
Oligopeptide Sortase Inhibitor Modulates Staphylococcus aureus Cell Adhesion and Biofilm Formation

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. LPRDA Synthesis and Characterization
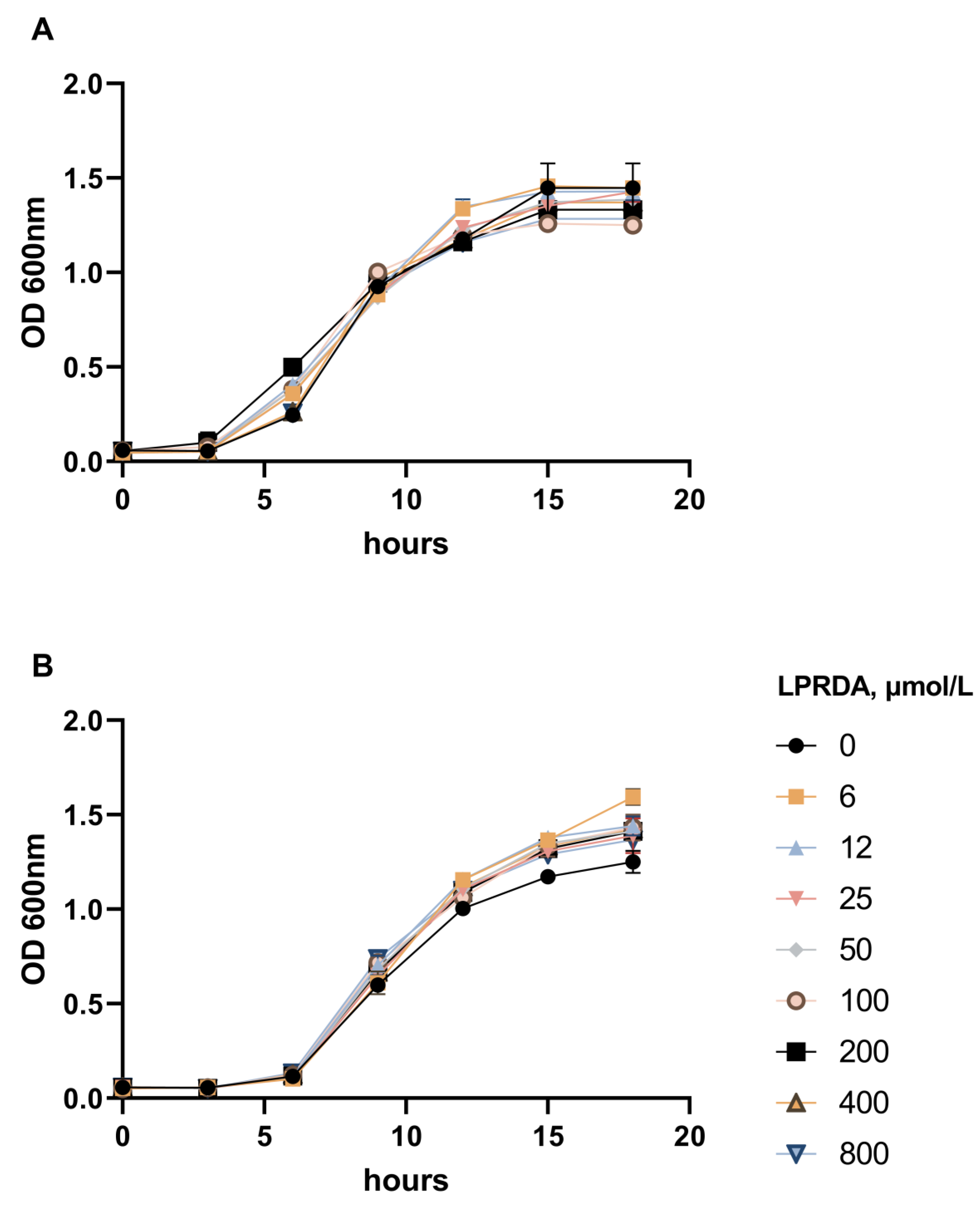
2.2. Study of LPRDA Bactericidal Activity
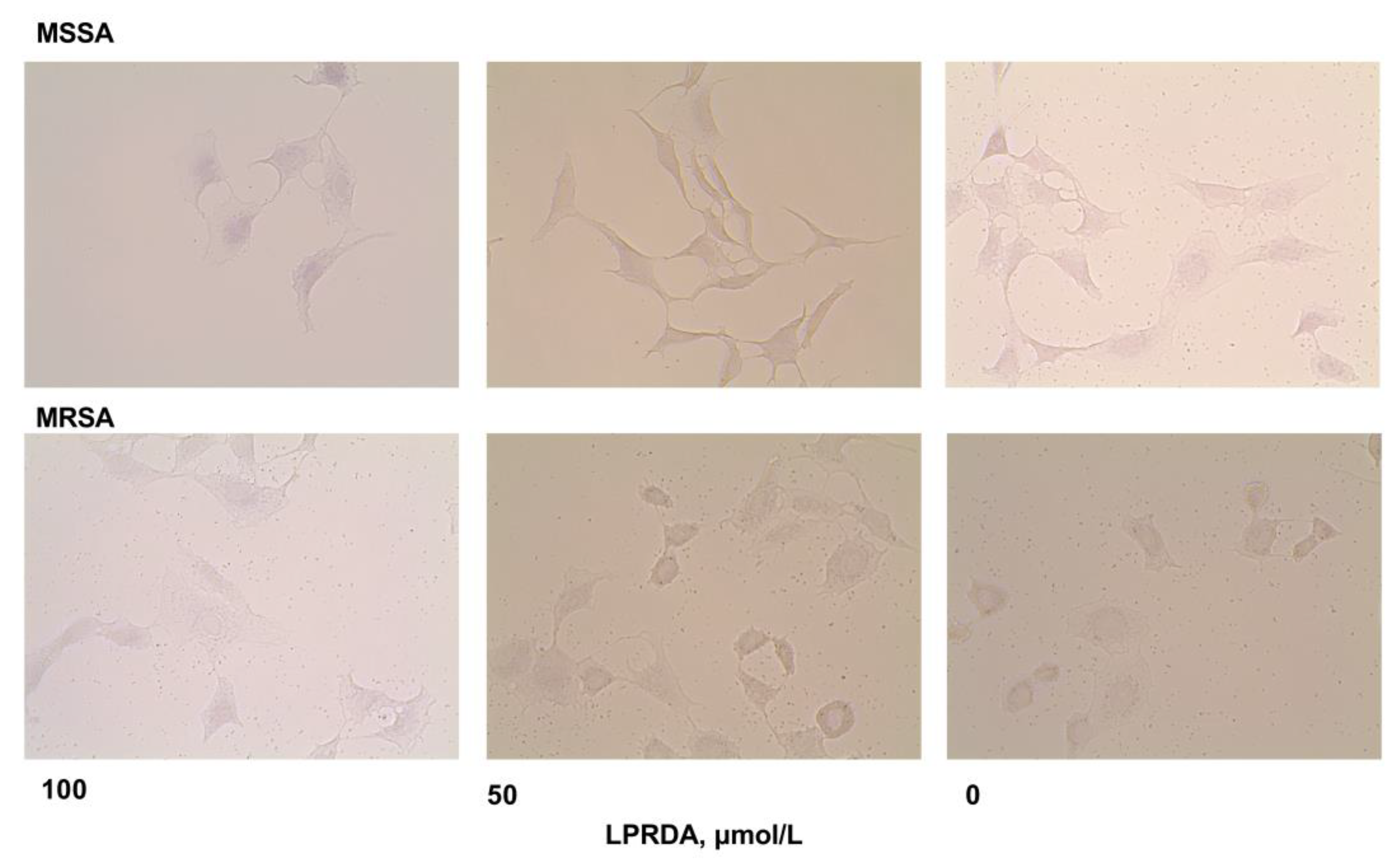
2.3. Adhesion of S. aureus Strains Modified by LPRDA to Vero Cells
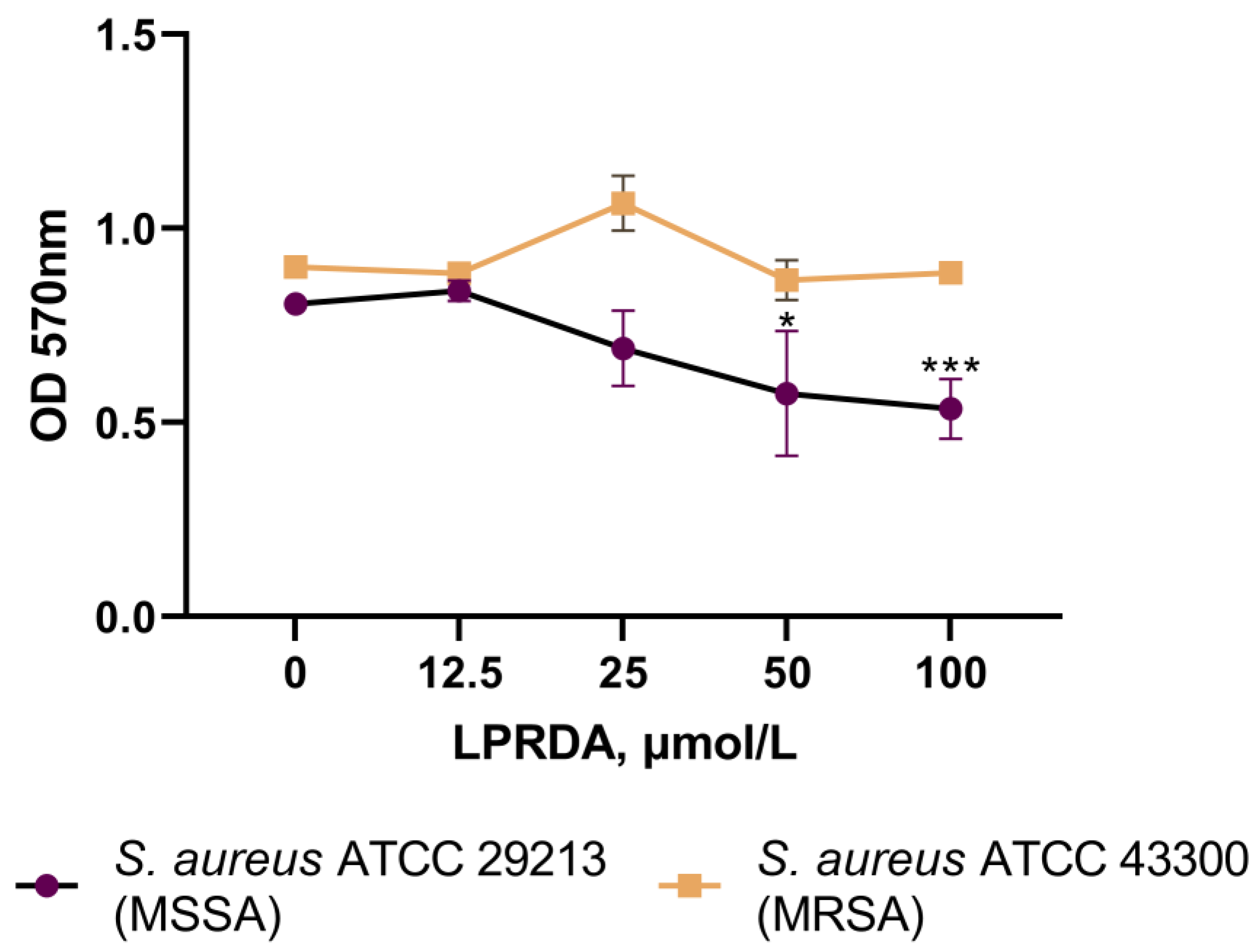
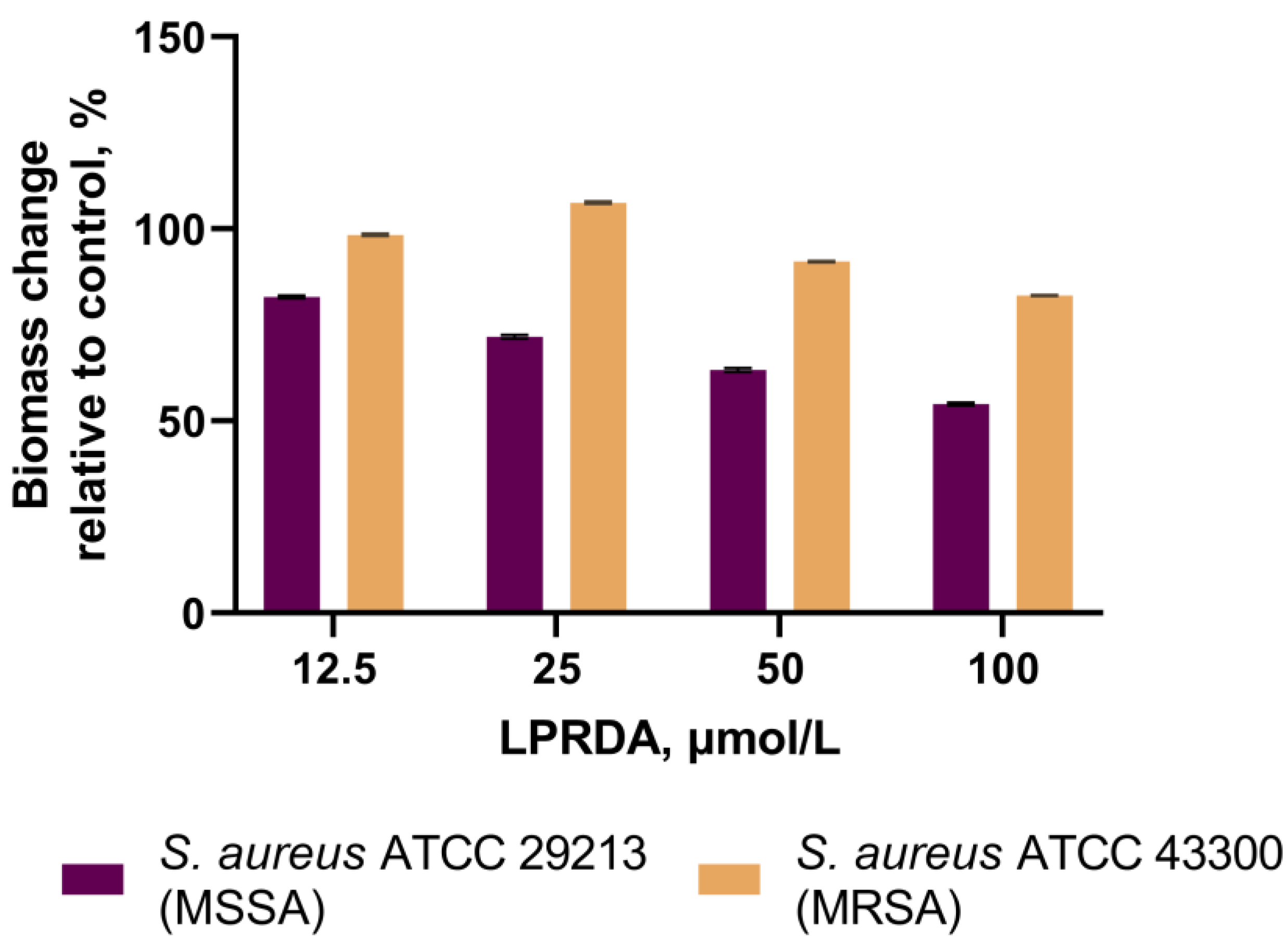
2.4. Biofilm Formation Assays
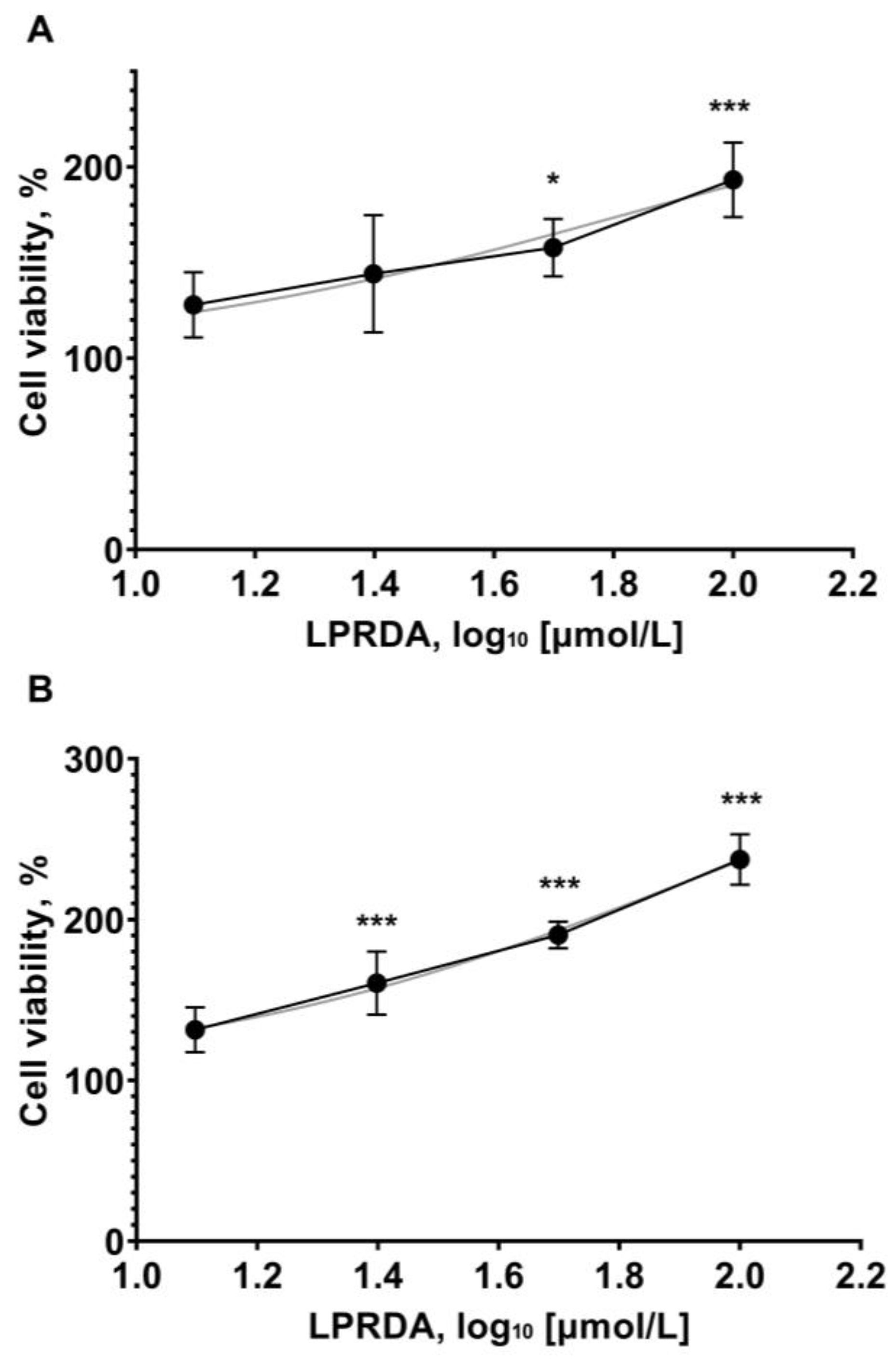
2.5. Viability of Vero Cells in the Presence of LPRDA
3. Discussion
4. Materials and Methods
4.1. Reagents, Cell Cultures, and Strains
4.2. S. aureus Viability in the Presence of LPRDA
4.3. Bacterial Adhesion to Vero Cells
4.4. Inhibition of Biofilm Formation Assay
4.5. Eukaryotic Cell Viability Assay
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dickey, S.; Cheung, G.; Otto, M. Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- Giormezis, N.; Doudoulakakis, A.; Tsilipounidaki, K.; Militsopoulou, M.; Kalogeras, G.; Stamouli, V.; Kolonitsiou, F.; Petinaki, E.; Lebessi, E.; Spiliopoulou, I. Emergence of a mupirocin-resistant, methicillin-susceptible Staphylococcus aureus clone associated with skin and soft tissue infections in Greece. BMC Microbiol. 2021, 21, 203. [Google Scholar] [CrossRef]
- Cheung, G.Y.C.; Bae, J.S.; Otto, M. Pathogenicity and virulence of Staphylococcus aureus. Virulence 2021, 12, 547–569. [Google Scholar] [CrossRef]
- Ofek, I.; Hasty, D.L.; Sharon, N. Anti-adhesion therapy of bacterial diseases: Prospects and problems. FEMS Immunol. Med. Microbiol. 2003, 38, 181–191. [Google Scholar] [CrossRef]
- Krachler, A.M.; Orth, K. Targeting the bacteria–host interface. Virulence 2013, 4, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; von Ossowski, I.; Krishnan, V. Exploiting pilus-mediated bacteria-host interactions for health benefits. Mol. Aspects Med. 2021, 81, 100998. [Google Scholar] [CrossRef]
- Foster, T.; Geoghegan, J.; Ganesh, V.; Höök, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.J. The MSCRAMM family of cell-wall-anchored surface proteins of gram-positive cocci. Trends Microbiol. 2019, 27, 927–941. [Google Scholar] [CrossRef]
- Leonard, A.C.; Petrie, L.E.; Cox, G. Bacterial anti-adhesives: Inhibition of Staphylococcus aureus nasal colonization. ACS Infect. Dis. 2019, 5, 1668–1681. [Google Scholar] [CrossRef]
- Bhat, A.H.; Nguyen, M.T.; Das, A.; Ton-That, H. Anchoring surface proteins to the bacterial cell wall by sortase enzymes: How it started and what we know now. Curr. Opin. Microbiol. 2021, 60, 73–79. [Google Scholar] [CrossRef]
- Sapra, R.; Rajora, A.K.; Kumar, P.; Maurya, G.P.; Pant, N.; Haridas, V. Chemical biology of sortase a inhibition: A gateway to anti-infective therapeutic agents. J. Med. Chem. 2021, 64, 13097–13130. [Google Scholar] [CrossRef]
- Nitulescu, G.; Margina, D.; Zanfirescu, A.; Olaru, O.T.; Nitulescu, G.M. Targeting bacterial sortases in search of anti-virulence therapies with low risk of resistance development. Pharmaceuticals 2021, 14, 415. [Google Scholar] [CrossRef]
- Ha, M.W.; Yi, S.W.; Paek, S.M. Design and synthesis of small molecules as potent Staphylococcus aureus sortase a inhibitors. Antibiotics 2020, 9, 706. [Google Scholar] [CrossRef]
- Alharthi, S.; Alavi, S.E.; Moyle, P.M.; Ziora, Z.M. Sortase A (SrtA) inhibitors as an alternative treatment for superbug infections. Drug Discov. Today 2021, 26, 2164–2172. [Google Scholar] [CrossRef]
- Kudryavtsev, K.V.; Fedotcheva, T.A.; Shimanovsky, N.L. Inhibitors of sortases of gram-positive bacteria and their role in the treatment of infectious diseases. Pharm. Chem. J. 2021, 55, 751–756. [Google Scholar] [CrossRef]
- Zrelovs, N.; Kurbatska, V.; Rudevica, Z.; Leonchiks, A.; Fridmanis, D. Sorting out the superbugs: Potential of sortase a inhibitors among other antimicrobial strategies to tackle the problem of antibiotic resistance. Antibiotics 2021, 10, 164. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.; Pan, J.; Dong, J.; Zhou, X.; Niu, X.; Deng, X. Oligopeptide targeting sortase a as potential anti-infective therapy for Staphylococcus aureus. Front. Microbiol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Bozhkova, S.A.; Gordina, E.M.; Labutin, D.V.; Sokolov, M.N.; Kudryavtsev, K.V. Synthetic low-molecular-mass compounds as potential inhibitors of Staphylococcus aureus adhesion in experiment. Pharm. Chem. J. 2022, 55, 1269–1275. [Google Scholar] [CrossRef]
- Shulga, D.A.; Kudryavtsev, K.V. Selection of promising novel fragment sized S. aureus SrtA noncovalent inhibitors based on QSAR and docking modeling studies. Molecules 2021, 26, 7677. [Google Scholar] [CrossRef]
- Shulga, D.A.; Kudryavtsev, K.V. Theoretical studies of Leu-Pro-Arg-Asp-Ala pentapeptide (LPRDA) binding to Sortase A of Staphylococcus aureus. Molecules 2022, 27, 8182. [Google Scholar] [CrossRef]
- Coin, I.; Beyermann, M.; Bienert, M. Solid-phase peptide synthesis: From standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2007, 2, 3247–3256. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Sig. Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Matano, L.M.; Morris, H.G.; Wood, B.M.; Meredith, T.C.; Walker, S. Accelerating the discovery of antibacterial compounds using pathway-directed whole cell screening. Bioorg. Med. Chem. 2016, 24, 6307–6314. [Google Scholar] [CrossRef] [Green Version]
- Fernandes de Oliveira, L.M.; Steindorff, M.; Darisipudi, M.N.; Mrochen, D.M.; Trübe, P.; Bröker, B.M.; Brönstrup, M.; Tegge, W.; Holtfreter, S. Discovery of Staphylococcus aureus adhesion inhibitors by automated imaging and their characterization in a mouse model of persistent nasal colonization. Microorganisms 2021, 9, 631. [Google Scholar] [CrossRef]
- Bozhkova, S.A.; Gordina, E.M.; Markov, M.A.; Afanasyev, A.V.; Artyukh, V.A.; Malafeev, K.V.; Ivan’kova, E.M. The effect of vancomycin and silver combination on the duration of antibacterial activity of bone cement and methicillin-resistant Staphylococcus aureus biofilm formation. Traumatol. Orthoped. Russia 2021, 27, 54–64. (In Russian) [Google Scholar] [CrossRef]
- Bozhkova, S.A.; Krasnova, M.V.; Polyakova, E.M.; Rukina, A.N.; Shabanova, V.V. Biofilm formation by clinical isolates of S. aureus and S. epidermidis in prosthetic joint infection. Clin. Microbiol. Antimicrob. Chemother. 2014, 16, 149–156. (In Russian) [Google Scholar]
- Gordina, E.M.; Bozhkova, S.A. Bacterial biofilms in orthopedics: The problem and possible prospects for prevention. Russ. Med. J. 2021, 8, 29–32. (In Russian) [Google Scholar]
- Houston, P.; Rowe, S.E.; Pozzi, C.; Waters, E.M.; O’Gara, J.P. Essential role for the major autolysin in the fibronectin-binding protein-mediated Staphylococcus aureus biofilm phenotype. Infect. Immun. 2011, 79, 1153–1165. [Google Scholar] [CrossRef] [Green Version]
- Estrela, A.B.; Abraham, W.-R. Combining biofilm-controlling compounds and antibiotics as a promising new way to control biofilm infections. Pharmaceuticals 2010, 3, 1374–1393. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, G.A. Microtiter dish biofilm formation assay. J. Vis. Exp. 2011, 47, 2437. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3182663/ (accessed on 17 May 2020). [CrossRef]
- Stepanović, S.; Vuković, D.; Hola, V.; Bonaventura, G.D.; Djukić, S.; Ćirković, I.; Ruzicka, F. Quantification of biofilm in microtiter plates: Overview of testing conditions and practical recommendations for assessment of biofilm production by staphylococci. APMIS 2007, 115, 891–899. [Google Scholar] [CrossRef]
- Koch, J.A.; Pust, T.M.; Cappellini, A.J.; Mandell, J.B.; Ma, D.; Shah, N.B.; Brothers, K.M.; Urish, K.L. Staphylococcus epidermidis biofilms have a high tolerance to antibiotics in periprosthetic joint infection. Life 2020, 10, 253. [Google Scholar] [CrossRef]
- Kumari, P.; Nath, Y.; Murty, U.S.; Ravichandiran, V.; Mohan, U. Sortase a mediated bioconjugation of common epitopes decreases biofilm formation in Staphylococcus aureus. Front. Microbiol. 2020, 11, 1702. [Google Scholar] [CrossRef]
- Oh, K.B.; Nam, K.W.; Ahn, H.; Shin, J.; Kim, S.; Mar, W. Therapeutic effect of (Z)-3-(2,5-dimethoxyphenyl)-2-(4-methoxyphenyl)acrylonitrile (DMMA) against Staphylococcus aureus infection in a murine model. Biochem. Biophys. Res. Commun. 2010, 396, 440–444. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, H.; Zhu, K.; Gong, S.; Dramsi, S.; Wang, Y.T.; Li, J.; Chen, F.; Zhang, R.; Zhou, L.; et al. Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase. Proc. Natl. Acad. Sci. USA 2014, 111, 13517–13522. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Bi, C.; Cai, H.; Liu, B.; Zhong, X.; Deng, X.; Wang, T.; Xiang, H.; Niu, X.; Wang, D. The therapeutic effect of chlorogenic acid against Staphylococcus aureus infection through sortase A inhibition. Front. Microbiol. 2015, 6, 1031. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, P.; He, X.; Yuan, Z.W.; Yin, Z.Q.; Fu, H.; Lin, J.; He, C.; Liang, X.; Lv, C.; Shu, G.; et al. Erianin against Staphylococcus aureus infection via inhibiting sortase A. Toxins 2018, 10, 385. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Zhang, T.; Guan, X.N.; Dong, Z.; Lan, L.; Yang, S.; Yang, C.G. Tideglusib and its analogues as inhibitors of Staphylococcus aureus SrtA. J. Med. Chem. 2020, 63, 8442–8457. [Google Scholar] [CrossRef]
- Song, W.; Wang, L.; Zhao, Y.; Lanzi, G.; Wang, X.; Zhang, C.; Guan, J.; Wang, W.; Guo, X.; Meng, Y.; et al. Hibifolin, a natural sortase a inhibitor, attenuates the pathogenicity of Staphylococcus aureus and enhances the antibacterial activity of cefotaxime. Microbiol. Spectrum 2022, 10, e00950-22. [Google Scholar] [CrossRef]
- Wang, X.; Luan, Y.; Hou, J.; Jiang, T.; Zhao, Y.; Song, W.; Wang, L.; Kong, X.; Guan, J.; Song, D.; et al. The protection effect of rhodionin against methicillin-resistant Staphylococcus aureus-induced pneumonia through sortase A inhibition. World J. Microbiol. Biotechnol. 2023, 39, 18. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. aureus Cell Strain | LPRDA Concentration (µmol/L) | AI (Mean ± SD); p * | PI (Mean ± SD,%); p * | ML (Mean ± SD); p * |
|---|---|---|---|---|
| ATCC 29213 (MSSA) | 100 | 3.4 ± 0.4; 0.021 | 84.0 ± 2.8; 0.021 | 286.9 ± 38.8; 0.009 |
| 50 | 2.9 ± 0.2; 0.01 | 85.0 ± 4.6; 0.054 | 240.8 ± 9.2; 0.002 | |
| 25 | 4.8 ± 0.2; 0.43 | 99.0 ± 0.8; 0.373 | 471.1 ± 26.5; 0.414 | |
| 12.5 | 5.4; 0.53 | 100; 0.53 | 542.9; 0.53 | |
| 0 (control) | 5.2 ± 0.3; – | 100; – | 516.7 ± 31.1; – | |
| ATCC 43300 (MRSA) | 100 | 6.8 ± 0.1; 0.01 | 100; – | 679.2 ± 13.6; 0.01 |
| 50 | 7.3 ± 0.2; 0.332 | 100; – | 727.4 ± 18.2; 0.33 | |
| 25 | 7.4 ± 0.1; 0.516 | 100; – | 744.2 ± 8.3; 0.516 | |
| 12.5 | 7.9; 0.056 | 100; – | 790.2; 0.056 | |
| 0 (control) | 7.6 ± 0.1; – | 100; – | 755.9 ± 10.6; – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozhkova, S.A.; Gordina, E.M.; Labutin, D.V.; Kudryavtsev, K.V. Oligopeptide Sortase Inhibitor Modulates Staphylococcus aureus Cell Adhesion and Biofilm Formation. Antibiotics 2022, 11, 1836. https://doi.org/10.3390/antibiotics11121836
Bozhkova SA, Gordina EM, Labutin DV, Kudryavtsev KV. Oligopeptide Sortase Inhibitor Modulates Staphylococcus aureus Cell Adhesion and Biofilm Formation. Antibiotics. 2022; 11(12):1836. https://doi.org/10.3390/antibiotics11121836
Chicago/Turabian StyleBozhkova, Svetlana A., Ekaterina M. Gordina, Dmitry V. Labutin, and Konstantin V. Kudryavtsev. 2022. "Oligopeptide Sortase Inhibitor Modulates Staphylococcus aureus Cell Adhesion and Biofilm Formation" Antibiotics 11, no. 12: 1836. https://doi.org/10.3390/antibiotics11121836
APA StyleBozhkova, S. A., Gordina, E. M., Labutin, D. V., & Kudryavtsev, K. V. (2022). Oligopeptide Sortase Inhibitor Modulates Staphylococcus aureus Cell Adhesion and Biofilm Formation. Antibiotics, 11(12), 1836. https://doi.org/10.3390/antibiotics11121836