
Antimicrobial Susceptibility and Molecular Features of Colonizing Isolates of Pseudomonas aeruginosa and the Report of a Novel Sequence Type (ST) 3910 from Thailand

 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Results
2.1. Antimicrobial Susceptibility Profiles
2.2. Genome Assembly Quality
2.3. Sequence Types and Serotypes
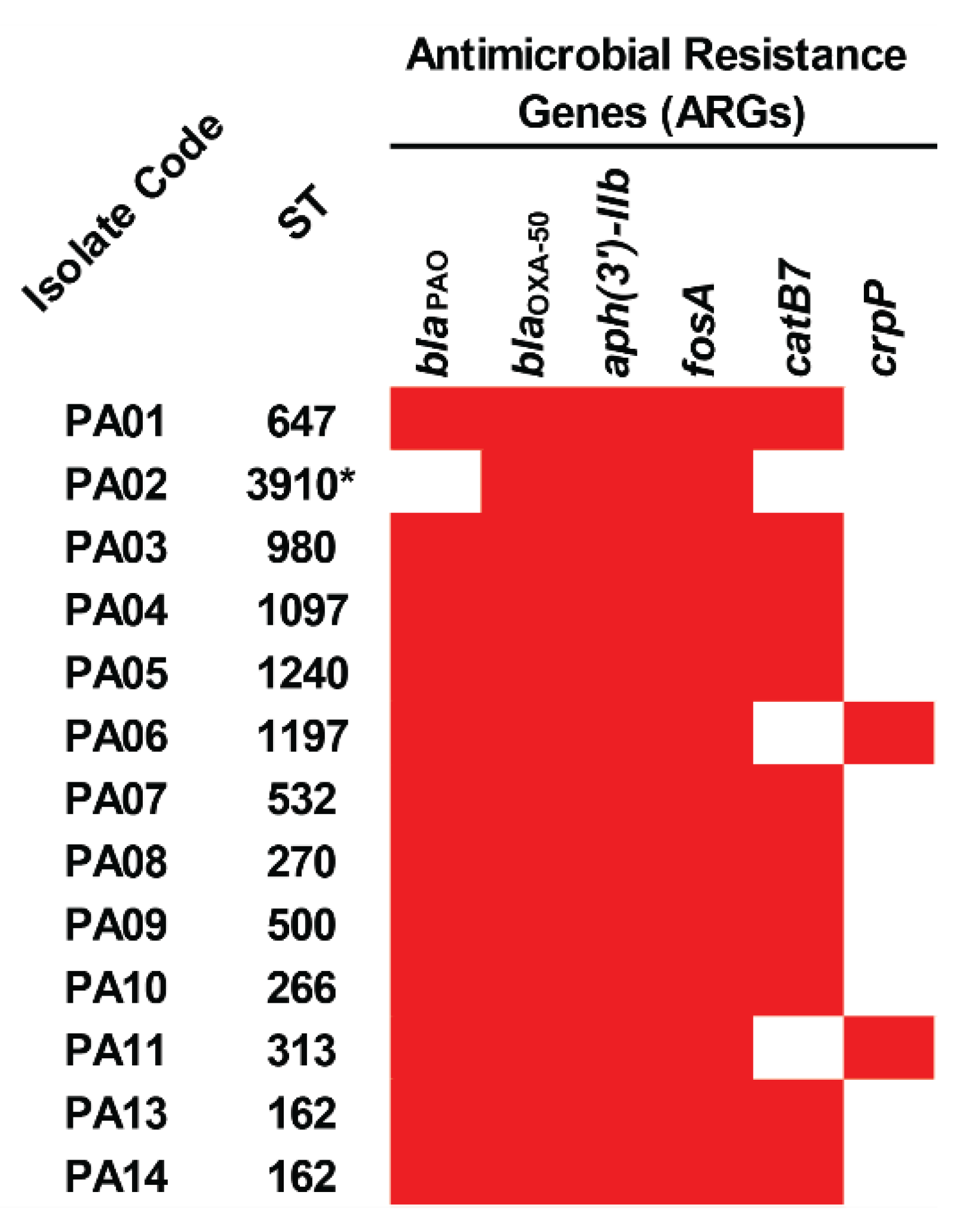
2.4. Acquired Antimicrobial Resistance Genes
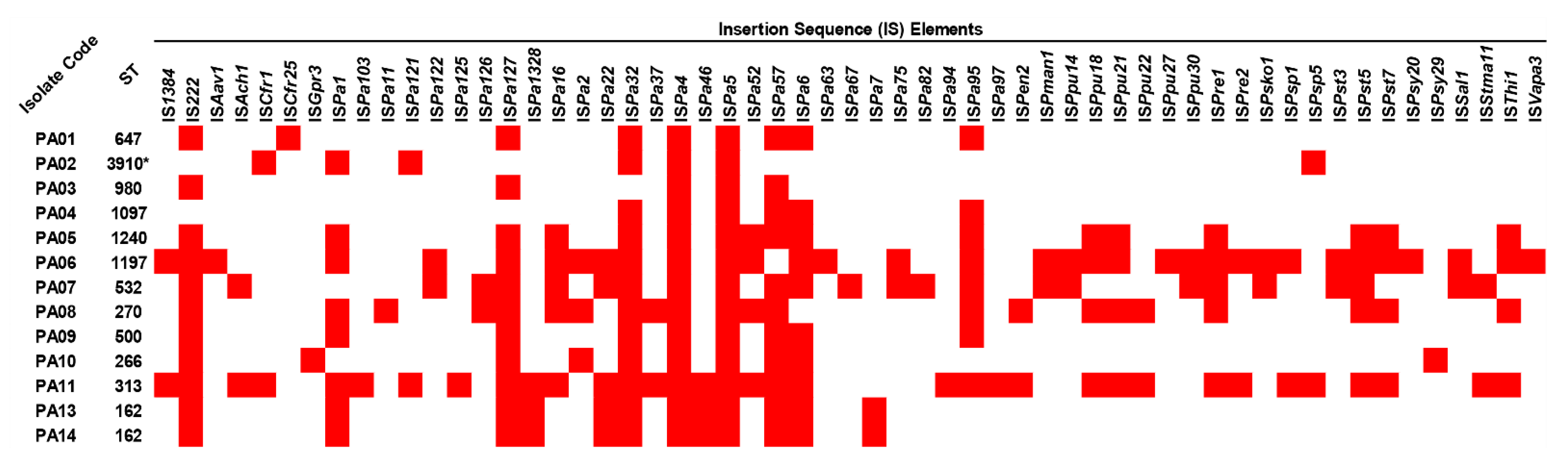
2.5. Insertion Sequence Elements
2.6. Virulence-Associated Genes
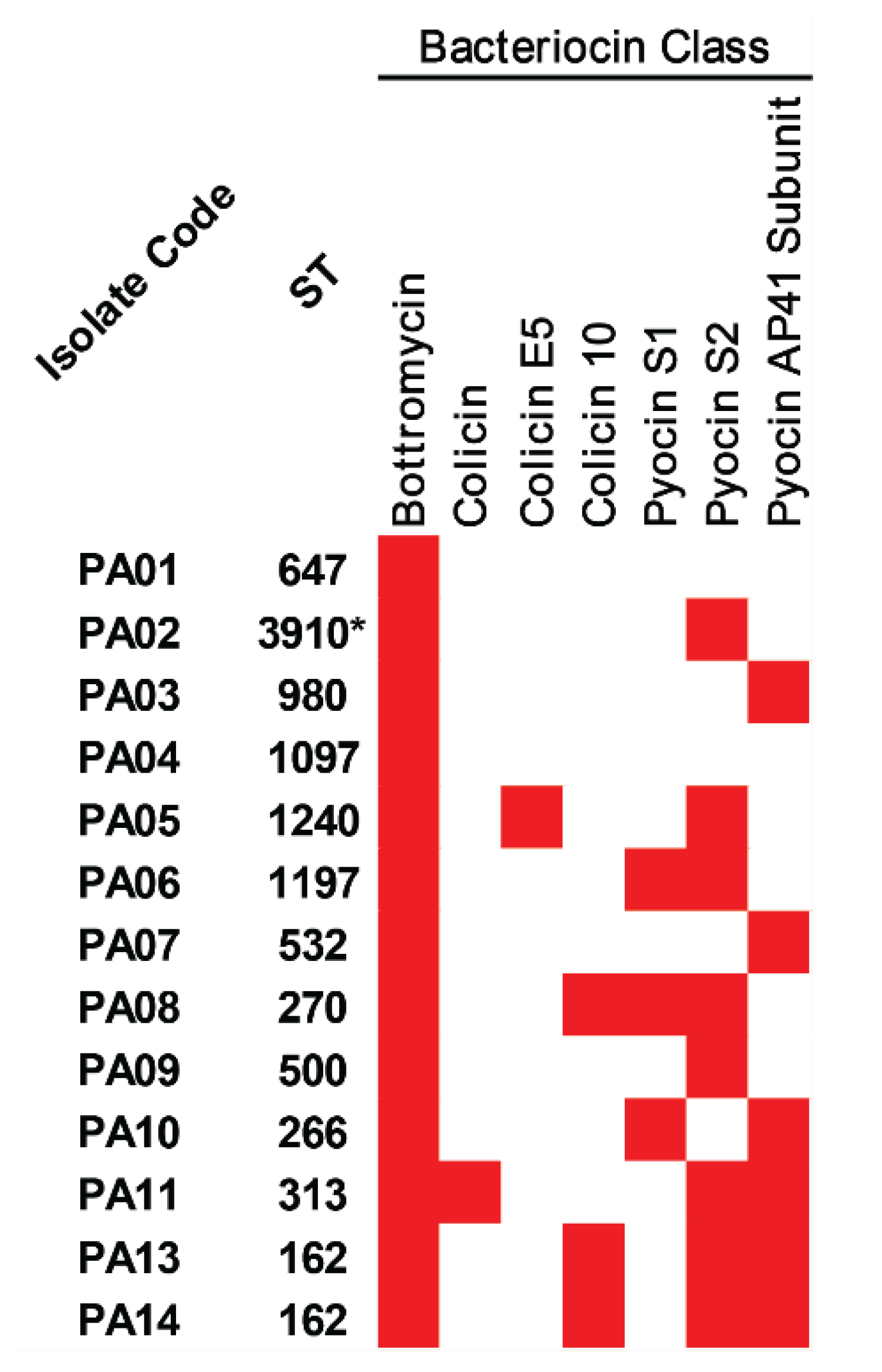
2.7. Bacteriocins
2.8. CRISPR-Cas System
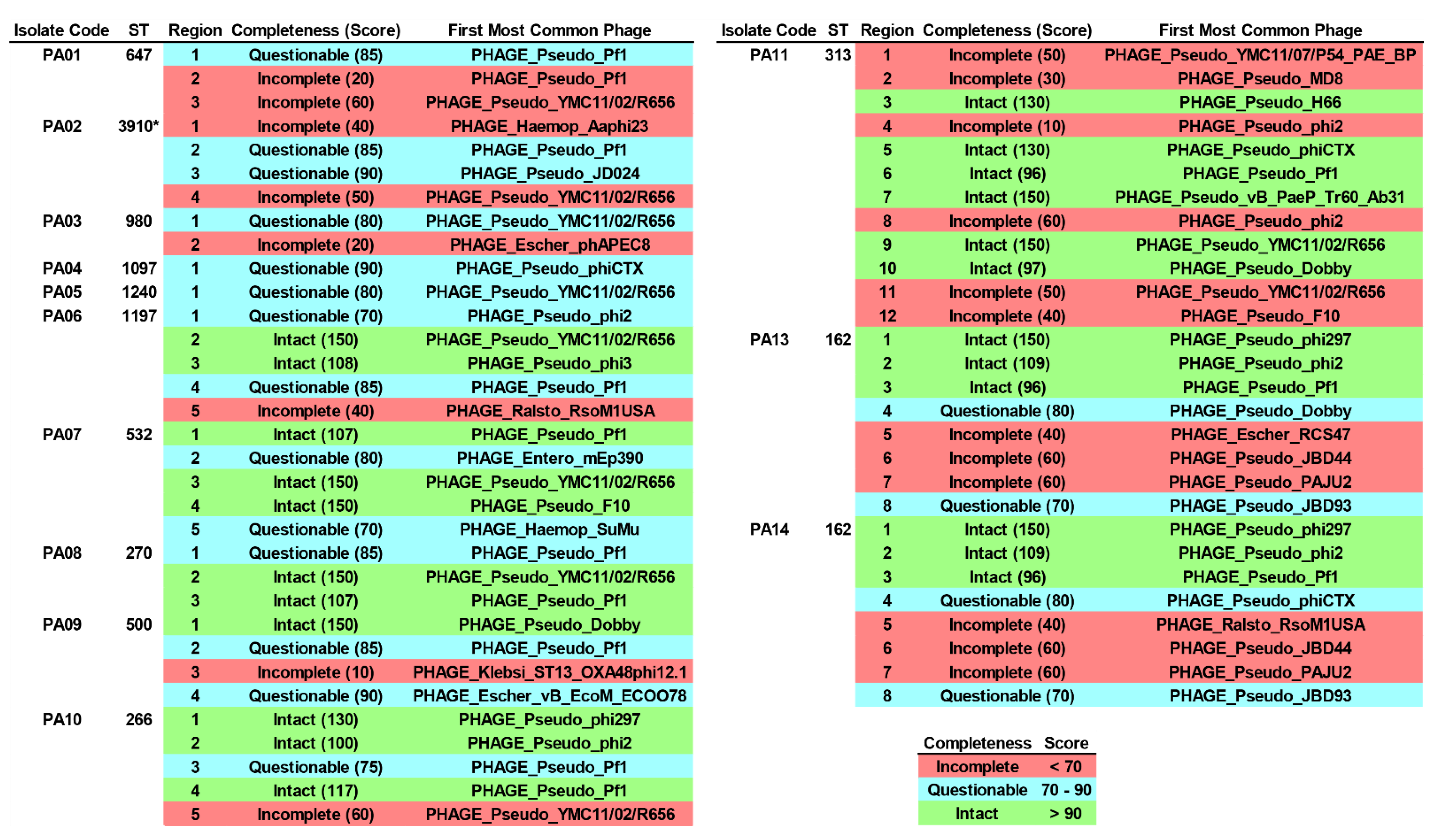
2.9. Integrated Bacteriophage Genomes
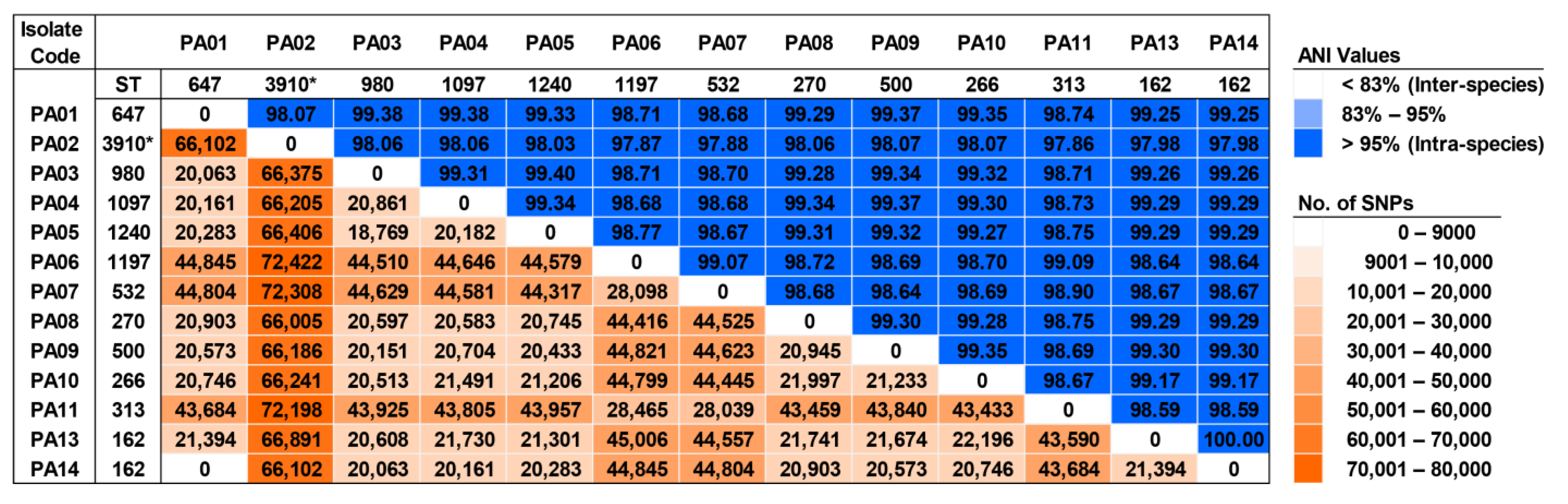
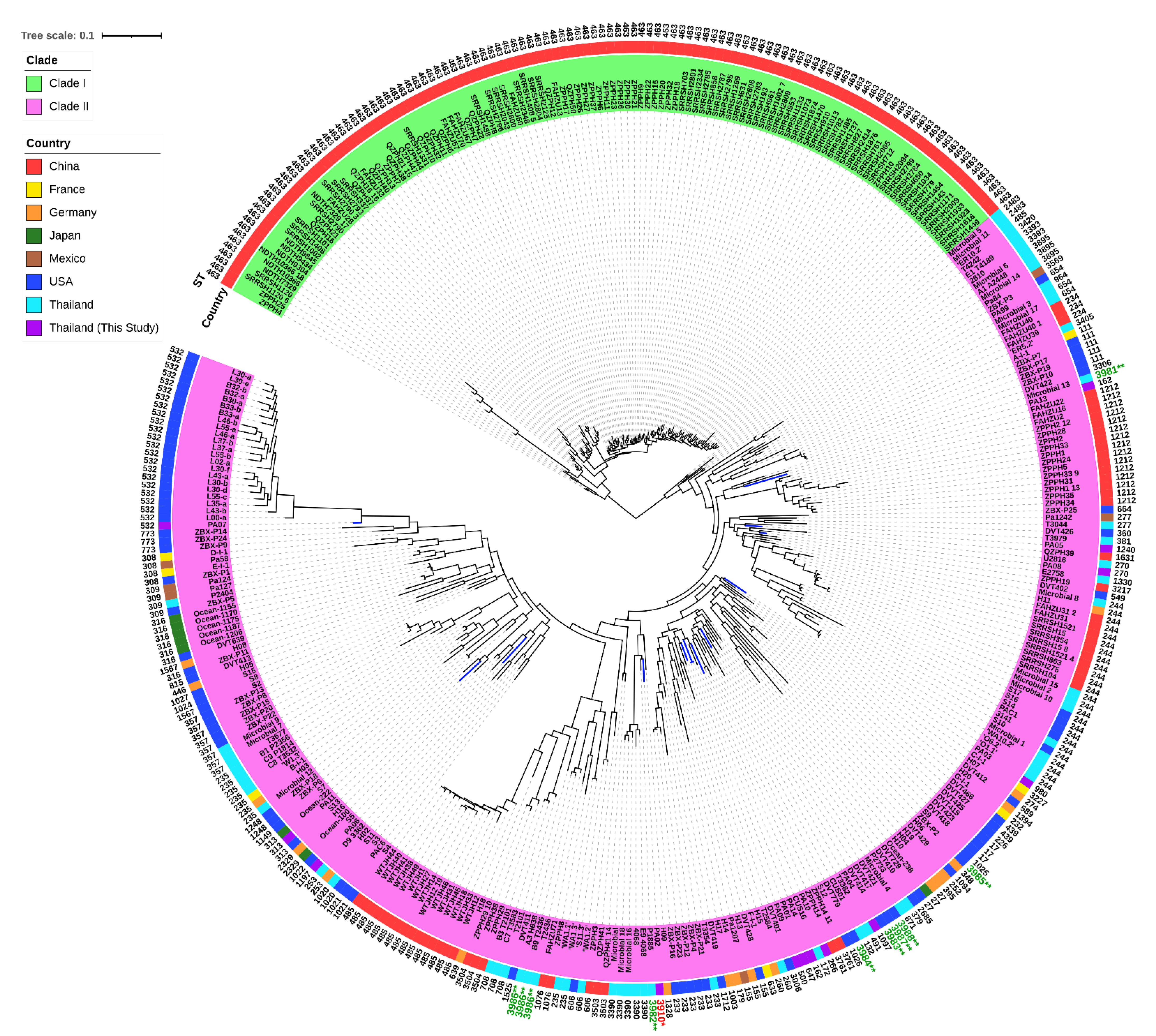
2.10. Genomic Diversity and Phylogenetic Relationship
3. Discussion
4. Materials and Methods
4.1. Colonizing Isolates of Pseudomonas aeruginosa
4.2. Antimicrobial Susceptibility Testing
4.3. DNA Extraction and Whole-Genome Sequencing
4.4. Genome Assembly and Annotation
4.5. Sequence Analysis
4.6. Genomic Diversity and Phylogenetic Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamada, K.; Aoki, K.; Nagasawa, T.; Sasaki, M.; Murakami, H.; Ishii, T.; Ujiie, S.; Morita, T.; Ishii, Y.; Tateda, K. Complete whole-genome sequence of the novel Pseudomonas species strain TUM18999, isolated from a patient with a burn wound in Japan. J. Glob. Antimicrob. Resist. 2021, 24, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Cabot, G.; López-Causapé, C.; Ocampo-Sosa, A.A.; Sommer, L.M.; Domínguez, M.Á.; Zamorano, L.; Juan, C.; Tubau, F.; Rodríguez, C.; Moyà, B. Deciphering the resistome of the widespread Pseudomonas aeruginosa sequence type 175 international high-risk clone through whole-genome sequencing. Antimicrob. Agents Chemother. 2016, 60, 7415–7423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cain, A.K.; Nolan, L.M.; Sullivan, G.J.; Whitchurch, C.B.; Filloux, A.; Parkhill, J. Complete genome sequence of Pseudomonas aeruginosa reference strain PAK. Microbiol. Resour. Announc. 2019, 8, e00865-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDC. Antibiotic Resistance Threats in the United States, 2019 (2019 AR Threats Report); Centers for Disease Control and Prevention (CDC): Atlanta, GA, USA, 2019. Available online: https://www.cdc.gov/drugresistance/Biggest-Threats.html (accessed on 4 January 2020).
- Lob, S.H.; Kazmierczak, K.M.; Chen, W.-T.; Siddiqui, F.; DeRyke, C.A.; Young, K.; Motyl, M.R.; Sahm, D.F. In vitro activity of ceftolozane/tazobactam against Gram-negative isolates collected from ICU patients with lower respiratory tract infections in seven Asian countries—SMART 2017–2019. J. Glob. Antimicrob. Resist. 2022, 29, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Cumley, N.; Wearn, C.M.; Niebel, M.; Constantinidou, C.; Thomas, C.M.; Pallen, M.J.; Moiemen, N.S.; Bamford, A.; Oppenheim, B. Seeking the source of Pseudomonas aeruginosa infections in a recently opened hospital: An observational study using whole-genome sequencing. BMJ Open 2014, 4, e006278. [Google Scholar] [CrossRef] [Green Version]
- Cottalorda, A.; Leoz, M.; Dahyot, S.; Gravey, F.; Grand, M.; Froidure, T.; Aujoulat, F.; Le Hello, S.; Jumas-Bilak, E.; Pestel-Caron, M. Within-host microevolution of Pseudomonas aeruginosa urinary isolates: A seven-patient longitudinal genomic and phenotypic study. Front. Microbiol. 2021, 11, 611246. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, J.; Shen, H.; Chen, Z.; Yang, Q.-w.; Zhu, J.; Li, X.; Yang, Q.; Zhao, F.; Ji, J. Emergence of ceftazidime-and avibactam-resistant Klebsiella pneumoniae carbapenemase-producing Pseudomonas aeruginosa in China. Msystems 2021, 6, e00787-21. [Google Scholar] [CrossRef]
- Cazares, A.; Moore, M.P.; Hall, J.P.; Wright, L.L.; Grimes, M.; Emond-Rhéault, J.-G.; Pongchaikul, P.; Santanirand, P.; Levesque, R.C.; Fothergill, J.L. A megaplasmid family driving dissemination of multidrug resistance in Pseudomonas. Nat. Commun. 2020, 11, 1370. [Google Scholar] [CrossRef] [Green Version]
- Soonthornsit, J.; Pimwaraluck, K.; Kongmuang, N.; Pratya, P.; Phumthanakorn, N. Molecular epidemiology of antimicrobial-resistant Pseudomonas aeruginosa in a veterinary teaching hospital environment. Vet. Res. Commun. 2022, 1, 1–14. [Google Scholar] [CrossRef]
- Irum, S.; Naz, K.; Ullah, N.; Mustafa, Z.; Ali, A.; Arslan, M.; Khalid, K.; Andleeb, S. Antimicrobial resistance and genomic characterization of six new sequence types in multidrug-resistant Pseudomonas aeruginosa clinical isolates from Pakistan. Antibiotics 2021, 10, 1386. [Google Scholar] [CrossRef]
- Ahmed, O.B. Detection of antibiotic resistance genes in Pseudomonas aeruginosa by whole genome sequencing. Infect. Drug Resist. 2022, 3, 6703–6709. [Google Scholar] [CrossRef]
- Olsson, A.; Wistrand-Yuen, P.; Nielsen, E.I.; Friberg, L.E.; Sandegren, L.; Lagerbäck, P.; Tängdén, T. Efficacy of antibiotic combinations against multidrug-resistant Pseudomonas aeruginosa in automated time-lapse microscopy and static time-kill experiments. Antimicrob. Agents Chemother. 2020, 64, e02111-19. [Google Scholar] [CrossRef] [Green Version]
- Sokol, P.A.; Luan, M.-Z.; Storey, D.G.; Thirukkumaran, P. Genetic rearrangement associated with in vivo mucoid conversion of Pseudomonas aeruginosa PAO is due to insertion elements. J. Bacteriol. 1994, 176, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Golovlev, E. The mechanism of formation of Pseudomonas aeruginosa biofilm, a type of structured population. Microbiology 2002, 71, 249–254. [Google Scholar] [CrossRef]
- Rumbaugh, K.P.; Hamood, A.N.; Griswold, J.A. Analysis of Pseudomonas aeruginosa Clinical isolates for possible variations within the virulence genes exotoxin a and exoenzymes. J. Surg. Res. 1999, 82, 95–105. [Google Scholar] [CrossRef]
- Rocchetta, H.L.; Lam, J.S. Identification and functional characterization of an ABC transport system involved in polysaccharide export of A-band lipopolysaccharide in Pseudomonas aeruginosa. J. Bacteriol. 1997, 179, 4713–4724. [Google Scholar] [CrossRef] [Green Version]
- Huszczynski, S.M.; Coumoundouros, C.; Pham, P.; Lam, J.S.; Khursigara, C.M. Unique regions of the polysaccharide copolymerase Wzz2 from Pseudomonas aeruginosa are essential for O-specific antigen chain length control. J. Bacteriol. 2019, 201, e00165-19. [Google Scholar] [CrossRef] [Green Version]
- Gellatly, S.L.; Hancock, R.E. Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog. Dis. 2013, 67, 159–173. [Google Scholar] [CrossRef] [Green Version]
- Oliver, A.; Mulet, X.; López-Causapé, C.; Juan, C. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist. Updat. 2015, 21, 41–59. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Ichioka, M.; Hirose, T.; Nagai, K.; Matsumoto, A.; Matsui, H.; Hanaki, H.; Masuma, R.; Takahashi, Y.; Ōmura, S. Bottromycin derivatives: Efficient chemical modifications of the ester moiety and evaluation of anti-MRSA and anti-VRE activities. Bioorganic Med. Chem. Lett. 2010, 20, 6116–6120. [Google Scholar] [CrossRef]
- Brown, D.G.; Lister, T.; May-Dracka, T.L. New natural products as new leads for antibacterial drug discovery. Bioorgan. Med. Chem. Lett. 2014, 24, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Hille, F.; Charpentier, E. CRISPR-Cas: Biology, mechanisms and relevance. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 496. [Google Scholar] [CrossRef] [Green Version]
- Fiedoruk, K.; Zakrzewska, M.; Daniluk, T.; Piktel, E.; Chmielewska, S.; Bucki, R. Two lineages of Pseudomonas aeruginosa filamentous phages: Structural uniformity over integration preferences. Genome Biol. Evol. 2020, 12, 1765–1781. [Google Scholar] [CrossRef] [PubMed]
- de Korne-Elenbaas, J.; Bruisten, S.M.; de Vries, H.J.; van Dam, A.P. Within-host genetic variation in Neisseria gonorrhoeae over the course of infection. Microbiol. Spectr. 2022, 9, e00313-22. [Google Scholar] [CrossRef] [PubMed]
- Chukamnerd, A.; Pomwised, R.; Phoo, M.T.P.; Terbtothakun, P.; Hortiwakul, T.; Charoenmak, B.; Chusri, S. In vitro synergistic activity of fosfomycin in combination with other antimicrobial agents against carbapenem-resistant Klebsiella pneumoniae isolated from patients in a hospital in Thailand. J. Infect. Chemother. 2021, 27, 507–514. [Google Scholar] [CrossRef]
- Chukamnerd, A.; Singkhamanan, K.; Chongsuvivatwong, V.; Palittapongarnpim, P.; Doi, Y.; Pomwised, R.; Sakunrang, C.; Jeenkeawpiam, K.; Yingkajorn, M.; Chusri, S. Whole-genome analysis of carbapenem-resistant Acinetobacter baumannii from clinical isolates in Southern Thailand. Comput. Struct. Biotechnol. J. 2022, 20, 545–558. [Google Scholar] [CrossRef]
- Kaewnirat, K.; Chuaychob, S.; Chukamnerd, A.; Pomwised, R.; Surachat, K.; Phoo, M.T.P.; Phaothong, C.; Sakunrang, C.; Jeenkeawpiam, K.; Hortiwakul, T. In vitro synergistic activities of fosfomycin in combination with other antimicrobial agents against carbapenem-resistant Escherichia coli harboring blaNDM-1 on the IncN2 plasmid and a study of the genomic characteristics of these pathogens. Infect. Drug Resist. 2022, 15, 1777. [Google Scholar] [CrossRef]
- Phoo, M.T.P.; Ngasaman, R.; Indoung, S.; Naknaen, A.; Chukamnerd, A.; Pomwised, R. Occurrence of NDM-5 and antibiotic resistance genes among Escherichia coli and Klebsiella pneumoniae in companion animals in Thailand. Southeast Asian J. Trop. Med. Public Health 2020, 51, 391–405. [Google Scholar]
- Chukamnerd, A.; Pomwised, R.; Jeenkeawpiam, K.; Sakunrang, C.; Chusri, S.; Surachat, K. Genomic insights into blaNDM-carrying carbapenem-resistant Klebsiella pneumoniae clinical isolates from a university hospital in Thailand. Microbiol. Res. 2022, 263, 127136. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 28th ed; Approved Standard M100; Clinical Laboratory Standard Institute (CLSI): Wayne, PA, USA, 2018; Volume 38. [Google Scholar]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comp. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [Green Version]
- Thrane, S.W.; Taylor, V.L.; Lund, O.; Lam, J.S.; Jelsbak, L. Application of whole-genome sequencing data for O-specific antigen analysis and in silico serotyping of Pseudomonas aeruginosa isolates. J. Clin. Microbiol. 2016, 54, 1782–1788. [Google Scholar] [CrossRef] [Green Version]
- Siguier, P.; Pérochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [Green Version]
- Cury, J.; Jové, T.; Touchon, M.; Néron, B.; Rocha, E.P. Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Res. 2016, 44, 4539–4550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- de Jong, A.; van Hijum, S.A.; Bijlsma, J.J.; Kok, J.; Kuipers, O.P. BAGEL: A web-based bacteriocin genome mining tool. Nucleic Acids Res. 2006, 34, W273–W279. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [Green Version]
- Nordstrom, H.R.; Evans, D.R.; Finney, A.G.; Westbrook, K.J.; Zamora, P.F.; Hofstaedter, C.E.; Yassin, M.H.; Pradhan, A.; Iovleva, A.; Ernst, R.K. Genomic characterization of lytic bacteriophages targeting genetically diverse Pseudomonas aeruginosa clinical isolates. iScience 2022, 1, 104372. [Google Scholar] [CrossRef]
- Botelho, J.; Tüffers, L.; Fuss, J.; Buchholz, F.; Utpatel, C.; Klockgether, J.; Niemann, S.; Tümmler, B.; Schulenburg, H. Phylogroup-specific variation shapes pangenome dynamics in Pseudomonas aeruginosa. bioRxiv 2022. [Google Scholar] [CrossRef]
- Espinosa-Camacho, L.F.; Delgado, G.; Miranda-Novales, G.; Soberón-Chávez, G.; Alcaraz, L.D.; Morales-Espinosa, R. Complete genome sequences of two Pseudomonas aeruginosa strains isolated from children with bacteremia. Genome Announc. 2017, 5, e00927-17. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-Camacho, L.F.; Delgado, G.; Soberón-Chávez, G.; Alcaraz, L.D.; Castañon, J.; Morales-Espinosa, R. Complete genome sequences of four extensively drug-resistant Pseudomonas aeruginosa strains, isolated from adults with ventilator-associated pneumonia at a tertiary referral hospital in Mexico city. Genome Announc. 2017, 5, e00925-17. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2016, 2, 2314. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate Code | Minimum Inhibitory Concentration (µg/mL) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| β-Lactam Combination Agents | Carbapenems | Lipopeptide | Aminoglycosides | Fluoroquinolones | |||||||
| TZP | C/T | IPM | MEM | DOR | CST | AMK | GEN | TOB | CIP | LVX | |
| PA01 | 16 (S) | 4 (S) | 16 (R) | 16 (R) | 8 (R) | 4 (R) | 2 (S) | 1 (S) | 2 (S) | 0.25 (S) | 1 (S) |
| PA02 | 4 (S) | 0.5 (S) | 64 (R) | 0.25 (S) | 0.25 (S) | 4 (R) | 8 (S) | 2 (S) | 4 (S) | 0.06 (S) | 0.25 (S) |
| PA03 | 8 (S) | 2 (S) | 4 (I) | 0.5 (S) | 0.5 (S) | 8 (R) | 2 (S) | 1 (S) | 1 (S) | 0.25 (S) | 0.5 (S) |
| PA04 | 8 (S) | 1 (S) | 4 (I) | 0.5 (S) | 0.5 (S) | 4 (R) | 4 (S) | 1 (S) | 2 (S) | 0.25 (S) | 0.5 (S) |
| PA05 | 8 (S) | 2 (S) | 4 (I) | 0.5 (S) | 0.5 (S) | 4 (R) | 2 (S) | 1 (S) | 1 (S) | 0.25 (S) | 0.25 (S) |
| PA06 | 16 (S) | 0.5 (S) | 4 (I) | 2 (S) | 1 (S) | 2 (I) | 2 (S) | 1 (S) | 2 (S) | 0.25 (S) | 0.5 (S) |
| PA07 | 8 (S) | 0.5 (S) | 4 (I) | 1 (S) | 0.5 (S) | 4 (R) | 2 (S) | 1 (S) | 0.5 (S) | 0.25 (S) | 0.5 (S) |
| PA08 | 8 (S) | 0.5 (S) | 2 (S) | 0.5 (S) | 0.5(S) | 2 (I) | 2 (S) | 1 (S) | 1 (S) | 0.25 (S) | 0.5 (S) |
| PA09 | 8 (S) | 1 (S) | 1 (S) | 0.5 (S) | 0.5 (S) | 2 (I) | 2 (S) | 1 (S) | 1 (S) | 0.25 (S) | 0.5 (S) |
| PA10 | 8 (S) | 2 (S) | 2 (S) | 0.5 (S) | 0.5 (S) | 4 (R) | 4 (S) | 1 (S) | 2 (S) | 0.25 (S) | 0.5 (S) |
| PA11 | 16 (S) | 0.5 (S) | 2 (S) | 0.5 (S) | 0.5 (S) | 2 (I) | 2 (S) | 1 (S) | 1 (S) | 0.25 (S) | 0.25 (S) |
| PA13 | 8 (S) | 0.5 (S) | 32 (R) | 8 (R) | 2 (S) | 4 (R) | 4 (S) | 1 (S) | 2 (S) | 0.25 (S) | 0.5 (S) |
| PA14 | 16 (S) | 1 (S) | 4 (I) | 0.5 (S) | 0.5 (S) | 8 (R) | 2 (S) | 1 (S) | 0.5 (S) | 0.25 (S) | 0.5 (S) |
| Isolate Code | Patient’s Sex | Specimen | Serotype | Multilocus Sequence Typing (MLST) | Accession No. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| acsA | aroE | guaA | mutL | nuoD | ppsA | trpE | ST | |||||
| PA01 | Female | Rectum | O6 | 17 | 5 | 6 | 3 | 4 | 15 | 10 | 647 | JANTQX000000000 |
| PA02 | Male | Rectum | O11 | 20 | 30 | 64 | 26 | 30 | 24 | 32 | 3910 * | JANTQW000000000 |
| PA03 | Female | Rectum | O3 | 16 | 5 | 11 | 3 | 4 | 12 | 3 | 980 | JANTQV000000000 |
| PA04 | Female | Throat | O6 | 6 | 5 | 1 | 30 | 4 | 6 | 27 | 1097 | JANTQU000000000 |
| PA05 | Female | Rectum | O3 | 28 | 5 | 11 | 7 | 1 | 6 | 61 | 1240 | JANTQT000000000 |
| PA06 | Male | Rectum | O10 | 5 | 8 | 119 | 6 | 12 | 6 | 3 | 1197 | JANTQS000000000 |
| PA07 | Female | Throat | O11 | 5 | 4 | 5 | 5 | 5 | 20 | 4 | 532 | JANTQR000000000 |
| PA08 | Female | Throat | O5 | 22 | 3 | 17 | 5 | 2 | 10 | 7 | 270 | JANTQQ000000000 |
| PA09 | Male | Rectum | O6 | 11 | 57 | 7 | 3 | 4 | 15 | 1 | 500 | JANTQP000000000 |
| PA10 | Male | Throat | O6 | 16 | 5 | 11 | 72 | 44 | 7 | 52 | 266 | JANTQO000000000 |
| PA11 | Male | Throat | O1 | 47 | 8 | 7 | 6 | 8 | 11 | 40 | 313 | JANTQN000000000 |
| PA13 | Male | Throat | O11 | 6 | 5 | 6 | 34 | 27 | 3 | 7 | 162 | JANTQM000000000 |
| PA14 | Male | Throat | O11 | 6 | 5 | 6 | 34 | 27 | 3 | 7 | 162 | JANTQL000000000 |
| Isolate Code | ST | CRISPR-Cas | ||||
|---|---|---|---|---|---|---|
| Region | Element | No. of Spacer | No. of cas Gene (Cas Type) | Direct Repeat (DR) Consensus/cas Gene | ||
| PA01 | 647 | 1 | CRISPR | 13 | TTTCTTAGCTGCCTATACGGCAGTGAAC | |
| 2 | CRISPR | 11 | GTTCACTGCCGTATAGGCAGCTAAGAAA | |||
| 3 | Cas cluster | 6 (IF) | cas6, csy3, csy2, csy1, cas3-cas2, cas1 | |||
| 4 | CRISPR | 7 | TTTCTTAGCTGCCTACACGGCAGTGAAC | |||
| PA02 | 3910 * | 1 | CRISPR | 10 | GTTCACTGCCGTGTAGGCAGCTAAGAAA | |
| 2 | Cas cluster | 6 (IF) | cas1, cas3-cas2, csy1, csy2, csy3, cas6 | |||
| 3 | CRISPR | 4 | TTTCTTAGCTGCCTATACGGCAGTGAAC | |||
| 4 | CRISPR | 7 | TTTCTTAGCTGCCTATACGGCAGTGAAC | |||
| 5 | CRISPR | 27 | CGGTTCATCCCCACGCATGTGGGGAACAC | |||
| 6 | Cas cluster | 8 (IE) | cas2, cas1, cas6, cas5, cas7, cse2, cse1, cas3 | |||
| 7 | CRISPR | 4 | CGGTTCATCCCCACACCCGTGGGGAACAC | |||
| 8 | CRISPR | 4 | TTTCTTAGCTGCCTATACGGCAGTGAAC | |||
| 9 | CRISPR | 6 | TTTCTTAGCTGCCTACACGGCAGTGAAC | |||
| 10 | CRISPR | 10 | GTTCACTGCCGTGTAGGCAGCTAAGAAA | |||
| 11 | CRISPR | 5 | TTTCTTAGCTGCCTATACGGCAGTGAAC | |||
| PA03 | 980 | 1 | CRISPR | 22 | GTTCACTGCCGTATAGGCAGCTAAGAAA | |
| 2 | CRISPR | 11 | GTTCACTGCCGTGTAGGCAGCTAAGAAA | |||
| 3 | Cas cluster | 6 (IF) | cas1, cas3-cas2, csy1, csy2, csy3, cas6 | |||
| 4 | CRISPR | 13 | TTTCTTAGCTGCCTATACGGCAGTGAAC | |||
| PA04 | 1097 | 1 | CRISPR | 19 | GTTCACTGCCGTGTAGGCAGCTAAGAAA | |
| 2 | Cas cluster | 6 (IF) | cas1, cas3-cas2, csy1, csy2, csy3, cas6 | |||
| 3 | CRISPR | 22 | TTTCTTAGCTGCCTATACGGCAGTGAAC | |||
| PA05 | 1240 | 1 | CRISPR | 7 | GTTCACTGCCGTATAGGCAGCTAAGAAA | |
| 2 | Cas cluster | 6 (IF) | cas6, csy3, csy2, csy1, cas3-cas2, cas1 | |||
| 3 | CRISPR | 14 | TTTCTTAGCTGCCTACACGGCAGTGAAC | |||
| PA06 | 1197 | ND | ND | ND | ND | |
| PA07 | 532 | 1 | CRISPR | 11 | GTGTTCCCCACGGGTGTGGGGATGAACCG | |
| 2 | Cas cluster | 8 (IE) | cas3, cse1, cse2, cas7, cas5, cas6, cas1, cas2 | |||
| 3 | CRISPR | 12 | GTGTTCCCCACATGCGTGGGGATGAACCG | |||
| PA08 | 270 | ND | ND | ND | ND | |
| PA09 | 500 | ND | ND | ND | ND | |
| PA10 | 266 | 1 | CRISPR | 9 | TTTCTTAGCTGCCTATACGGCAGTGAAC | |
| PA11 | 313 | ND | ND | ND | ND | |
| PA13 | 162 | ND | ND | ND | ND | |
| PA14 | 162 | ND | ND | ND | ND | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chukamnerd, A.; Pomwised, R.; Chusri, S.; Singkhamanan, K.; Chumtong, S.; Jeenkeawpiam, K.; Sakunrang, C.; Saroeng, K.; Saengsuwan, P.; Wonglapsuwan, M.; et al. Antimicrobial Susceptibility and Molecular Features of Colonizing Isolates of Pseudomonas aeruginosa and the Report of a Novel Sequence Type (ST) 3910 from Thailand. Antibiotics 2023, 12, 165. https://doi.org/10.3390/antibiotics12010165
Chukamnerd A, Pomwised R, Chusri S, Singkhamanan K, Chumtong S, Jeenkeawpiam K, Sakunrang C, Saroeng K, Saengsuwan P, Wonglapsuwan M, et al. Antimicrobial Susceptibility and Molecular Features of Colonizing Isolates of Pseudomonas aeruginosa and the Report of a Novel Sequence Type (ST) 3910 from Thailand. Antibiotics. 2023; 12(1):165. https://doi.org/10.3390/antibiotics12010165
Chicago/Turabian StyleChukamnerd, Arnon, Rattanaruji Pomwised, Sarunyou Chusri, Kamonnut Singkhamanan, Sanicha Chumtong, Kongpop Jeenkeawpiam, Chanida Sakunrang, Kuwanhusna Saroeng, Phanvasri Saengsuwan, Monwadee Wonglapsuwan, and et al. 2023. "Antimicrobial Susceptibility and Molecular Features of Colonizing Isolates of Pseudomonas aeruginosa and the Report of a Novel Sequence Type (ST) 3910 from Thailand" Antibiotics 12, no. 1: 165. https://doi.org/10.3390/antibiotics12010165
APA StyleChukamnerd, A., Pomwised, R., Chusri, S., Singkhamanan, K., Chumtong, S., Jeenkeawpiam, K., Sakunrang, C., Saroeng, K., Saengsuwan, P., Wonglapsuwan, M., & Surachat, K. (2023). Antimicrobial Susceptibility and Molecular Features of Colonizing Isolates of Pseudomonas aeruginosa and the Report of a Novel Sequence Type (ST) 3910 from Thailand. Antibiotics, 12(1), 165. https://doi.org/10.3390/antibiotics12010165