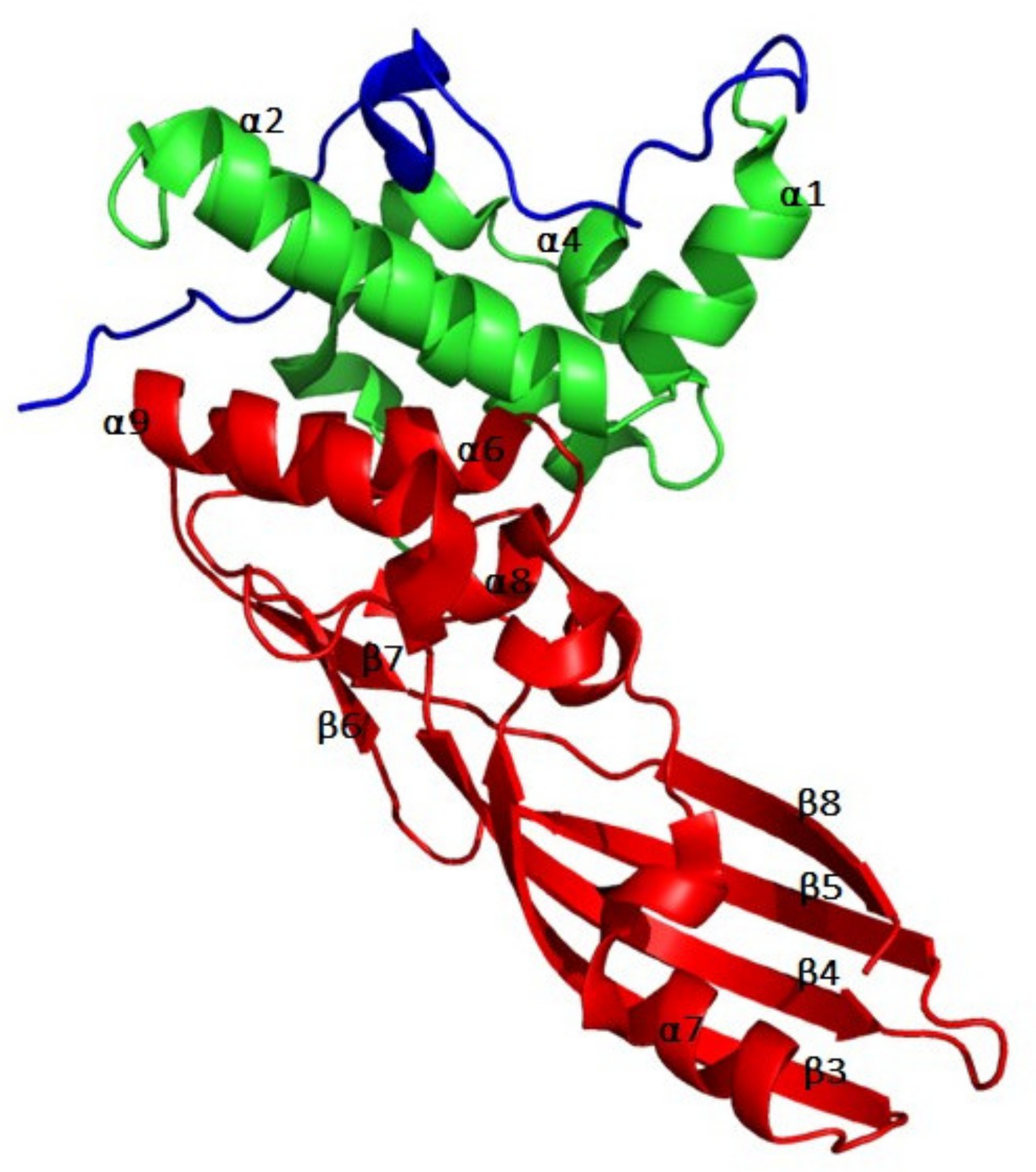
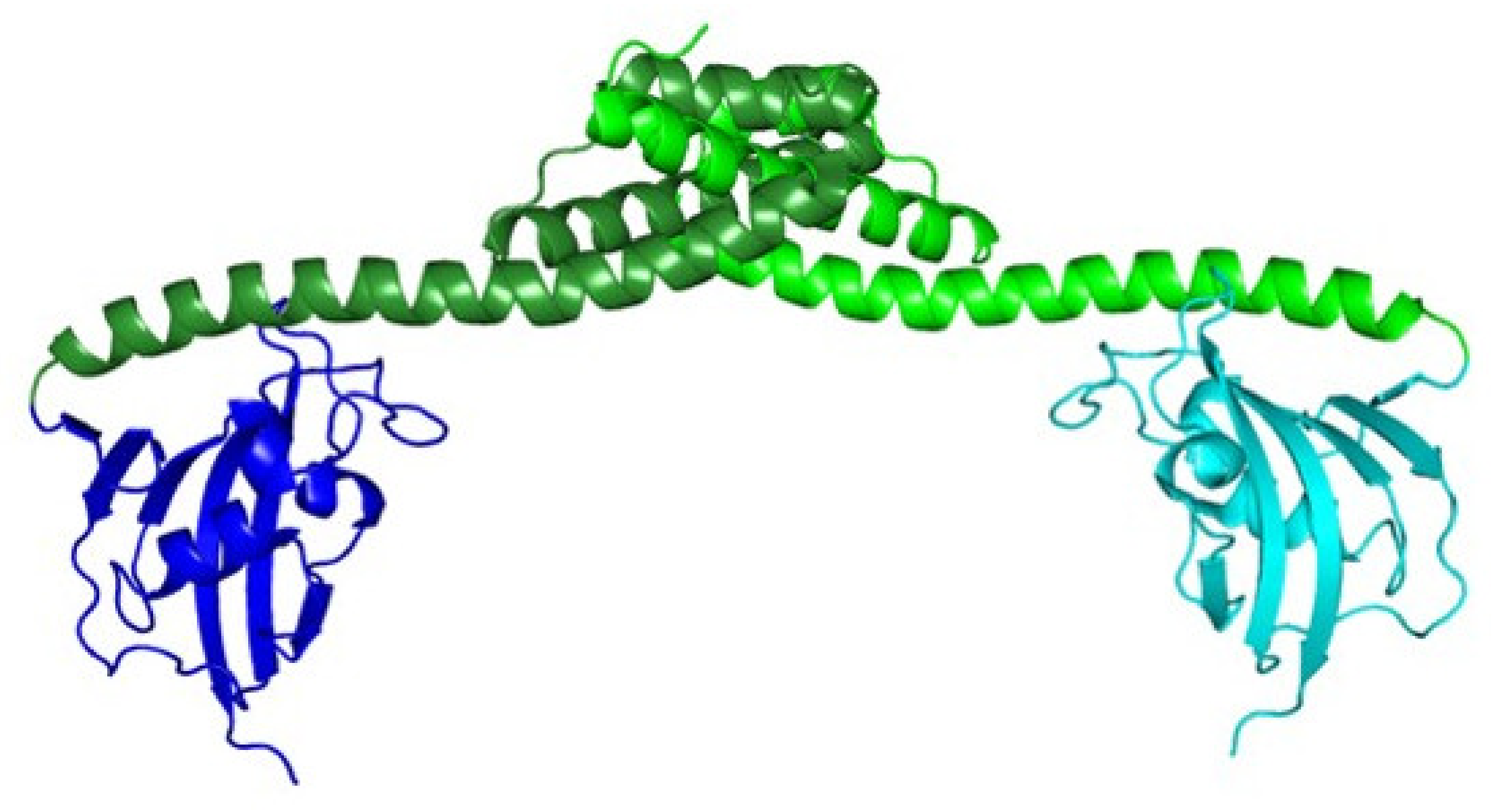
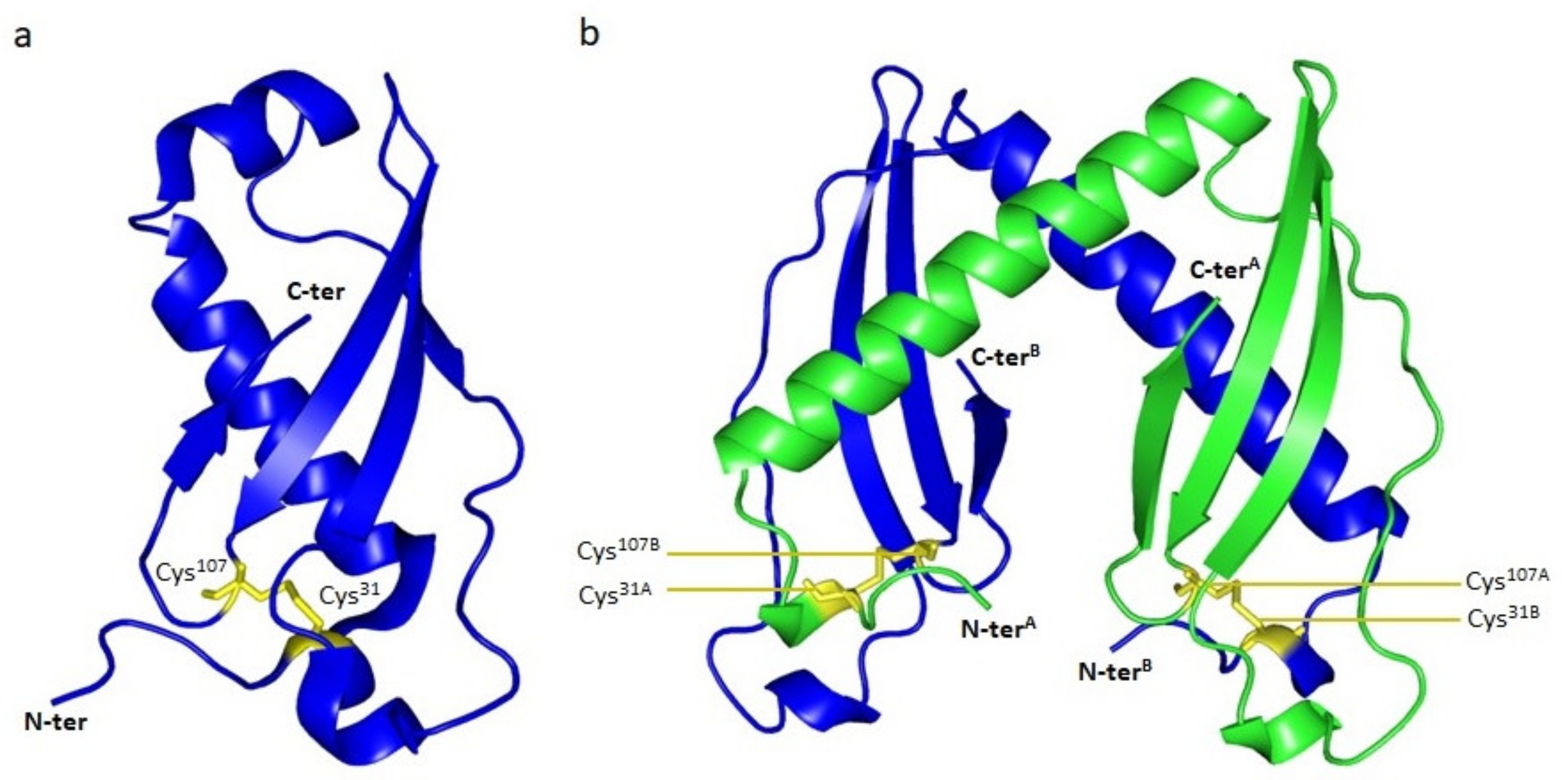
2.2. Tridimensional Structure
2.3. Mode of Action
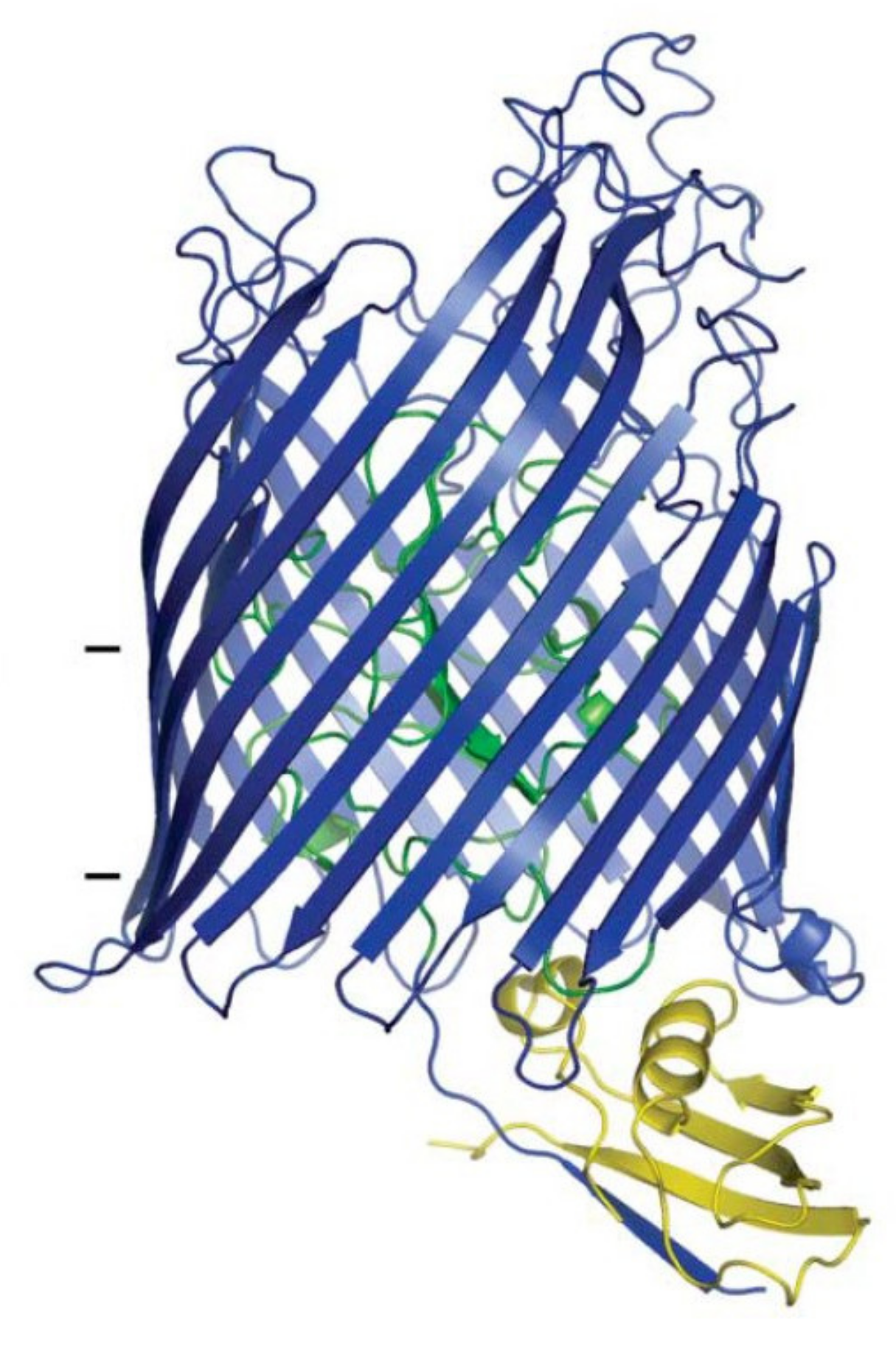
2.3.1. Binding to FhuA
2.3.2. Translocation
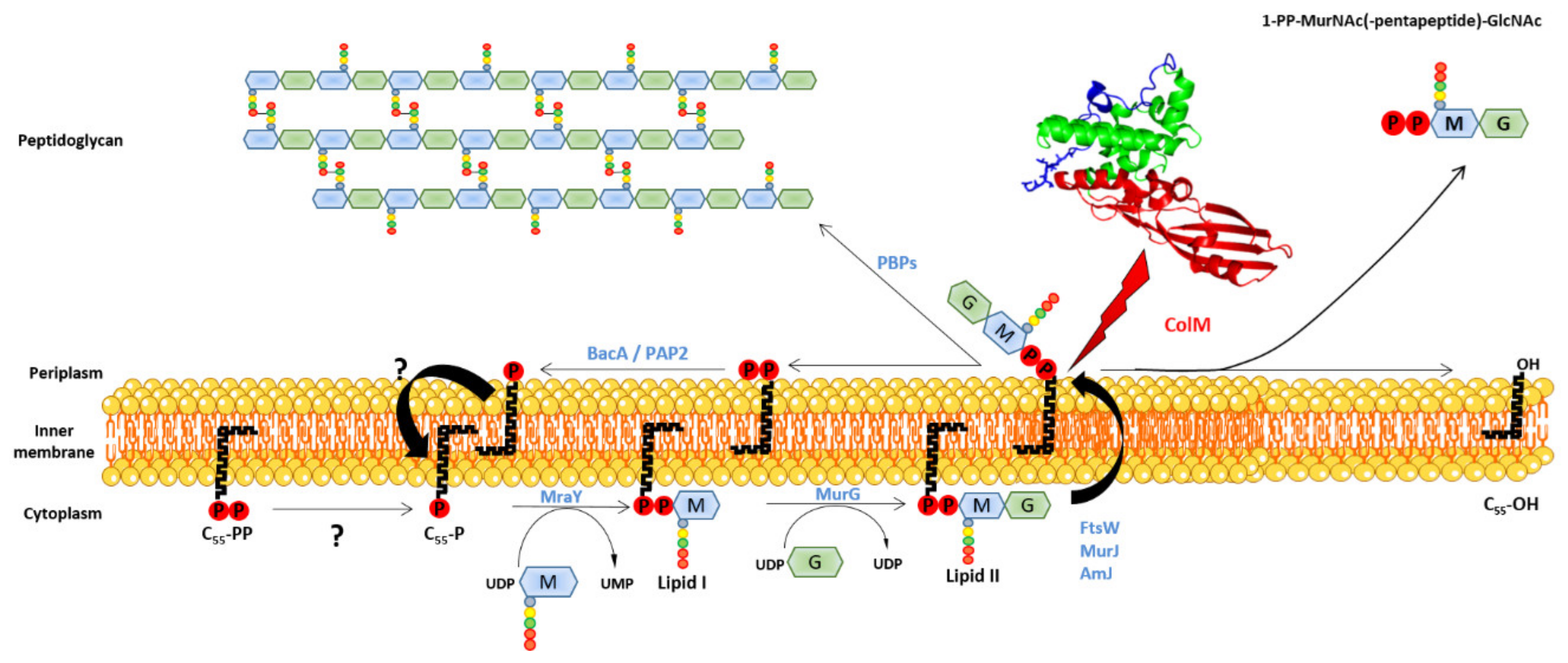
2.3.3. Maturation
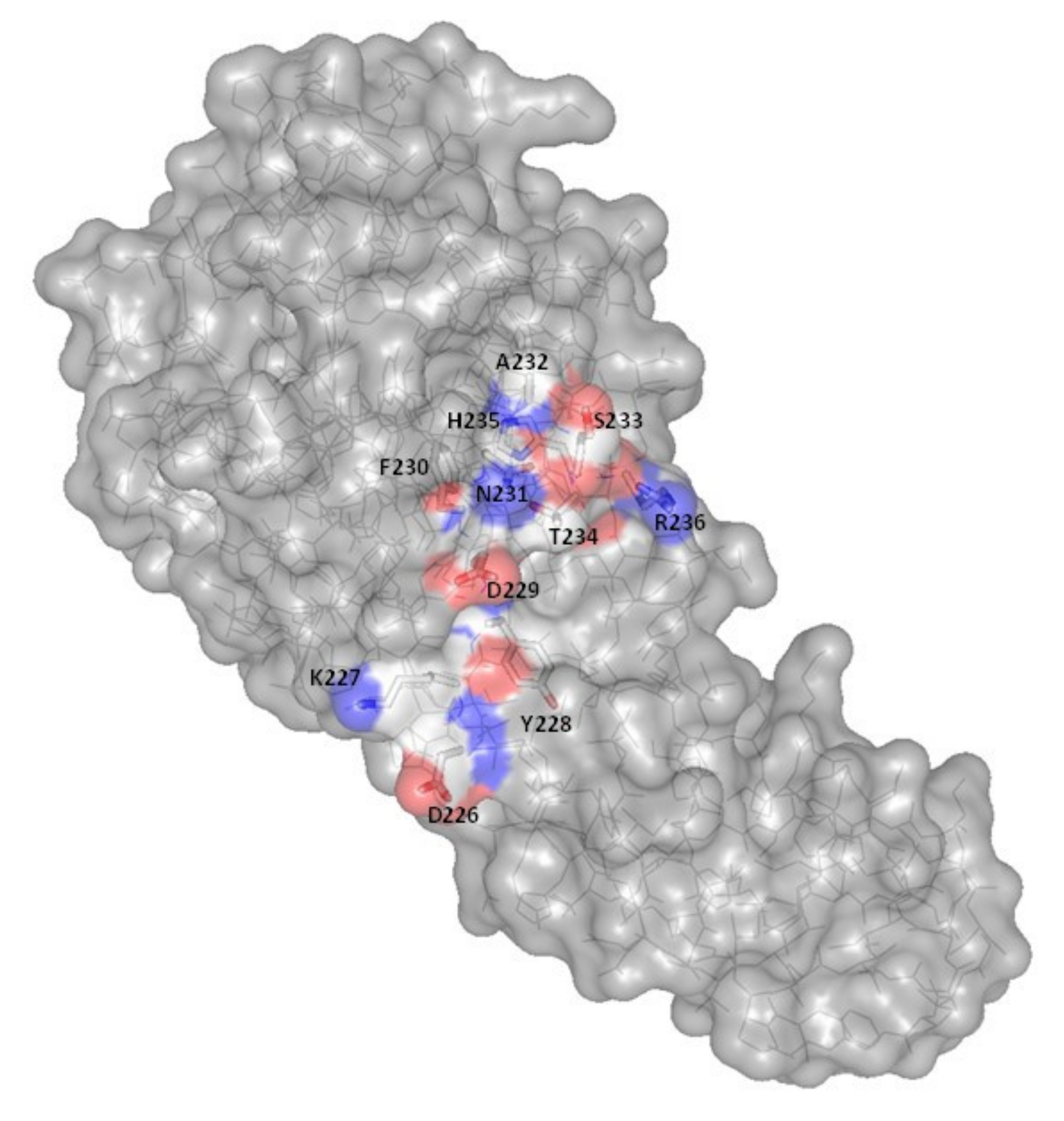
2.3.4. Activity
2.4. Immunity
3. The Orthologs of ColM
3.1. From Pseudomonas spp.
3.1.1. General Features
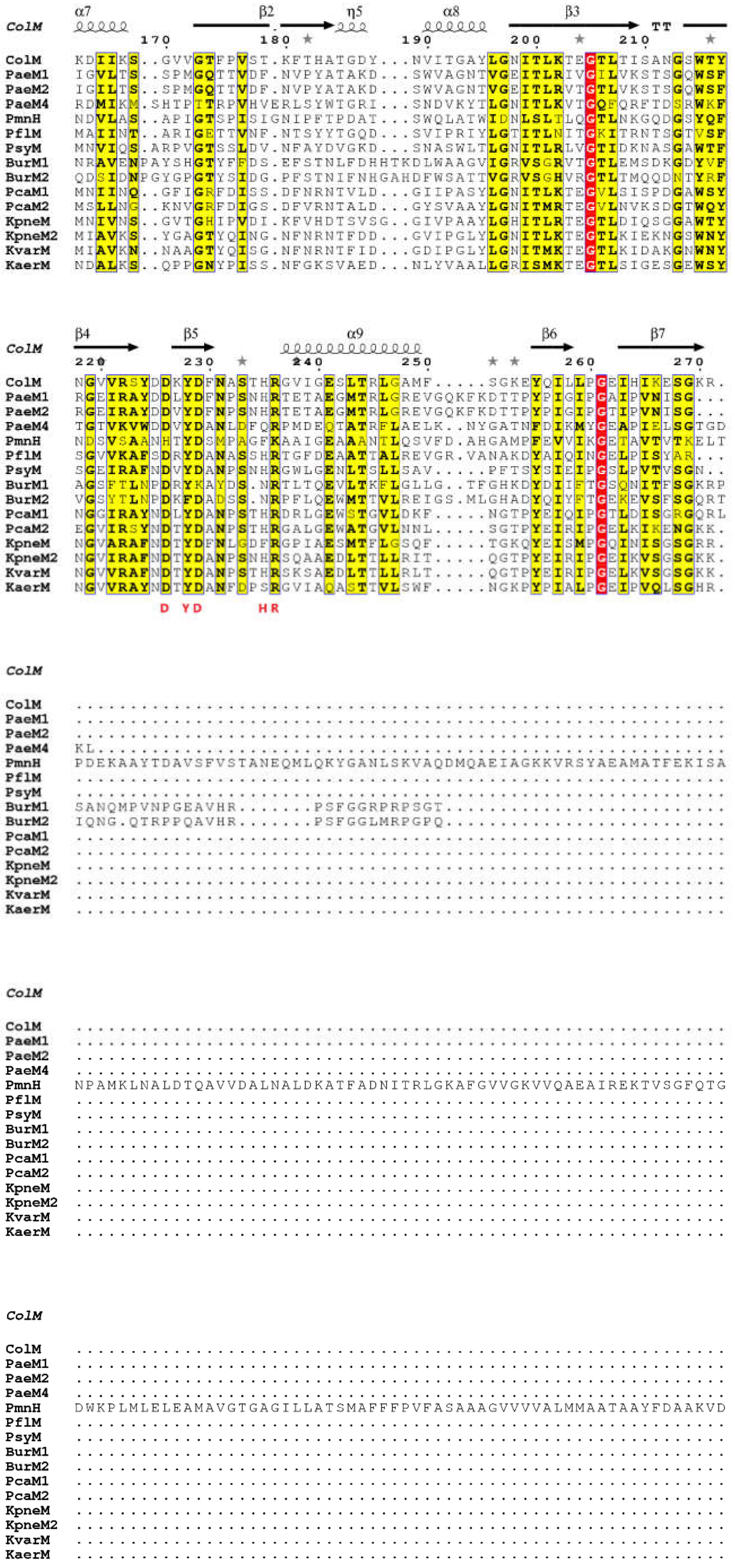
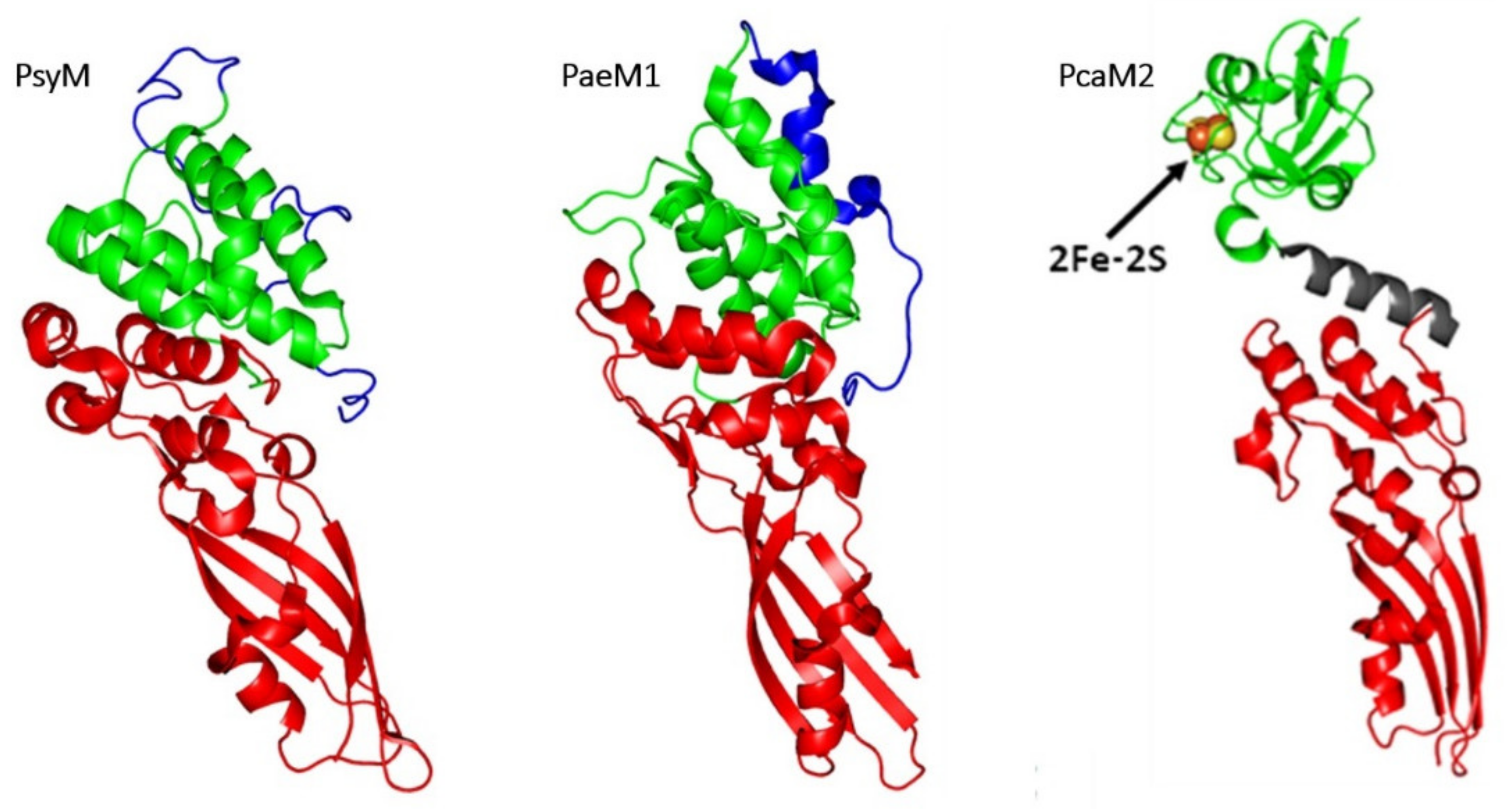
3.1.2. Comparison of 3D Structures
3.1.3. Diversity of Immunity Proteins
3.1.4. Hybrid Proteins Exhibiting ColM Catalytic Domain
3.2. From Burkholderia spp.
3.3. From Pectobacterium carotovorum
3.4. From Klebsiella spp.
4. Possible Applications and Uses
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Jack, R.W.; Tagg, J.R.; Ray, B. Bacteriocins of gram-positive bacteria. Microbiol. Rev. 1995, 59, 171–200. [Google Scholar] [CrossRef]
- Simons, A.; Alhanout, K.; Duval, R.E. Bacteriocins, Antimicrobial Peptides from Bacterial Origin: Overview of Their Biology and Their Impact against Multidrug-Resistant Bacteria. Microorganisms 2020, 8, 639. [Google Scholar] [CrossRef]
- Mavridou, D.; Gonzalez, D.; Kim, W.; West, S.A.; Foster, K.R. Bacteria Use Collective Behavior to Generate Diverse Combat Strategies. Curr. Boil. 2018, 28, 345–355.e4. [Google Scholar] [CrossRef]
- Granato, E.T.; Foster, K.R. The Evolution of Mass Cell Suicide in Bacterial Warfare. Curr. Biol. 2020, 30, 2836–2843. [Google Scholar] [CrossRef]
- Zacharof, M.P.; Lovitt, R.W. Investigation of Shelf Life of Potency and Activity of the Lactobacilli Produced Bacteriocins Through Their Exposure to Various Physicochemical Stress Factors. Probiotics Antimicrob. Proteins 2012, 4, 187–197. [Google Scholar] [CrossRef]
- Kolter, R.; Moreno, F. Genetics of ribosomally synthesized peptide antibiotics. Annu. Rev. Microbiol. 1992, 46, 141–163. [Google Scholar] [CrossRef]
- Duquesne, S.; Petit, V.; Peduzzi, J.; Rebuffat, S. Structural and functional diversity of microcins, gene-encoded antibacterial peptides from enterobacteria. J. Mol. Microbiol. Biotechnol. 2007, 13, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Cramer, W.A. Intramembrane helix-helix interactions as the basis of inhibition of the colicin E1 ion channel by its immunity protein. J. Biol. Chem. 1993, 268, 10176–10184. [Google Scholar] [CrossRef]
- Cascalès, E.; Buchanan, S.K.; Duché, D.; Kleanthous, C.; Lloubès, R.; Postle, K.; Riley, M.; Slatin, S.; Cavard, D. Colicin biology. Microbiol. Mol. Biol. Rev. 2007, 71, 158–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamet, A.; Nassif, X. New players in the toxin field: Polymorphic toxin systems in bacteria. MBio 2015, 6, e00285-15. [Google Scholar] [CrossRef] [Green Version]
- Gillor, O.; Vriezen, J.; Riley, M.A. The role of SOS boxes in enteric bacteriocin regulation. Microbiololgy 2008, 154 Pt 6, 1783–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šmarda, J.; Šmajs, D. Colicins—Exocellular lethal proteins of Escherichia coli. Folia Microbiol. 1998, 43, 563–582. [Google Scholar] [CrossRef] [PubMed]
- Braun, V.; Schaller, K.; Wabl, M.R. Isolation, characterization, and action of colicin M. Antimicrob. Agents Chemother. 1974, 5, 520–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köck, J.; Ölschläger, T.; Kamp, R.M.; Braun, V. Primary structure of colicin M, an inhibitor of murein biosynthesis. J. Bacteriol. 1987, 169, 3358–3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ölschläger, T.; Schramm, E.; Braun, V. Cloning and expression of the activity and immunity genes of colicins B and M on ColBM plasmids. Mol. Gen. Genet. 1984, 196, 482–487. [Google Scholar] [CrossRef]
- Christenson, J.K.; Gordon, D.M. Evolution of colicin BM plasmids: The loss of the colicin B activity gene. Microbiology 2009, 155 Pt 5, 1645–1655. [Google Scholar] [CrossRef] [Green Version]
- Braun, V.; Schaller, K.; Wolff, H. A common receptor protein for phage T5 and colicin M in the outer membrane of Escherichia coli B. Biochim. Biophys. Acta 1973, 323, 87–97. [Google Scholar] [CrossRef]
- Braun, V.; Hancock, R.E.; Hantke, K.; Hartmann, A. Functional organization of the outer membrane of escherichia coli: Phage and colicin receptors as components of iron uptake systems. J. Supramol. Struct. 1976, 5, 37–58. [Google Scholar] [CrossRef]
- Coulton, J.W.; Mason, P.; DuBow, M.S. Molecular cloning of the ferrichrome-iron receptor of Escherichia coli K-12. J. Bacteriol. 1983, 156, 1315–1321. [Google Scholar] [CrossRef] [Green Version]
- Pilsl, H.; Glaser, C.; Gross, P.; Killmann, H.; Ölschläger, T.; Braun, V. Domains of colicin M involved in uptake and activity. Mol. Gen. Genet 1993, 240, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Braun, V.; Patzer, S.I.; Hantke, K. Ton-dependent colicins and microcins: Modular design and evolution. Biochimie 2002, 84, 365–380. [Google Scholar] [CrossRef]
- Schaller, K.; Höltje, J.V.; Braun, V. Colicin M is an inhibitor of murein biosynthesis. J. Bacteriol. 1982, 152, 994–1000. [Google Scholar] [CrossRef]
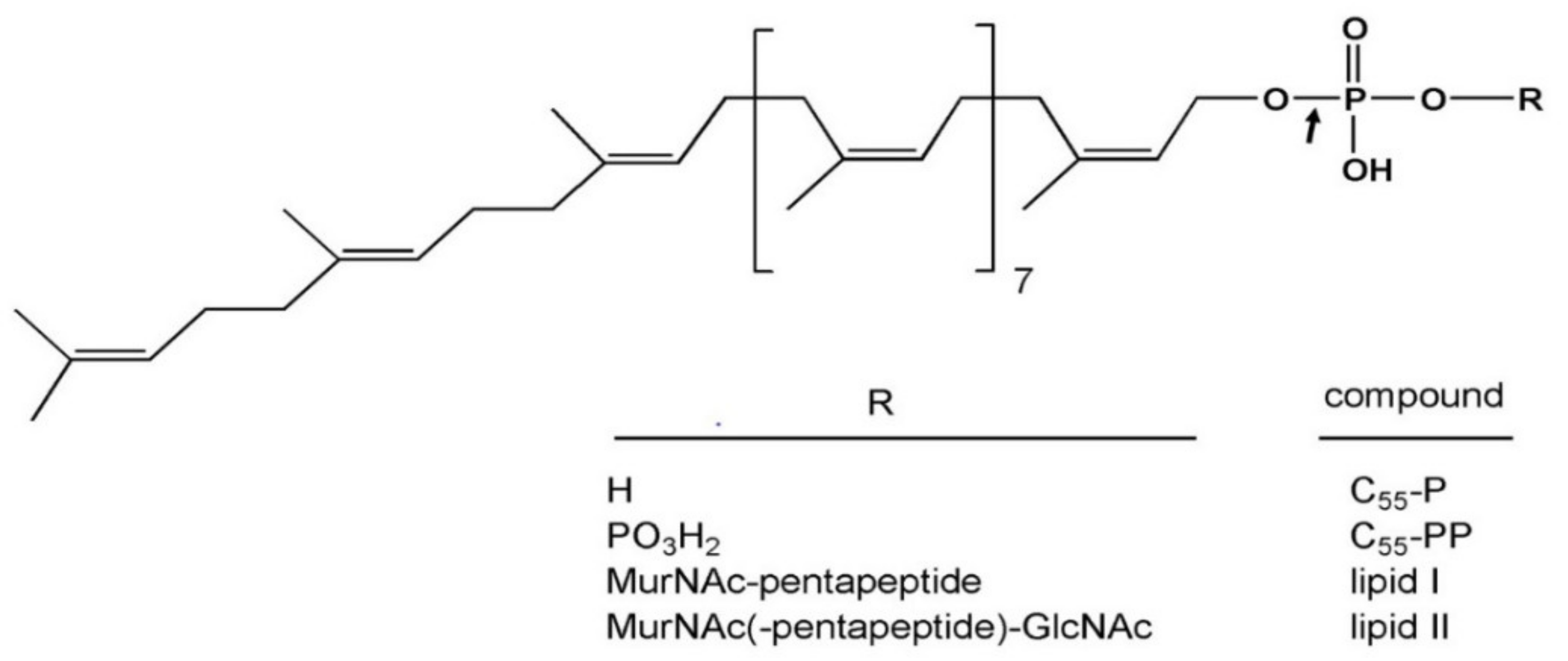
- El Ghachi, M.; Bouhss, A.; Barreteau, H.; Touzé, T.; Auger, G.; Blanot, D.; Mengin-Lecreulx, D. Colicin M exerts its bacteriolytic effect via enzymatic degradation of undecaprenyl phosphate-linked peptidoglycan precursors. J. Biol. Chem. 2006, 281, 22761–22772. [Google Scholar] [CrossRef] [Green Version]
- Zeth, K.; Römer, C.; Patzer, S.I.; Braun, V. Crystal structure of colicin M, a novel phosphatase specifically imported by Escherichia coli. J. Biol. Chem. 2008, 283, 25324–25331. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Surana, N.K.; St Geme, J.W., 3rd; Waksman, G. Structure of the outer membrane translocator domain of the Haemophilus influenzae Hia trimeric autotransporter. EMBO J. 2006, 25, 2297–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreteau, H.; Bouhss, A.; Gérard, F.; Duché, D.; Boussaid, B.; Blanot, D.; Lloubès, R.; Mengin-Lecreulx, D.; Touzé, T. Deciphering the catalytic domain of colicin M, a peptidoglycan lipid II-degrading enzyme. J. Biol. Chem. 2010, 285, 12378–12389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, A.D.; Deisenhofer, J. Metal import through microbial membranes. Cell 2004, 116, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Helbig, S.; Braun, V. Mapping functional domains of colicin M. J. Bacteriol. 2011, 193, 815–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endriss, F.; Braun, V. Loop deletions indicate regions important for FhuA transport and receptor functions in Escherichia coli. J. Bacteriol. 2004, 186, 4818–4823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakes, K.S.; Finkelstein, A. The colicin Ia receptor, Cir, is also the translocator for colicin Ia. Mol. Microbiol. 2010, 75, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Jakes, K.S. Translocation trumps receptor binding in colicin entry into Escherichia coli. Biochem. Soc. Trans. 2012, 40, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Dreher, R.; Braun, V.; Wittmann-Liebold, B. Functional domains of colicin M. Arch. Microbiol. 1985, 140, 343–346. [Google Scholar] [CrossRef]
- Schöffler, H.; Braun, V. Transport across the outer membrane of Escherichia coli K12 via the FhuA receptor is regulated by the TonB protein of the cytoplasmic membrane. Mol. Gen. Genet. 1989, 217, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, P.D.; Croteau, N.; Ng-Thow-Hing, C.; Khursigara, C.M.; Moiseeva, N.; Allaire, M.; Coulton, J.W. Structure of TonB in complex with FhuA, E. coli outer membrane receptor. Science 2006, 312, 1399–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, V.; Frenz, J.; Hantke, K.; Schaller, K. Penetration of colicin M into cells of Escherichia coli. J. Bacteriol. 1980, 142, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Hullmann, J.; Patzer, S.I.; Römer, C.; Hantke, K.; Braun, V. Periplasmic chaperone FkpA is essential for imported colicin M toxicity. Mol. Microbiol. 2008, 69, 926–937. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, J.E.; Otzen, D.E. Interactions between folding factors and bacterial outer membrane proteins. Mol. Microbiol. 2005, 57, 326–346. [Google Scholar] [CrossRef]
- Saul, F.A.; Arié, J.P.; Vulliez-le Normand, B.; Kahn, R.; Betton, J.M.; Bentley, G.A. Structural and functional studies of FkpA from Escherichia coli, a cis/trans peptidyl-prolyl isomerase with chaperone activity. J. Mol. Biol. 2004, 335, 595–608. [Google Scholar] [CrossRef]
- Barnéoud-Arnoulet, A.; Barreteau, H.; Touzé, T.; Mengin-Lecreulx, D.; Lloubès, R.; Duché, D. Toxicity of the colicin M catalytic domain exported to the periplasm is FkpA independent. J. Bacteriol. 2010, 192, 5212–5219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramm, K.; Plückthun, A. The periplasmic Escherichia coli peptidylprolyl cis,trans-isomerase FkpA. II. Isomerase-independent chaperone activity in vitro. J. Biol. Chem. 2000, 275, 17106–17113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helbig, S.; Patzer, S.I.; Schiene-Fischer, C.; Zeth, K.; Braun, V. Activation of colicin M by the FkpA prolyl cis-trans isomerase/chaperone. J. Biol. Chem. 2011, 286, 6280–6290. [Google Scholar] [CrossRef] [Green Version]
- Barreteau, H.; Kovač, A.; Boniface, A.; Sova, M.; Gobec, S.; Blanot, D. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 168–207. [Google Scholar] [CrossRef] [Green Version]
- Bouhss, A.; Trunkfield, A.E.; Bugg, T.D.; Mengin-Lecreulx, D. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol. Rev. 2008, 32, 208–233. [Google Scholar] [CrossRef] [Green Version]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef] [Green Version]
- Barreteau, H.; Blanot, D.; Mengin-Lecreulx, D.; Touzé, T. Lipid intermediates in bacterial peptidoglycan biosynthesis. In Biogenesis of Fatty Acids, Lipids and Membranes, Handbook of Hydrocarbon and Lipid Microbiology; Geiger, O., Ed.; Springer International Publishing AG: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Harkness, R.E.; Braun, V. Inhibition of lipopolysaccharide O-antigen synthesis by colicin M. J. Biol. Chem. 1989, 264, 14716–14722. [Google Scholar] [CrossRef]
- Harkness, R.E.; Braun, V. Colicin M inhibits peptidoglycan biosynthesis by interfering with lipid carrier recycling. J. Biol. Chem. 1989, 264, 6177–6182. [Google Scholar] [CrossRef]
- Harkness, R.E.; Braun, V. Colicin M is only bactericidal when provided from outside the cell. Mol. Gen. Genet. 1990, 222, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Gérard, F.; Brooks, M.A.; Barreteau, H.; Touzé, T.; Graille, M.; Bouhss, A.; Blanot, D.; van Tilbeurgh, H.; Mengin-Lecreulx, D. X-ray structure and site-directed mutagenesis analysis of the Escherichia coli colicin M immunity protein. J. Bacteriol. 2011, 193, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Barreteau, H.; Bouhss, A.; Fourgeaud, M.; Mainardi, J.L.; Touzé, T.; Gérard, F.; Blanot, D.; Arthur, M.; Mengin-Lecreulx, D. Human- and plant-pathogenic Pseudomonas species produce bacteriocins exhibiting colicin M-like hydrolase activity towards peptidoglycan precursors. J. Bacteriol. 2009, 191, 3657–3664. [Google Scholar] [CrossRef] [Green Version]
- Grinter, R.; Milner, J.; Walker, D. Ferredoxin containing bacteriocins suggest a novel mechanism of iron uptake in Pectobacterium spp. PLoS ONE 2012, 7, e33033. [Google Scholar] [CrossRef] [Green Version]
- Ghequire, M.G.; De Mot, R. Distinct colicin M-like bacteriocin-immunity pairs in Burkholderia. Sci. Rep. 2015, 5, 17368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghequire, M.; Buchanan, S.K.; De Mot, R. The ColM Family, Polymorphic Toxins Breaching the Bacterial Cell Wall. MBio 2018, 9, e02267-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patin, D.; Barreteau, H.; Auger, G.; Magnet, S.; Crouvoisier, M.; Bouhss, A.; Touzé, T.; Arthur, M.; Mengin-Lecreulx, D.; Blanot, D. Colicin M hydrolyses branched lipids II from Gram-positive bacteria. Biochimie 2012, 94, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Ölschläger, T.; Braun, V. Sequence, expression, and localization of the immunity protein for colicin M. J. Bacteriol. 1987, 169, 4765–4769. [Google Scholar] [CrossRef] [Green Version]
- Ölschläger, T.; Turba, A.; Braun, V. Binding of the immunity protein inactivates colicin M. Mol. Microbiol. 1991, 5, 1105–1111. [Google Scholar] [CrossRef]
- Gross, P.; Braun, V. Colicin M is inactivated during import by its immunity protein. Mol. Gen. Genet. 1996, 251, 388–396. [Google Scholar] [CrossRef]
- Usón, I.; Patzer, S.I.; Rodríguez, D.D.; Braun, V.; Zeth, K. The crystal structure of the dimeric colicin M immunity protein displays a 3D domain swap. J. Struct. Biol. 2012, 178, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Helbig, S.; Hantke, K.; Ammelburg, M.; Braun, V. CbrA is a flavin adenine dinucleotide protein that modifies the Escherichia coli outer membrane and confers specific resistance to Colicin M. J. Bacteriol. 2012, 194, 4894–4903. [Google Scholar] [CrossRef] [Green Version]
- Barreteau, H.; Patin, D.; Bouhss, A.; Blanot, D.; Mengin-Lecreulx, D.; Touzé, T. CbrA Mediates Colicin M Resistance in Escherichia coli through Modification of Undecaprenyl-Phosphate-Linked Peptidoglycan Precursors. J. Bacteriol. 2020, 202, e00436-20. [Google Scholar] [CrossRef]
- Barreteau, H.; Tiouajni, M.; Graille, M.; Josseaume, N.; Bouhss, A.; Patin, D.; Blanot, D.; Fourgeaud, M.; Mainardi, J.L.; Arthur, M.; et al. Functional and structural characterization of PaeM, a colicin M-like bacteriocin produced by Pseudomonas aeruginosa. J. Biol. Chem. 2012, 287, 37395–37405. [Google Scholar] [CrossRef] [Green Version]
- Grinter, R.; Roszak, A.W.; Cogdell, R.J.; Milner, J.J.; Walker, D. The crystal structure of the lipid II-degrading bacteriocin syringacin M suggests unexpected evolutionary relationships between colicin M-like bacteriocins. J. Biol. Chem. 2012, 287, 38876–38888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinter, R.; Josts, I.; Zeth, K.; Roszak, A.W.; McCaughey, L.C.; Cogdell, R.J.; Milner, J.J.; Kelly, S.M.; Byron, O.; Walker, D. Structure of the atypical bacteriocin pectocin M2 implies a novel mechanism of protein uptake. Mol. Microbiol. 2014, 93, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Ghequire, M.G.; Kemland, L.; De Mot, R. Novel Immunity Proteins Associated with Colicin M-like Bacteriocins Exhibit Promiscuous Protection in Pseudomonas. Front. Microbiol. 2017, 8, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghequire, M.G.; Kemland, L.; Anoz-Carbonell, E.; Buchanan, S.K.; De Mot, R. A Natural Chimeric Pseudomonas Bacteriocin with Novel Pore-Forming Activity Parasitizes the Ferrichrome Transporter. MBio 2017, 8, e01961-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denkovskienė, E.; Paškevičius, Š.; Misiūnas, A.; Stočkūnaitė, B.; Starkevič, U.; Vitkauskienė, A.; Hahn-Löbmann, S.; Schulz, S.; Giritch, A.; Gleba, Y.; et al. Broad and Efficient Control of Klebsiella Pathogens by Peptidoglycan-Degrading and Pore-Forming Bacteriocins Klebicins. Sci. Rep. 2019, 9, 15422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghequire, M.G.; De Mot, R. Ribosomally encoded antibacterial proteins and peptides from Pseudomonas. FEMS Microbiol. Rev. 2014, 38, 523–568. [Google Scholar] [CrossRef] [Green Version]
- Latino, L.; Patin, D.; Chérier, D.; Touzé, T.; Pourcel, C.; Barreteau, H.; Mengin-Lecreulx, D. Impact of FiuA Outer Membrane Receptor Polymorphism on the Resistance of Pseudomonas aeruginosa toward Peptidoglycan Lipid II-Targeting PaeM Pyocins. J. Bacteriol. 2019, 201, e00164-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chérier, D. Caractérisation Biochimique et Structurale de Bactériocines Ciblant le Métabolisme du Peptidoglycane Bactérien, Alternative Potentielle aux Antibiotiques. Ph.D. Thesis, Université Paris-Saclay, Gif-sur-Yvette, France, 2017. [Google Scholar]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Ghequire, M.; Öztürk, B. A Colicin M-Type Bacteriocin from Pseudomonas aeruginosa Targeting the HxuC Heme Receptor Requires a Novel Immunity Partner. Appl. Environ. Microbiol. 2018, 84, e00716-18. [Google Scholar] [CrossRef] [Green Version]
- Hannauer, M.; Barda, Y.; Mislin, G.L.; Shanzer, A.; Schalk, I.J. The ferrichrome uptake pathway in Pseudomonas aeruginosa involves an iron release mechanism with acylation of the siderophore and recycling of the modified desferrichrome. J. Bacteriol. 2010, 192, 1212–1220. [Google Scholar] [CrossRef] [Green Version]
- Chérier, D.; Giacomucci, S.; Patin, D.; Bouhss, A.; Touzé, T.; Blanot, D.; Mengin-Lecreulx, D.; Barreteau, H. Pectocin M1 (PcaM1) Inhibits Escherichia coli Cell Growth and Peptidoglycan Biosynthesis through Periplasmic Expression. Antibiotics 2016, 5, 36. [Google Scholar] [CrossRef]
- Grinter, R.; Josts, I.; Mosbahi, K.; Roszak, A.W.; Cogdell, R.J.; Bonvin, A.M.; Milner, J.J.; Kelly, S.M.; Byron, O.; Smith, B.O.; et al. Structure of the bacterial plant-ferredoxin receptor FusA. Nat. Commun. 2016, 7, 13308. [Google Scholar] [CrossRef]
- Hansen, J.N. Nisin as a model food preservative. Crit. Rev. Food Sci. Nutr. 1994, 34, 69–93. [Google Scholar] [CrossRef]
- Schulz, S.; Stephan, A.; Hahn, S.; Bortesi, L.; Jarczowski, F.; Bettmann, U.; Paschke, A.K.; Tusé, D.; Stahl, C.H.; Giritch, A.; et al. Broad and efficient control of major foodborne pathogenic strains of Escherichia coli by mixtures of plant-produced colicins. Proc. Natl. Acad. Sci. USA 2015, 112, E5454–E5460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paškevičius, Š.; Starkevič, U.; Misiūnas, A.; Vitkauskienė, A.; Gleba, Y.; Ražanskienė, A. Plant-expressed pyocins for control of Pseudomonas aeruginosa. PLoS ONE 2017, 12, e0185782. [Google Scholar] [CrossRef] [Green Version]
- Hahn-Löbmann, S.; Stephan, A.; Schulz, S.; Schneider, T.; Shaverskyi, A.; Tusé, D.; Giritch, A.; Gleba, Y. Colicins and Salmocins-New Classes of Plant-Made Non-antibiotic Food Antibacterials. Front. Plant Sci. 2019, 10, 437. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://fda.report/media/93655/GRAS-Notice-000593 (accessed on 29 July 2021).
- Gordon, D.M.; O’Brien, C.L. Bacteriocin diversity and the frequency of multiple bacteriocin production in Escherichia coli. Microbiology 2006, 152, 3239–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotter, P.D.; Ross, R.P.; Hill, C. Bacteriocins-a viable alternative to antibiotics? Nat. Rev. Microbiol. 2013, 11, 95–105. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Enzymatic Activity (pmol·min−1·mg−1) | Cytotoxicity a (ng) |
|---|---|---|
| Wild-type | 630 ± 57 (100%) | 0.4 |
| D226A | 2 ± 0.9 (0.3%) | >10,000 |
| Y228A | ND | >10,000 |
| D229A | 12 ± 5 (1.9%) | >10,000 |
| H235A | 9 ± 4 (1.4%) | >10,000 |
| R236A | ND | >10,000 |
| Composition of the Peptide Part of Lipid II | SA ColM (%) a |
|---|---|
| L-Ala-γ-D-Glu-meso-A2pm-D-Ala-D-Ala (Gram-negative) | 100 b |
| L-Ala-γ-D-Glu-L-Lys-D-Ala-D-Ala (Gram-positive) | 90 ± 6 |
| L-Ala-γ-D-Glu-L-Lys-(L-Ala)-D-Ala-D-Ala (E. faecalis) | 87 ± 11 |
| L-Ala-γ-D-Glu-L-Lys-(L-Ala)2-D-Ala-D-Ala (E. faecalis) | 77 ± 5 |
| L-Ala-γ-D-Glu-L-Lys-(D-iAsn)-D-Ala-D-Ala (E. faecium) | 94 ± 8 |
| L-Ala-γ-D-Glu-L-Lys-(Gly)-D-Ala-D-Ala (S. aureus) | 79 ± 8 |
| L-Ala-γ-D-Glu-L-Lys-(Gly)3-D-Ala-D-Ala (S. aureus) | 89 ± 8 |
| L-Ala-γ-D-Glu-L-Lys-(Gly)5-D-Ala-D-Ala (S. aureus) | 81 ± 11 |
| Protein | Specific Activity (nmol·min−1·mg−1) |
|---|---|
| ColM | 0.4 |
| PaeM1 | 219 |
| PflM | 0.017 |
| PsyM | 1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chérier, D.; Patin, D.; Blanot, D.; Touzé, T.; Barreteau, H. The Biology of Colicin M and Its Orthologs. Antibiotics 2021, 10, 1109. https://doi.org/10.3390/antibiotics10091109
Chérier D, Patin D, Blanot D, Touzé T, Barreteau H. The Biology of Colicin M and Its Orthologs. Antibiotics. 2021; 10(9):1109. https://doi.org/10.3390/antibiotics10091109
Chicago/Turabian StyleChérier, Dimitri, Delphine Patin, Didier Blanot, Thierry Touzé, and Hélène Barreteau. 2021. "The Biology of Colicin M and Its Orthologs" Antibiotics 10, no. 9: 1109. https://doi.org/10.3390/antibiotics10091109
APA StyleChérier, D., Patin, D., Blanot, D., Touzé, T., & Barreteau, H. (2021). The Biology of Colicin M and Its Orthologs. Antibiotics, 10(9), 1109. https://doi.org/10.3390/antibiotics10091109