
Next-Generation Polymyxin Class of Antibiotics: A Ray of Hope Illuminating a Dark Road



Abstract
:1. Introduction
2. Old Polymyxins (Colistin and Polymyxin B)
2.1. General Features
2.2. Pharmacokinetic/Pharmacodynamic (PK/PD) Properties
2.3. Antibiofilm Activity
2.4. Toxicity
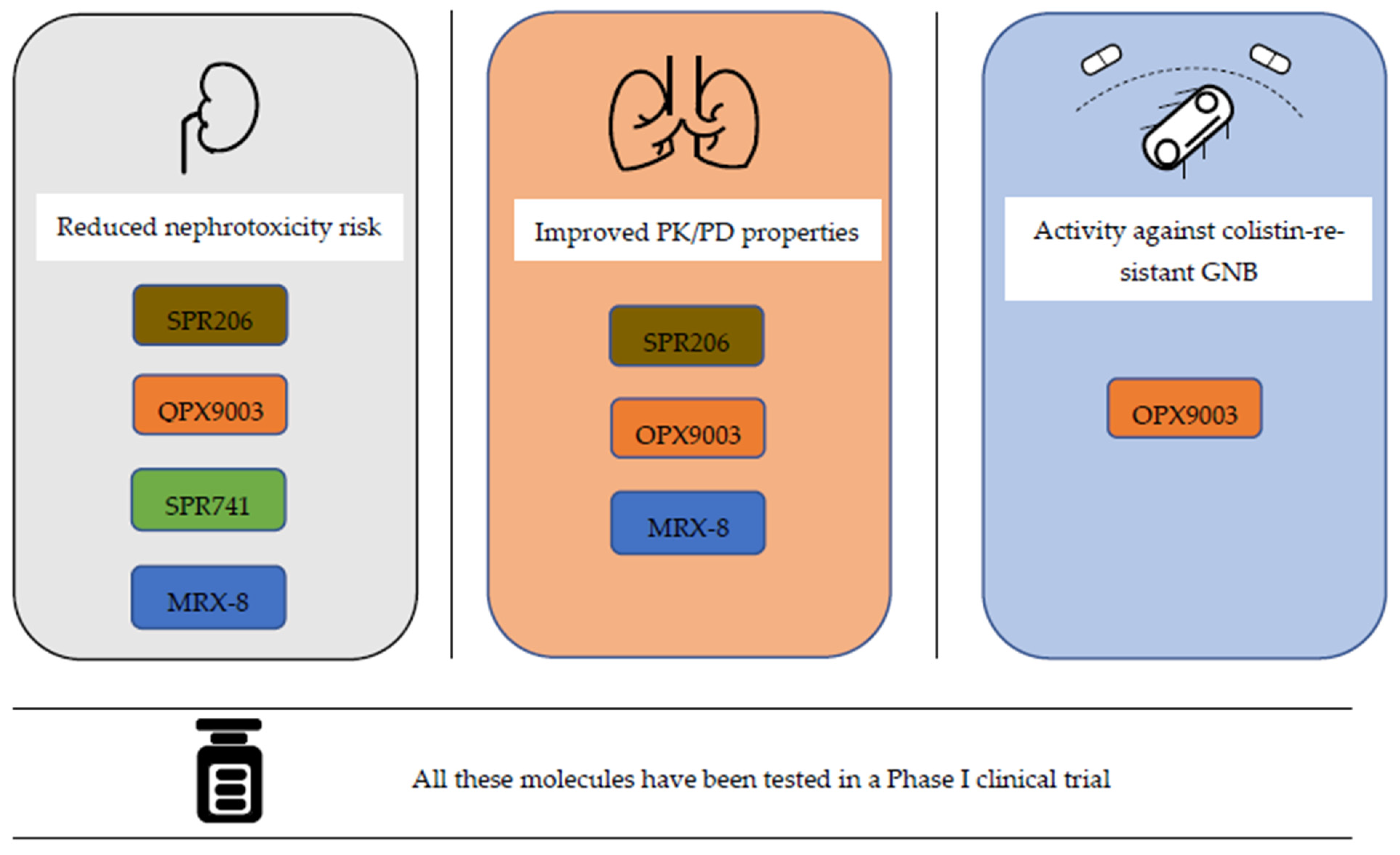
3. Why Do We Need to Develop Next-Generation Polymyxins?
4. Novel Polymyxin Molecules
4.1. SPR206
4.2. QPX9003
4.3. MRX-8
5. PBT2
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Global Antimicrobial Resistance Surveillance System (GLASS) Report: Early Implementation 2017–2018; WHO: Geneva, Switzerland, 2019. Available online: https://www.who.int/publications/i/item/9789241515061 (accessed on 13 January 2022).
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States; CDC: Atlanta, GA, USA, 2019. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed on 13 January 2022).
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Linden, K.P.; Kusne, S.; Coley, K.; Fontes, P.; Kramer, D.J.; Paterson, D. Use of parenteral colistin for the treatment of serious infections due to antimicrobial-resistant Pseudomonas aeruginosa. Clin. Infect. Dis. 2003, 37, e154–e160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Nation, R.L.; Turnidge, J.D.; Milne, R.W.; Coulthard, K.; Rayner, C.R.; Paterson, D.L. Colistin: The re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect. Dis. 2006, 6, 589–601. [Google Scholar] [CrossRef] [PubMed]
- van Duin, D.; Lok, J.J.; Earley, M.; Cober, E.; Richter, S.S.; Perez, F.; Salata, R.A.; Kalayjian, R.C.; Watkins, R.R.; Doi, Y.; et al. Colistin versus ceftazidime-avibactam in the treatment of infections due to carbapenem-resistant Enterobacteriaceae. Clin. Infect. Dis. 2018, 66, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, R.K.; Nguyen, M.H.; Chen, L.; Press, E.G.; Potoski, B.A.; Marini, R.V.; Doi, Y.; Kreiswirth, B.N.; Clancy, C.J. Ceftazidime-avibactam is superior to other treatment regimens against carbapenem-resistant Klebsiella pneumoniae bacteremia. Antimicrob. Agents Chemother. 2017, 25, 61. [Google Scholar] [CrossRef] [Green Version]
- Motsch, J.; Murta de Oliveira, C.; Stus, V.; Köksal, I.; Lyulko, O.; Boucher, H.W.; Kaye, K.S.; File, T.M.; Brown, M.L.; Khan, I.; et al. RESTORE-IMI 1: A Multicenter, Randomized, Double-blind Trial Comparing Efficacy and Safety of Imipenem/Relebactam vs Colistin Plus Imipenem in Patients with Imipenem-nonsusceptible Bacterial Infections. Clin. Infect. Dis. 2020, 70, 1799–1808. [Google Scholar] [CrossRef] [Green Version]
- Wunderink, R.G.; Giamarellos-Bourboulis, E.J.; Rahav, G.; Mathers, A.J.; Bassetti, M.; Vazquez, J.; Cornely, O.A.; Solomkin, J.; Bhowmick, T.; Bishara, J.; et al. Effect and safety of meropenem-vaborbactam versus best-available therapy in patients with carbapenem-resistant Enterobacteriaceae infections: The TANGO II randomized clinical trial. Infect. Dis. Ther. 2018, 7, 439–455. [Google Scholar] [CrossRef] [Green Version]
- Pogue, J.M.; Kaye, K.S.; Veve, M.P.; Patel, T.S.; Gerlach, A.T.; Davis, S.L.; Puzniak, L.A.; File, T.M.; Olson, S.; Dhar, S.; et al. Ceftolozane/tazobactam vs polymyxin or aminoglycoside-based regimens for the treatment of drug-resistant Pseudomonas aeruginosa. Clin. Infect. Dis. 2020, 71, 304–310. [Google Scholar] [CrossRef]
- Paul, M.; Daikos, G.L.; Durante-Mangoni, E.; Yahav, D.; Carmeli, Y.; Benattar, Y.D.; Skiada, A.; Andini, R.; Eliakim-Raz, N.; Nutman, A.; et al. Colistin alone versus colistin plus meropenem for treatment of severe infections caused by carbapenem-resistant Gram-negative bacteria: An open-label, randomised controlled trial. Lancet Infect. Dis. 2018, 18, 391–400. [Google Scholar] [CrossRef]
- Kaye, K.S.; Marchaim, D.; Thamlikitkul, V.; Carmeli, Y.; Chiu, C.H.; Daikos, G.; Dhar, S.; Durante-Mangoni, E.; Gikas, A.; Kotanidou, A.; et al. Results from the OVERCOME trial: Colistin monotherapy versus combination therapy for the treatment of pneumonia or bloodstream infection due to extensively drug resistant Gram-negative bacilli. In Proceedings of the 31st European Congress of Clinical Microbiology and Infectious Diseases, Vienna, Austria, 9–12 July 2021. [Google Scholar]
- Gutiérrez-Gutiérrez, B.; Salamanca, E.; de Cueto, M.; Hsueh, P.-R.; Viale, P.; Paño-Pardo, J.R.; Venditti, M.; Tumbarello, M.; Daikos, G.; Cantón, R.; et al. Effect of appropriate combination therapy on mortality of patients with bloodstream infections due to carbapenemase-producing Enterobacteriaceae (INCREMENT): A retrospective cohort study. Lancet Infect. Dis. 2017, 17, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Isler, B.; Aslan, A.T.; Akova, M.; Harris, P.; Paterson, D.L. Treatment strategies for OXA-48-like and NDM producing Klebsiella pneumoniae infections. Expert Rev. Anti-Infect. Ther. 2022, 20, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Carrara, E.; Retamar, P.; Tängdén, T.; Bitterman, R.; Bonomo, R.A.; de Waele, J.; Daikos, G.L.; Akova, M.; Harbarth, S.; et al. European Society of Clinical Microbiology and Infectious Diseases (ESCMID) guidelines for the treatment of infections caused by multidrug-resistant Gram-negative bacilli (endorsed by European society of intensive care medicine). Clin. Microbiol. Infect. 2022, 28, 521–547. [Google Scholar] [CrossRef] [PubMed]
- Shields, R.K.; Nguyen, M.H.; Chen, L.; Press, E.G.; Kreiswirth, B.N.; Clancy, C.J. Pneumonia and Renal Replacement Therapy Are Risk Factors for Ceftazidime-Avibactam Treatment Failures and Resistance among Patients with Carbapenem-Resistant Enterobacteriaceae Infections. Antimicrob. Agents Chemother. 2018, 62, e02497-17. [Google Scholar] [CrossRef] [Green Version]
- Tumbarello, M.; Raffaelli, F.; Giannella, M.; Mantengoli, E.; Mularoni, A.; Venditti, M.; De Rosa, F.G.; Sarmati, L.; Bassetti, M.; Brindicci, G.; et al. Ceftazidime-Avibactam Use for Klebsiella pneumoniae Carbapenemase-Producing K. pneumoniae Infections: A Retrospective Observational Multicenter Study. Clin. Infect. Dis. 2021, 73, 1664–1676. [Google Scholar] [CrossRef]
- Lomovskaya, O.; Sun, D.; Rubio-Aparicio, D.; Nelson, K.; Tsivkovski, R.; Griffith, D.C.; Dudley, M.N. Vaborbactam: Spectrum of β-lactamase inhibition and impact of resistance mechanisms on activity in Enterobacteriaceae. Antimicrob. Agents Chemother. 2017, 61, e01443-17. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Rubio-Aparicio, D.; Nelson, K.; Dudley, M.N.; Lomovskaya, O. Meropenem-vaborbactam resistance selection, resistance prevention, and molecular mechanisms in mutants of KPC-producing klebsiella pneumoniae. Antimicrob. Agents Chemother. 2017, 61, e01694-17. [Google Scholar] [CrossRef] [Green Version]
- Lapuebla, A.; Abdallah, M.; Olafisoye, O.; Cortes, C.; Urban, C.; Quale, J.; Landman, D. Activity of meropenem combined with RPX7009, a novel β-lactamase inhibitor, against Gram-negative clinical isolates in New York City. Antimicrob. Agents Chemother. 2015, 59, 4856–4860. [Google Scholar] [CrossRef] [Green Version]
- Castanheira, M.; Rhomberg, P.R.; Flamm, R.K.; Jones, R.N. Effect of the β-lactamase inhibitor vaborbactam combined with meropenem against serine carbapenemase-producing Enterobacteriaceae. Antimicrob. Agents Chemother. 2016, 60, 5454–5458. [Google Scholar] [CrossRef] [Green Version]
- Livermore, D.M.; Warner, M.; Mushtaq, S. Activity of MK-7655 combined with imipenem against Enterobacteriaceae and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2013, 68, 2286–2290. [Google Scholar] [CrossRef]
- Aslan, A.T.; Akova, M. The Role of Colistin in the Era of New β-Lactam/β-Lactamase Inhibitor Combinations. Antibiotics 2022, 11, 277. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guan, D.; Chen, F.; Shi, W.; Lan, L.; Huang, W. Total and Semisyntheses of Polymyxin Analogues with 2-Thr or 10-Thr Modifications to Decipher the Structure-Activity Relationship and Improve the Antibacterial Activity. J. Med. Chem. 2021, 64, 5746–5765. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Godoy, A.; Hansford, K.A.; Muldoon, C.; Becker, B.; Elliott, A.G.; Huang, J.X.; Pelingon, R.; Butler, M.S.; Blaskovich, M.A.T.; Cooper, M.A. Structure-Function Studies of Polymyxin B Lipononapeptides. Molecules 2019, 24, 553. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.; Dawson, M.J. Development of new polymyxin derivatives for multi-drug resistant Gram-negative infections. J. Antibiot. 2017, 70, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaara, M. Polymyxins and Their Potential Next Generation as Therapeutic Antibiotics. Front. Microbiol. 2019, 10, 1689. [Google Scholar] [CrossRef] [PubMed]
- Gales, A.C.; Jones, R.N.; Sader, H.S. Contemporary activity of colistin and polymyxin B against a worldwide collection of Gram-negative pathogens: Results from the SENTRY antimicrobial surveillance program (2006-09). J. Antimicrob. Chemother. 2011, 66, 2070–2074. [Google Scholar] [CrossRef] [Green Version]
- Gales, A.C.; Seifert, H.; Gur, D.; Castanheira, M.; Jones, R.N.; Sader, H.S. Antimicrobial susceptibility of Acinetobacter calcoaceticus—Acinetobacter baumannii complex and Stenotrophomonas maltophilia clinical isolates: Results from the SENTRY antimicrobial surveillance program. Open Forum Infect. Dis. 2019, 6, S34–S46. [Google Scholar] [CrossRef] [Green Version]
- Mena-Bueno, S.; Poveda-Urkixo, I.; Asensio, D.; Echarte, I.; Zabalza-Baranguá, A.; Grilló, M.J. Bru SIC: A novel selective medium for the primary isolation of Brucella in veterinary samples. Microbiol. Spectr. 2022, e01759-22. [Google Scholar] [CrossRef]
- Malott, R.J.; Wu, C.H.; Lee, T.D.; Hird, T.J.; Dalleska, N.F.; Zlosnik, J.E.; Newman, D.K.; Speert, D.P. Fosmidomycin Decreases Membrane Hopanoids and Potentiates the Effects of Colistin on Burkholderia multivorans Clinical Isolates. Antimicrob. Agents Chemother. 2014, 58, 5211–5219. [Google Scholar] [CrossRef] [Green Version]
- Gogry, F.A.; Siddiqui, M.T.; Sultan, I.; Haq, Q.M.R. Current Update on Intrinsic and Acquired Colistin Resistance Mechanisms in Bacteria. Front. Med. 2021, 8, 677720. [Google Scholar] [CrossRef]
- Falagas, M.E.; Kasiakou, S.K.; Saravolatz, L.D. Colistin: The revival of polymyxins for the management of multidrug-resistant Gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landman, D.; Georgescu, C.; Martin, D.A.; Quale, J. Polymyxins revisited. Clin Microbiol Rev. 2008, 21, 449–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Qin, W.; Lin, J.; Fang, S.; Qiu, J. Antimicrobial Mechanisms of Polymyxin and Bacterial Resistance. Biomed Res. Int. 2015, 2015, 679109. [Google Scholar] [CrossRef]
- Li, J.; Nation, R.L.; Milne, R.W.; Turnidge, J.D.; Coulthard, K. Evaluation of colistin as an agent against multi-resistant Gram-negative bacteria. Int. J. Antimicrob. Agents 2005, 25, 11–25. [Google Scholar] [CrossRef]
- Deris, Z.Z.; Akter, J.; Sivanesan, S.; Roberts, K.D.; Thompson, P.E.; Nation, R.L.; Li, J.; Velkov, T. A secondary mode of action of polymyxins against Gram-negative bacteria involves the inhibition of NADH-quinone oxidoreductase activity. J. Antibiot. 2013, 67, 147–151. [Google Scholar] [CrossRef] [Green Version]
- French, S.; Farha, M.; Ellis, M.J.; Sameer, Z.; Côté, J.P.; Cotroneo, N.; Lister, T.; Rubio, A.; Brown, E.D. Potentiation of Antibiotics against Gram-Negative Bacteria by Polymyxin B Analogue SPR741 from Unique Perturbation of the Outer Membrane. ACS Infect. Dis. 2020, 6, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Bergen, P.J.; Landersdorfer, C.B.; Zhang, J.; Zhao, M.; Lee, H.J.; Nation, R.L.; Li, J. Pharmacokinetics and pharmacodynamics of ‘old’ polymyxins: What is new? Diagn. Microbiol. Infect. Dis. 2012, 74, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Cheah, S.E.; Wang, J.; Nguyen, V.T.T.; Turnidge, J.D.; Li, J.; Nation, R.L. New pharmacokinetic/pharmacodynamic studies of systemically administered colistin against Pseudomonas aeruginosa and Acinetobacter baumannii in mouse thigh and lung infection models: Smaller response in lung infection. J. Antimicrob. Chemother. 2015, 70, 3291–3297. [Google Scholar] [CrossRef] [Green Version]
- Garonzik, S.M.; Li, J.; Thamlikitkul, V.; Paterson, D.L.; Shoham, S.; Jacob, J.; Silveira, F.P.; Forrest, A.; Nation, R.L. Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob. Agents Chemother. 2011, 55, 3284–3294. [Google Scholar] [CrossRef] [Green Version]
- Nation, R.L.; Velkov, T.; Li, J. Colistin and polymyxin B: Peas in a pod, or chalk and cheese? Clin. Infect. Dis. 2014, 59, 88–94. [Google Scholar] [CrossRef]
- Plachouras, D.; Karvanen, M.; Friberg, L.E.; Papadomichelakis, E.; Antoniadou, A.; Tsangaris, I.; Karaiskos, I.; Poulakou, G.; Kontopidou, F.; Armaganidis, A.; et al. Population pharmacokinetic analysis of colistin methanesulfonate and colistin after intravenous administration in critically ill patients with infections caused by gram-negative bacteria. Antimicrob. Agents Chemother. 2009, 53, 3430–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grégoire, N.; Mimoz, O.; Mégarbane, B.; Comets, E.; Chatelier, D.; Lasocki, S.; Gauzit, R.; Balayn, D.; Gobin, P.; Marchand, S.; et al. New colistin population pharmacokinetic data in critically ill patients suggesting an alternative loading dose rationale. Antimicrob. Agents Chemother. 2014, 58, 7324–7330. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Milne, R.W.; Nation, R.L.; Turnidge, J.D.; Smeaton, T.C.; Coulthard, K. Pharmacokinetics of colistin methanesulphonate and colistin in rats following an intravenous dose of colistin methanesulphonate. J. Antimicrob. Chemother. 2004, 53, 837–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Milne, R.W.; Nation, R.L.; Turnidge, J.D.; Smeaton, T.C.; Coulthard, K. Use of high-performance liquid chromatography to study the pharmacokinetics of colistin sulfate in rats following intravenous administration. Antimicrob. Agents Chemother. 2003, 47, 1766–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, B.T.; Pogue, J.M.; Zavascki, A.P.; Paul, M.; Daikos, G.L.; Forrest, A.; Giacobbe, D.R.; Viscoli, C.; Giamarellou, H.; Karaiskos, I.; et al. International Consensus Guidelines for the Optimal Use of the Polymyxins: Endorsed by the American College of Clinical Pharmacy (ACCP), European Society of Clinical Microbiology and Infectious Diseases (ESCMID), Infectious Diseases Society of America (IDSA), International Society for Anti-infective Pharmacology (ISAP), Society of Critical Care Medicine (SCCM), and Society of Infectious Diseases Pharmacists (SIDP). Pharmacotherapy 2019, 39, 10–39. [Google Scholar] [CrossRef] [Green Version]
- Nation, R.L.; Garonzik, S.M.; Thamlikitkul, V.; Giamarellos-Bourboulis, E.J.; Forrest, A.; Paterson, D.L.; Li, J.; Silveira, F.P. Dosing guidance for intravenous colistin in critically-ill patients. Clin. Infect. Dis. 2017, 64, 565–571. [Google Scholar] [CrossRef]
- Nation, R.L.; Garonzik, S.M.; Li, J.; Thamlikitkul, V.; Giamarellos-Bourboulis, E.J.; Paterson, D.L.; Turnidge, J.D.; Forrest, A.; Silveira, F.P. Updated US and European dose recommendations for intravenous colistin: How do they perform? Clin. Infect. Dis. 2016, 62, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Lee, W.; Kwa, A.L. Polymyxin B versus colistin: An update. Expert Rev. Anti-Infect. Ther. 2015, 13, 1481–1497. [Google Scholar] [CrossRef]
- Phe, K.; Lee, Y.; McDaneld, P.M.; Prasad, N.; Yin, T.; Figueroa, D.A.; Musick, W.L.; Cottreau, J.M.; Hu, M.; Tam, V.H. In vitro assessment and multicenter cohort study of comparative nephrotoxicity rates associated with colistimethate versus polymyxin B therapy. Antimicrob. Agents Chemother. 2014, 58, 2740–2746. [Google Scholar] [CrossRef] [Green Version]
- Kwa, A.L.; Abdelraouf, K.; Low, J.G.; Tam, V.H. Pharmacokinetics of polymyxin B in a patient with renal insufficiency: A case report. Clin. Infect. Dis. 2011, 52, 1280–1281. [Google Scholar] [CrossRef]
- Manchandani, P.; Thamlikitkul, V.; Dubrovskaya, Y.; Babic, J.T.; Lye, D.C.; Lee, L.S.; Tam, V.H. Population Pharmacokinetics of Polymyxin B. Clin. Pharmacol. Ther. 2018, 104, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Miglis, C.; Rhodes, N.J.; Avedissian, S.N.; Kubin, C.J.; Yin, M.T.; Nelson, B.C.; Pai, M.P.; Scheetz, M.H. Population Pharmacokinetics of Polymyxin B in Acutely Ill Adult Patients. Antimicrob. Agents Chemother. 2018, 62, e01475-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelraouf, K.; He, J.; Ledesma, K.R.; Hu, M.; Tam, V.H. Pharmacokinetics and renal disposition of polymyxin B in an animal model. Antimicrob. Agents Chemother. 2012, 56, 5724–5727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, A.M.; Landersdorfer, C.B.; Jacob, J.; Boniatti, M.M.; Dalarosa, M.G.; Falci, D.R.; Behle, T.F.; Bordinhao, R.C.; Wang, J.; Forrest, A.; et al. Population pharmacokinetics of intravenous polymyxin B in critically ill patients: Implications for selection of dosage regimens. Clin. Infect. Dis. 2013, 57, 524–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavascki, A.P.; Goldani, L.Z.; Cao, G.; Superti, S.V.; Lutz, L.; Barth, A.L.; Ramos, F.; Boniatti, M.M.; Nation, R.L.; Li, J. Pharmacokinetics of intravenous polymyxin B in critically ill patients. Clin. Infect. Dis. 2008, 47, 1298–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manchandani, P.; Zhou, J.; Ledesma, K.R.; Truong, L.D.; Chow, D.S.; Eriksen, J.L.; Tam, V.H. Characterization of Polymyxin B Biodistribution and Disposition in an Animal Model. Antimicrob. Agents Chemother. 2016, 60, 1029–1034. [Google Scholar] [CrossRef] [Green Version]
- Herrera, K.M.S.; Silva, F.K.D.; Oliveira, M.E.; Paiva, M.C.; Soares, A.C.; Siqueira Ferreira, J.M. First report of polymyxin B activity in Klebsiella pneumoniae biofilm. J. Chemother. 2019, 31, 127–131. [Google Scholar] [CrossRef]
- Boncompagni, S.R.; Micieli, M.; Di Maggio, T.; Aiezza, N.; Antonelli, A.; Giani, T.; Padoani, G.; Vailati, S.; Pallecchi, L.; Rossolini, G.M. Activity of fosfomycin/colistin combinations against planktonic and biofilm Gram-negative pathogens. J. Antimicrob. Chemother. 2022, 77, 2199–2208. [Google Scholar] [CrossRef]
- Hengzhuang, W.; Wu, H.; Ciofu, O.; Song, Z.; Høiby, N. In vivo pharmacokinetics/pharmacodynamics of colistin and imipenem in Pseudomonas aeruginosa biofilm infection. Antimicrob. Agents Chemother. 2012, 56, 2683–2690. [Google Scholar] [CrossRef] [Green Version]
- Brodt, A.M.; Stovold, E.; Zhang, L. Inhaled antibiotics for stable non-cystic fibrosis bronchiectasis: A systematic review. Eur. Respir. J. 2014, 44, 382–393. [Google Scholar] [CrossRef]
- Karagoz, G.; Kadanali, A.; Dede, B.; Sahin, O.T.; Comoglu, S.; Altug, S.B.; Naderi, S. Extensively drug-resistant Pseudomonas aeruginosa ventriculitis and meningitis treated with intrathecal colistin. Int. J. Antimicrob. Agents. 2014, 43, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Ordooei Javan, A.; Shokouhi, S.; Sahraei, Z. A review on colistin nephrotoxicity. Eur. J. Clin. Pharmacol. 2015, 71, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Kasiakou, S.K. Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit. Care 2006, 10, R27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landersdorfer, C.B.; Nation, R.L. Colistin: How should it be dosed for the critically ill? Semin. Respir. Crit. Care Med. 2015, 36, 126–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doremus, C.; Marcella, S.W.; Cai, B.; Echols, R.M. Utilization of Colistin Versus β-Lactam and β-Lactamase Inhibitor Agents in Relation to Acute Kidney Injury in Patients with Severe Gram-Negative Infections. Infect. Dis. Ther. 2021, 11, 187–199. [Google Scholar] [CrossRef]
- Durante-Mangoni, E.; Andini, R.; Signoriello, S.; Cavezza, G.; Murino, P.; Buono, S.; De Cristofaro, M.; Taglialatela, C.; Bassetti, M.; Malacarne, P.; et al. Acute kidney injury during colistin therapy: A prospective study in patients with extensively-drug resistant Acinetobacter baumannii infections. Clin. Microbiol. Infect. 2016, 22, 984–989. [Google Scholar] [CrossRef]
- Lyu, C.; Zhang, Y.; Liu, X.; Wu, J.; Zhang, J. Clinical efficacy and safety of polymyxins based versus non-polymyxins based therapies in the infections caused by carbapenem-resistant Acinetobacter baumannii: A systematic review and meta-analysis. BMC Infect. Dis. 2020, 20, 296. [Google Scholar] [CrossRef] [Green Version]
- Aslan, A.T.; Kırbaş, E.; Sancak, B.; Tanrıverdi, E.S.; Otlu, B.; Gürsoy, N.C.; Yılmaz, Y.A.; Tozluyurt, A.; Liste, Ü.; Bıçakcıgil, A.; et al. A retrospective observational cohort study of the clinical epidemiology of bloodstream infections due to carbapenem-resistant Klebsiella pneumoniae in an OXA-48 endemic setting. Int. J. Antimicrob. Agents 2022, 59, 106554. [Google Scholar] [CrossRef]
- Chien, H.T.; Lin, Y.C.; Sheu, C.C.; Hsieh, K.P.; Chang, J.S. Is colistin-associated acute kidney injury clinically important in adults? A systematic review and meta-analysis. Int. J. Antimicrob. Agents. 2020, 55, 105889. [Google Scholar] [CrossRef]
- Phe, K.; Shields, R.K.; Tverdek, F.P.; Aitken, S.L.; Guervil, D.J.; Lam, W.M.; Musgrove, R.J.; Luce, A.M.; Tam, V.H. Predicting the risk of nephrotoxicity in patients receiving colistimethate sodium: A multicentre, retrospective, cohort study. J. Antimicrob. Chemother. 2016, 71, 3585–3587. [Google Scholar] [CrossRef]
- Rigatto, M.H.; Behle, T.F.; Falci, D.R.; Freitas, T.; Lopes, N.T.; Nunes, M.; Costa, L.W.; Zavascki, A.P. Risk factors for acute kidney injury (AKI) in patients treated with polymyxin B and influence of AKI on mortality: A multicentre prospective cohort study. J. Antimicrob. Chemother. 2015, 70, 1552–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, R.; Dewan, A. Comparison of nephrotoxicity of colistin with polymyxin B administered in currently recommended doses: A prospective study. Ann. Clin. Microbiol. Antimicrob. 2018, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Ngamprasertchai, T.; Boonyasiri, A.; Charoenpong, L.; Nimitvilai, S.; Lorchirachoonkul, N.; Wattanamongkonsil, L.; Thamlikitkul, V. Effectiveness and safety of polymyxin B for the treatment of infections caused by extensively drug-resistant Gram-negative bacteria in Thailand. Infect. Drug Resist. 2018, 11, 1219–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniara, B.P.; Healy, L.E.; Doan, T.L. Risk of nephrotoxicity associated with nonrenally adjusted intravenous polymyxin B compared to traditional dosing. J. Pharm. Pract. 2020, 33, 287–292. [Google Scholar] [CrossRef]
- Mattos, K.P.H.; Gouvêa, I.R.; Quintanilha, J.C.F.; Cursino, M.A.; Vasconcelos, P.E.N.S.; Moriel, P. Polymyxin B clinical outcomes: A prospective study of patients undergoing intravenous treatment. J. Clin. Pharm. Ther. 2019, 44, 415–419. [Google Scholar] [CrossRef]
- Kamel, N.A.; Elsayed, K.M.; Awad, M.F.; Aboshanab, K.M.; El Borhamy, M.I. Multimodal Interventions to Prevent and Control Carbapenem-Resistant Enterobacteriaceae and Extended-Spectrum β-Lactamase Producer-Associated Infections at a Tertiary Care Hospital in Egypt. Antibiotics 2021, 10, 509. [Google Scholar] [CrossRef]
- Nelson, B.C.; Eiras, D.P.; Gomez-Simmonds, A.; Loo, A.S.; Satlin, M.J.; Jenkins, S.G.; Whittier, S.; Calfee, D.P.; Furuya, E.Y.; Kubin, C.J. Clinical outcomes associated with polymyxin B dose in patients with bloodstream infections due to carbapenem-resistant Gram-negative rods. Antimicrob. Agents Chemother. 2015, 59, 7000–7006. [Google Scholar] [CrossRef] [Green Version]
- Rigatto, M.H.; Falci, D.R.; Lopes, N.T.; Zavascki, A.P. Clinical features and mortality of patients on renal replacement therapy receiving polymyxin B. Int. J. Antimicrob. Agents. 2016, 47, 146–150. [Google Scholar] [CrossRef]
- Ismail, B.; Shafei, M.N.; Harun, A.; Ali, S.; Omar, M.; Deris, Z.Z. Predictors of polymyxin B treatment failure in Gram-negative healthcare-associated infections among critically ill patients. J. Microbiol. Immunol. Infect. 2018, 51, 763–769. [Google Scholar] [CrossRef]
- Teo, J.Q.; Chang, C.W.; Leck, H.; Tang, C.Y.; Lee, S.J.; Cai, Y.; Ong, R.T.; Koh, T.H.; Tan, T.T.; Kwa, A.L. Risk factors and outcomes associated with the isolation of polymyxin B and carbapenem-resistant Enterobacteriaceae spp.: A case-control study. Int. J. Antimicrob. Agents. 2019, 53, 657–662. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute Performance Standards for Antimicrobial Susceptibility Testing; 24th informational supplement; CLSI document M100-S30. Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2020. Available online: https://www.nih.org.pk/wp-content/uploads/2021/02/CLSI-2020.pdf (accessed on 13 January 2022).
- EUCAST Colistin Breakpoints Guidance Document 2022. Available online: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Guidance_documents/Colistin_guidance_2022.pdf (accessed on 4 November 2022).
- Roberts, J.A.; Lippman, J. Pharmacokinetic issues for antibiotics in the critically ill patients. Crit. Care Med. 2009, 37, 840–851. [Google Scholar] [CrossRef] [Green Version]
- Piperaki, E.T.; Tzouvelekis, L.S.; Miriagou, V.; Daikos, G.L. Carbapenem-resistant Acinetobacter baumannii: In pursuit of an effective treatment. Clin. Microbiol. Infect. 2019, 25, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Isler, B.; Özer, B.; Çınar, G.; Aslan, A.T.; Vatansever, C.; Falconer, C.; Dolapçı, İ.; Şimşek, F.; Tülek, N.; Demirkaya, H.; et al. Characteristics and outcomes of carbapenemase harbouring carbapenem-resistant Klebsiella spp. bloodstream infections: A multicentre prospective cohort study in an OXA-48 endemic setting. Eur. J. Clin. Microbiol. Infect. Dis. 2022, 41, 841–847. [Google Scholar] [CrossRef]
- Paterson, D.L.; Harris, P.N.A. Colistin resistance: A major breach in our last line of defence. Lancet Infect. Dis. 2016, 16, 132–133. [Google Scholar] [CrossRef] [PubMed]
- Magee, T.V.; Brown, M.F.; Starr, J.T.; Ackley, D.C.; Abramite, J.A.; Aubrecht, J.; Butler, A.; Crandon, J.L.; Dib-Hajj, F.; Flanagan, M.E.; et al. Discovery of Dap-3 polymyxin analogues for the treatment of multidrug-resistant Gram-negative nosocomial infections. J. Med. Chem. 2013, 56, 5079–5093. [Google Scholar] [CrossRef] [PubMed]
- Akhoundsadegh, N.; Belanger, C.R.; Hancock, R.E.W. Outer Membrane Interaction Kinetics of New Polymyxin B Analogs in Gram-Negative Bacilli. Antimicrob. Agents Chemother. 2019, 63, e00935-19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, C.; Wang, Q.; Wang, X.; Chen, H.; Li, H.; Zhang, F.; Wang, H. Evaluation of the in vitro activity of new polymyxin B analogue SPR206 against clinical MDR, colistin-resistant and tigecycline-resistant Gram-negative bacilli. J. Antimicrob. Chemother. 2020, 75, 2609–2615. [Google Scholar] [CrossRef]
- Mendes, R.E.; Sader, H.S.; Arends, S.J.R.; Cotroneo, N.; Critchley, I.A.; Castanheira, M. In vitro activity of SPR206 and comparator compounds against Enterobacterales isolates responsible for infections in United States Hospitals. In Proceedings of the ID Week 2022, Washington, DC, USA, 19–23 October 2022. [Google Scholar]
- Mendes, R.E.; Sader, H.S.; Arends, S.J.R.; Cotroneo, N.; Critchley, I.A.; Castanheira, M. Activity of SPR206 and comparator agents against Pseudomonas aeruginosa and Acinetobacter baumannii causing infections in United States Hospitals. In Proceedings of the ID Week 2022, Washington, DC, USA, 19–23 October 2022. [Google Scholar]
- Karvanen, M.; Malmberg, C.; Lagerbäck, P.; Friberg, L.E.; Cars, O. Colistin Is Extensively Lost during Standard in Vitro Experimental Conditions. Antimicrob. Agents Chemother. 2017, 61, e00857-17. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.; Abbott, E.; Abdulle, O.; Boakes, S.; Coleman, S.; Divall, N.; Duperchy, E.; Moss, S.; Rivers, D.; Simonovic, M.; et al. Design of Next Generation Polymyxins with Lower Toxicity: The Discovery of SPR206. ACS Infect. Dis. 2019, 5, 1645–1656. [Google Scholar] [CrossRef]
- Bowers, D.R.; Cao, H.; Zhou, J.; Ledesma, K.R.; Sun, D.; Lomovskaya, O.; Tam, V.H. Assessment of minocycline and polymyxin B combination against Acinetobacter baumannii. Antimicrob. Agents Chemother. 2015, 59, 2720–2725. [Google Scholar] [CrossRef]
- He, J.; Gao, S.; Hu, M.; Chow, D.S.; Tam, V.H. A validated ultra-performance liquid chromatography-tandem mass spectrometry method for the quantification of polymyxin B in mouse serum and epithelial lining fluid: Application to pharmacokinetic studies. J. Antimicrob. Chemother. 2013, 68, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Lister, L.; Utley, L.; Bleavins, M. A GLP 14 day repeat dose toxicology study of SPR206 in monkeys, poster 146. In Proceedings of the ASM ESCMID 2018, Lisbon, Portugal, 4–7 September 2018. [Google Scholar]
- Grosser, L.; Heang, K.; Teague, J.; Warn, P.; Corbett, D.; Dawson, M.J.; Rubio, A. In vivo efficacy of SPR206 in murine lung and thigh infection models caused by multidrug resistant pathogens Pseudomonas aeruginosa and Acinetobacter baumannii, poster 139. In Proceedings of the ASM ESCMID 2018, Lisbon, Portugal, 4–7 September 2018. [Google Scholar]
- Pinchman, J.R.; Boger, D.L. Probing the role of the vancomycin E-ring aryl chloride: Selective divergent synthesis and evaluation of alternatively substituted E-ring analogues. J. Med. Chem. 2013, 56, 4116–4124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruss, J.; Lister, T.; Gupta, V.K.; Stone, E.; Morelli, L.; Lei, Y.; Melnick, D. Single- and Multiple-Ascending-Dose Study of the Safety, Tolerability, and Pharmacokinetics of the Polymyxin Derivative SPR206. Antimicrob. Agents Chemother. 2021, 65, e0073921. [Google Scholar] [CrossRef]
- Rodvold, K.A.; Bader, J.; Gupta, V.; Lister, T.; Srivastava, P.; Bruss, J. SPR206 Pharmacokinetics (PK) in Plasma, Epithelial Lining Fluid (ELF), and Alveolar Macrophages (AM) in Healthy Adult Subjects. In Proceedings of the ID Week 2022, Washington, DC, USA, 19–23 October 2022. [Google Scholar]
- Roberts, K.D.; Zhu, Y.; Azad, M.A.K.; Han, M.L.; Wang, J.; Wang, L.; Yu, H.H.; Horne, A.S.; Pinson, J.A.; Rudd, D. A synthetic lipopeptide targeting top-priority multidrug-resistant Gram-negative pathogens. Nat. Commun. 2022, 13, 1625. [Google Scholar] [CrossRef] [PubMed]
- Griffith, D.C.; Carmeli, Y.; Gehrke, S.; Morgan, E.E.; Dudley, M.N.; Loutit, J.S. A Phase 1 Study of the Safety, Tolerability, and Pharmacokinetics of Multiple Doses of the Lipopeptide QPX9003 in Healthy Adult Subjects. In Proceedings of the ID Week 2022, Washington, DC, USA, 19–23 October 2022. [Google Scholar]
- Wu, S.; Yin, D.; Zhi, P.; Guo, Y.; Yang, Y.; Zhu, D.; Hu, F. In Vitro Activity of MRX-8 and Comparators against Clinical Isolated Gram-Negative Bacilli in China. Front. Cell Infect. Microbiol. 2022, 12, 829592. [Google Scholar] [CrossRef]
- Duncan, L.R.; Wang, W.; Sader, H.S. In Vitro Potency and Spectrum of the Novel Polymyxin MRX-8 Tested against Clinical Isolates of Gram-Negative Bacteria. Antimicrob. Agents Chemother. 2022, 66, e0013922. [Google Scholar] [CrossRef] [PubMed]
- Lepak, A.J.; Wang, W.; Andes, D.R. Pharmacodynamic Evaluation of MRX-8, a Novel Polymyxin, in the Neutropenic Mouse Thigh and Lung Infection Models against Gram-Negative Pathogens. Antimicrob. Agents Chemother. 2020, 64, e01517–e01520. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Bohlmann, L.; Conroy, T.; Jen, F.E.; Everest-Dass, A.; Hansford, K.A.; Bolisetti, R.; El-Deeb, I.M.; Forde, B.M.; Phan, M.D.; et al. Repurposing a neurodegenerative disease drug to treat Gram-negative antibiotic-resistant bacterial sepsis. Sci. Transl. Med. 2020, 12, eabb3791. [Google Scholar] [CrossRef]
- Huntington Study Group Reach2HD Investigators. Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015, 14, 39–47. [Google Scholar] [CrossRef]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Keller, B.; Hayes, A.J.; Ong, C.Y.; Harbison-Price, N.; El-Deeb, I.M.; Li, G.; Keller, N.; Bohlmann, L.; Brouwer, S.; et al. Neurodegenerative Disease Treatment Drug PBT2 Breaks Intrinsic Polymyxin Resistance in Gram-Positive Bacteria. Antibiotics 2022, 11, 449. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
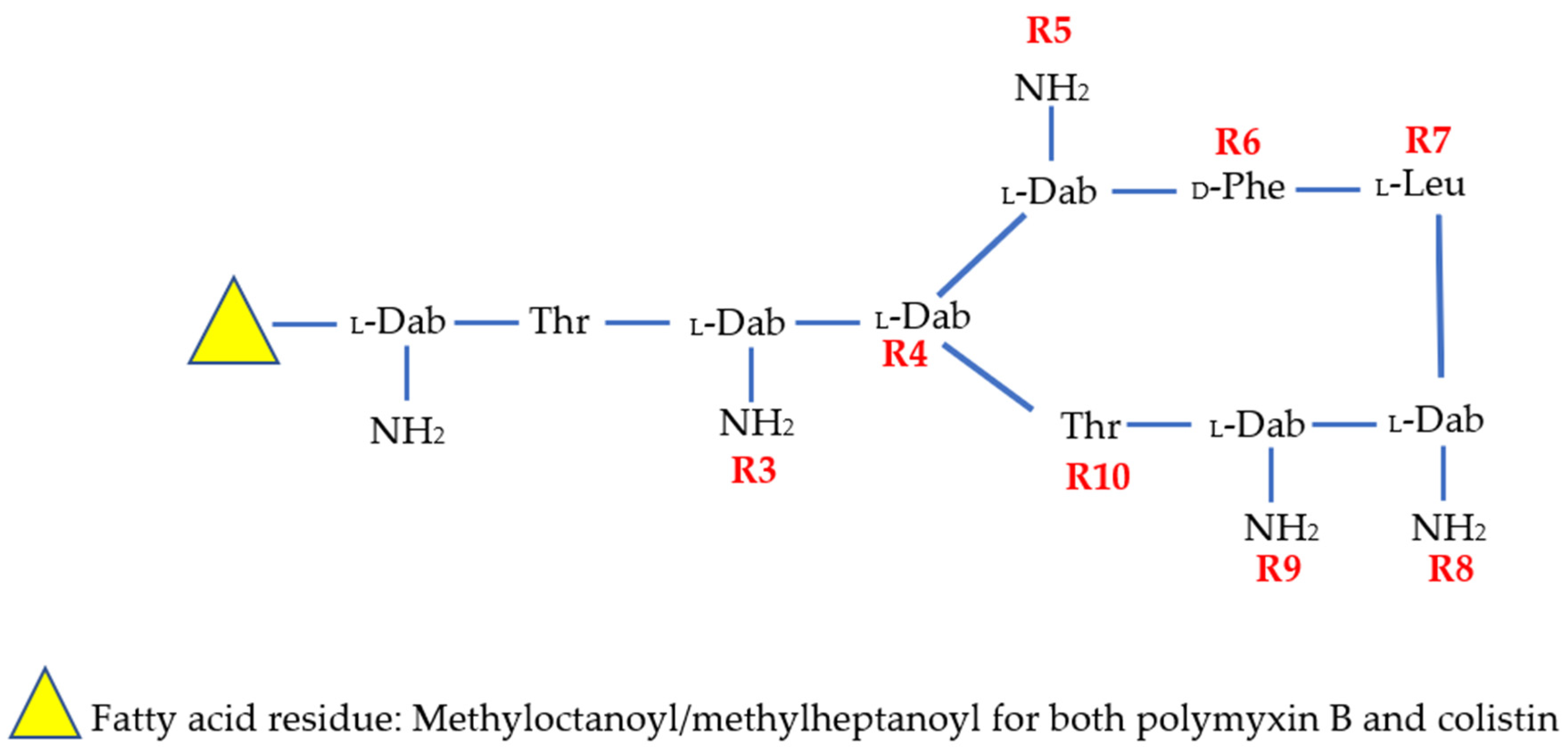
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | R9 | R10 | N-Terminus |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Colistin | -Dab | -Thr | -Dab | -Dab | -Dab | -DLeu | -Leu | -Dab | -Dab | -Thr | Methyloctanoyl/methylheptanoyl |
| PMB | -Dab | -Thr | -Dab | -Dab | -Dab | -DPhe | -Leu | -Dab | -Dab | -Thr | Methyloctanoyl/methylheptanoyl |
| SPR206 | - | -Thr | -Dab | -Dab | -Dab | -DPhe | -Leu | -Dab | -Dab | -Thr | (3S)-4-amino-3-(3-cholorophenyl)butanoyl |
| MRX-8 | -Dab | -Thr | -Dab | -Dab | -Dab | -DPhe | -Leu | -Dab | -Dab | -Thr | 3-(2,2-Dimethyl-butanoyloxy)-propanoyl (ester bond) |
| SPR741 | - | -Thr | -DSer | -Dab | -Dab | -DPhe | -Leu | -Dab | -Dab | -Thr | Acetyl |
| QPX9003 | -Dab | -Thr | -Dap | -Dab | -Dab | -DLeu | -Abu | -Dab | -Dab | -Thr | 2,4 Dicholorobenzoyl |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslan, A.T.; Akova, M.; Paterson, D.L. Next-Generation Polymyxin Class of Antibiotics: A Ray of Hope Illuminating a Dark Road. Antibiotics 2022, 11, 1711. https://doi.org/10.3390/antibiotics11121711
Aslan AT, Akova M, Paterson DL. Next-Generation Polymyxin Class of Antibiotics: A Ray of Hope Illuminating a Dark Road. Antibiotics. 2022; 11(12):1711. https://doi.org/10.3390/antibiotics11121711
Chicago/Turabian StyleAslan, Abdullah Tarık, Murat Akova, and David L. Paterson. 2022. "Next-Generation Polymyxin Class of Antibiotics: A Ray of Hope Illuminating a Dark Road" Antibiotics 11, no. 12: 1711. https://doi.org/10.3390/antibiotics11121711
APA StyleAslan, A. T., Akova, M., & Paterson, D. L. (2022). Next-Generation Polymyxin Class of Antibiotics: A Ray of Hope Illuminating a Dark Road. Antibiotics, 11(12), 1711. https://doi.org/10.3390/antibiotics11121711