


Kaurane-Type Diterpenoids as Potential Inhibitors of Dihydrofolate Reductase-Thymidylate Synthase in New World Leishmania Species

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Kauranes 302 and Its Derivative 302a Have Dual In Vitro Enzymatic Activity against L. major PTR1/DHFR-TS
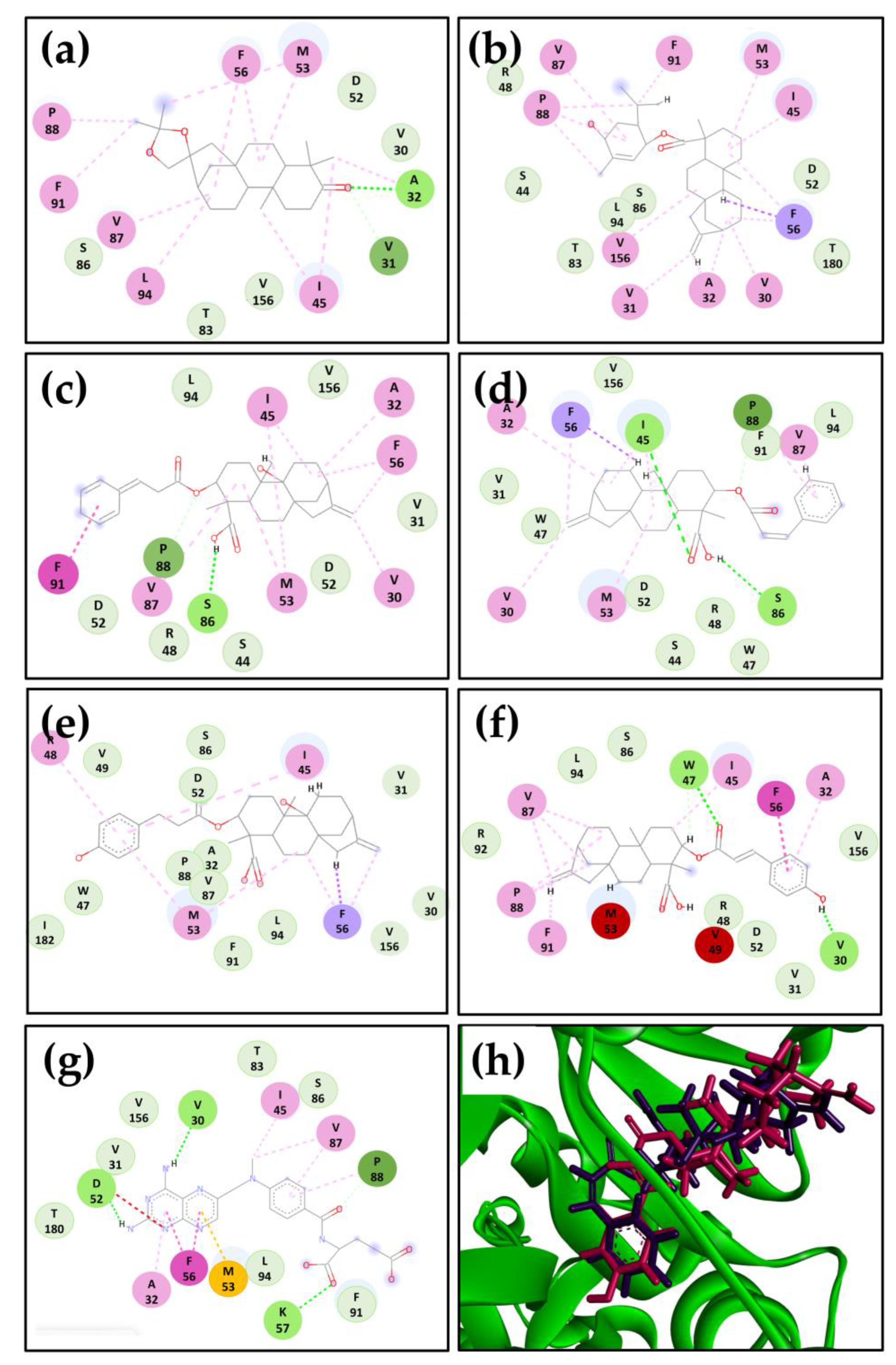
2.2. Hybrid Model of L. major DHFR-TS and Molecular Docking Calculations
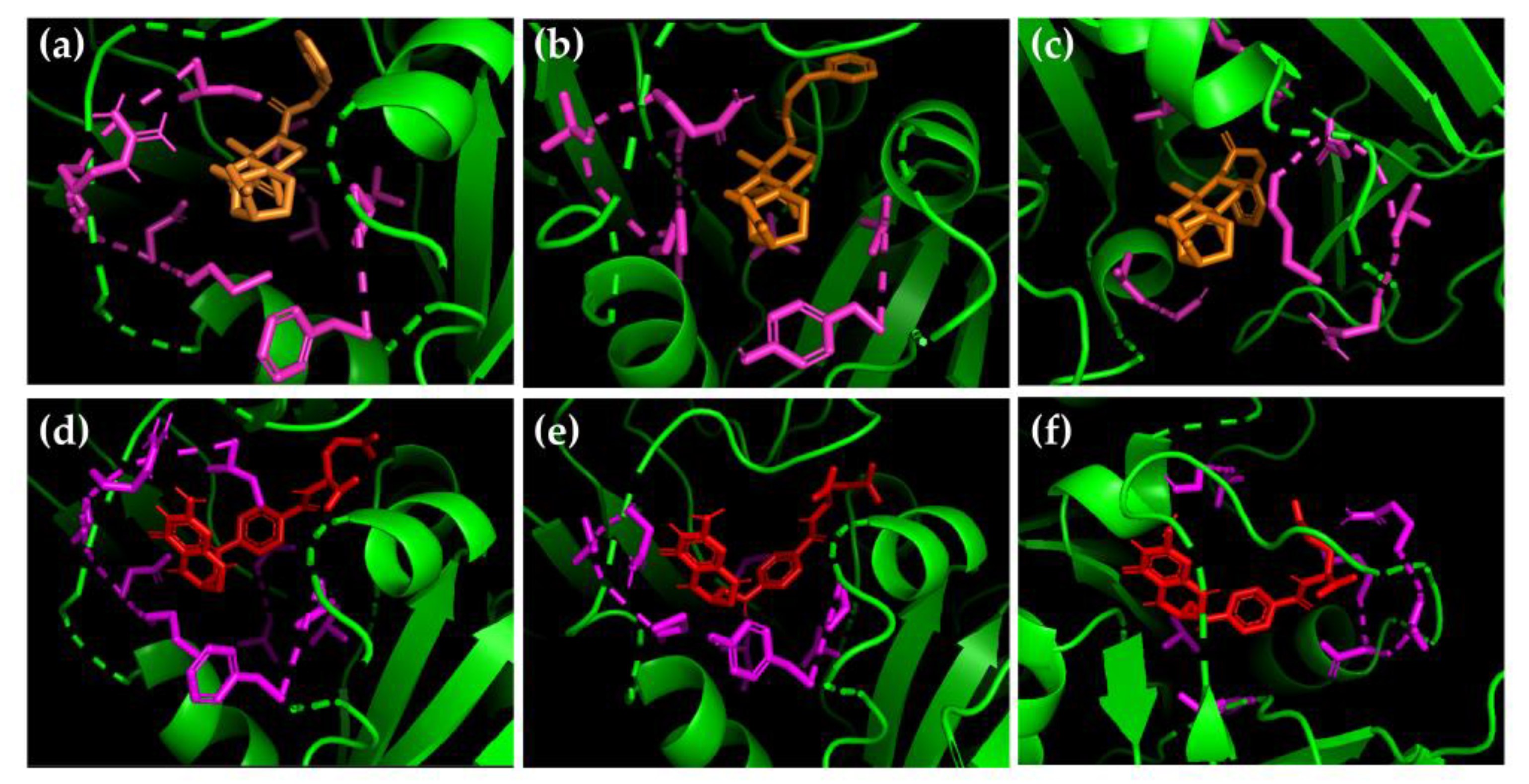
2.3. Kaurane 302 and Its Derivative 302a May Have the Potential to Inhibit DHFR-TS in Different Species of Leishmania from the New World
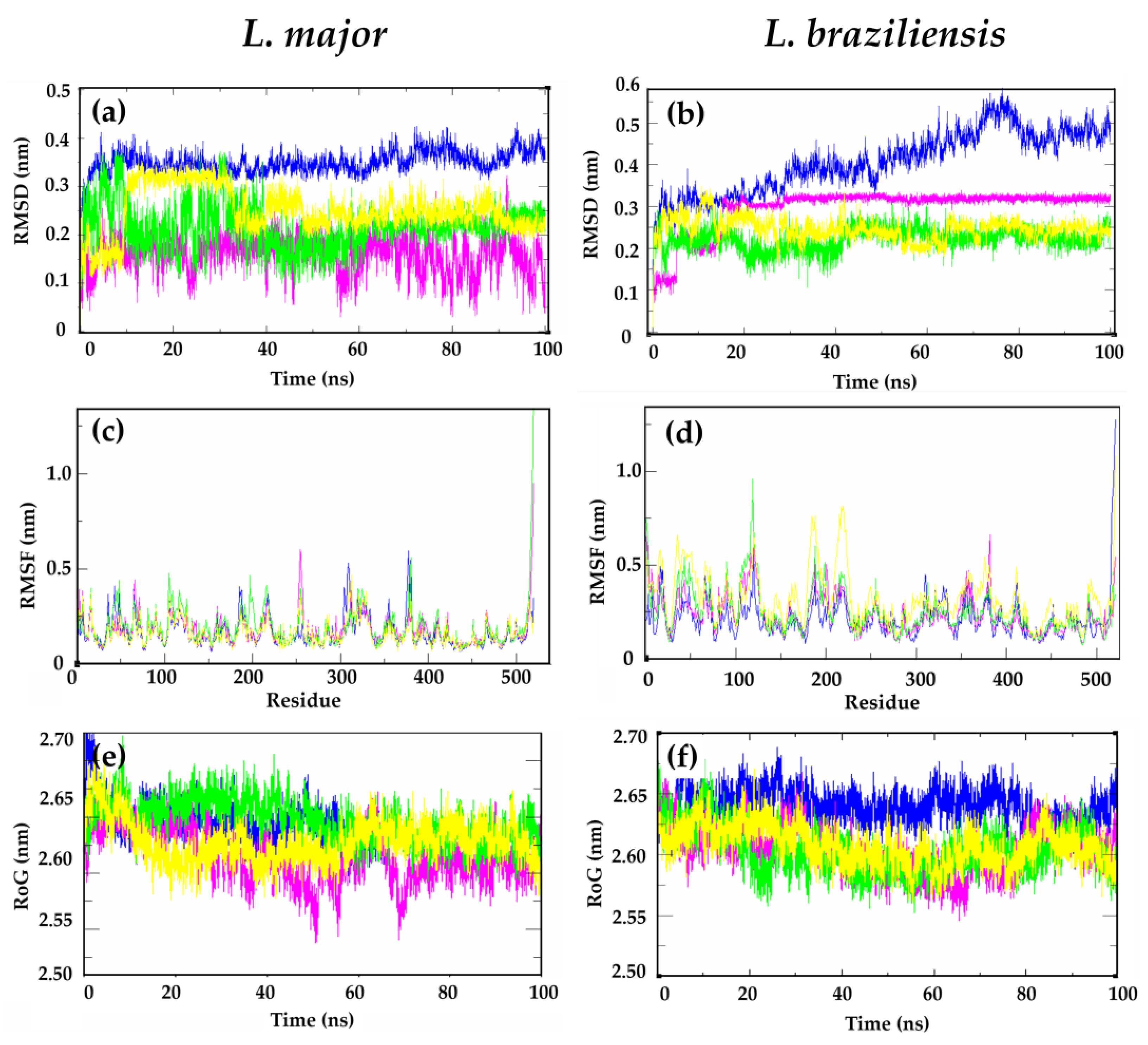
2.4. Molecular Dynamics Simulations for L. major and L. braziliensis DHFR-TS Interacting with 302 and MTX
2.5. Free Energy Calculations by the Molecular Mechanics Poisson–Boltzmann Surface Area Approach (MM/PBSA) Method
3. Materials and Methods
3.1. LmDHFR-TS Enzyme Inhibition Assay
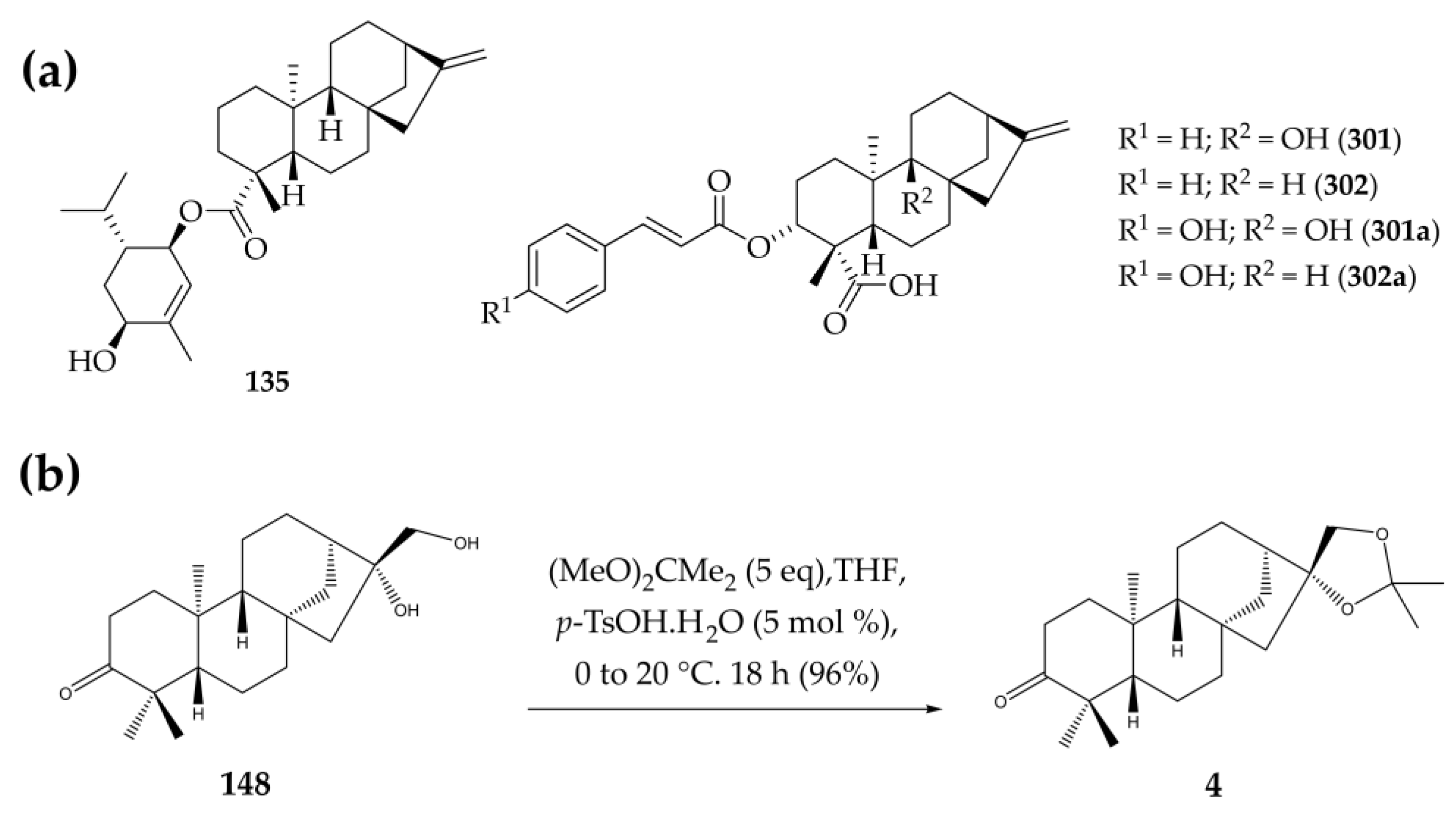
3.2. Isolation of Compound 148
3.3. Synthesis of 16ß,17-Isopropylidenedioxy-ent-kauran-3-one (4)
3.4. Hybrid Models of Leishmania DHFR-TS
3.5. Molecular Docking Calculations
3.6. Molecular Dynamics Simulations
3.7. Binding Free Energies Using the Molecular Mechanics Poisson–Boltzmann Surface Area (MM/PBSA) Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Soni, M.; Pratap, J.V. Development of Novel Anti-Leishmanials: The Case for Structure-Based Approaches. Pathogens 2022, 11, 950. [Google Scholar] [CrossRef] [PubMed]
- Gouri, V.; Upreti, S.; Samant, M. Evaluation of target-specific natural compounds for drug discovery against leishmaniasis. Parasitol. Int. 2022, 91, 102622. [Google Scholar] [CrossRef] [PubMed]
- Salari, S.; Bamorovat, M.; Sharifi, I.; Almani, P.G.N. Global distribution of treatment resistance gene markers for leishmaniasis. J. Clin. Lab. Anal. 2022, 36, e24599. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan, S.; Saudagar, P. Leishmaniasis: Where are we and where are we heading? Parasitol. Res. 2021, 120, 1541–1554. [Google Scholar] [CrossRef]
- Gupta, D.; Singh, P.K.; Yadav, P.K.; Narender, T.; Patil, U.K.; Jain, S.K.; Chourasia, M.K. Emerging strategies and challenges of molecular therapeutics in antileishmanial drug development. Int. Immunopharmacol. 2023, 115, 109649. [Google Scholar] [CrossRef]
- Frézard, F.; Aguiar, M.M.G.; Ferreira, L.A.M.; Ramos, G.S.; Santos, T.T.; Borges, G.S.M.; Vallejos, V.M.R.; De Morais, H.L.O. Liposomal Amphotericin B for Treatment of Leishmaniasis: From the Identification of Critical Physicochemical Attributes to the Design of Effective Topical and Oral Formulations. Pharmaceutics 2023, 15, 99. [Google Scholar] [CrossRef]
- Uliana, S.R.B.; Trinconi, C.T.; Coelho, A.C. Chemotherapy of leishmaniasis: Present challenges. Parasitology 2018, 145, 464–480. [Google Scholar] [CrossRef]
- Brindha, J.; Balamurali, M.M.; Chanda, K. An Overview on the Therapeutics of Neglected Infectious Diseases—Leishmaniasis and Chagas Diseases. Front. Chem. 2021, 9, 622286. [Google Scholar]
- Vickers, T.J.; Beverley, S.M. Folate metabolic pathways in Leishmania. Essays Biochem. 2011, 51, 63–80. [Google Scholar]
- Gilbert, I.H. Inhibitors of dihydrofolate reductase in leishmania and trypanosomes. Biochim. Biophys. Acta Mol. Basis Dis. 2002, 1587, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Ivanetich, K.M.; Santi, D.V. Thymidylate synthase-dihydrofolate reductase in protozoa. Exp. Parasitol. 1990, 70, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Shamshad, H.; Bakri, R.; Mirza, A.Z. Dihydrofolate reductase, thymidylate synthase, and serine hydroxy methyltransferase: Successful targets against some infectious diseases. Mol. Biol. Rep. 2022, 49, 6659–6691. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.B.; Sienkiewicz, N.; Wyllie, S.; Fairlamb, A.H. Dissecting the metabolic roles of pteridine reductase 1 in Trypanosoma brucei and Leishmania major. J. Biol. Chem. 2011, 286, 10429–10438. [Google Scholar] [CrossRef] [Green Version]
- Nare, B.; Luba, J.; Hardy, L.W.; Beverley, S. New approaches to Leishmania chemotherapy: Pteridine Reductase 1 (PTR1) as a target and modulator of antifolate sensitivity. Parasitology 1997, 114, S101–S110. [Google Scholar] [CrossRef] [Green Version]
- Panecka-Hofman, J.; Poehner, I.; Wade, R.C. Anti-trypanosomatid structure-based drug design—Lessons learned from targeting the folate pathway. Expert Opin. Drug Discov. 2022, 17, 1029–1045. [Google Scholar] [CrossRef] [PubMed]
- Kheirandish, F.; Bandehpour, M.; Haghighi, A.; Mahboudi, F.; Mohebali, M.; Kazemi, B. Inhibition of Leishmania major PTR1 Gene Expression by Antisense in Escherichia coli. Iran. J. Public Health 2012, 41, 65–71. [Google Scholar]
- Das Neves, G.M.H.; Kagami, L.P.; Gonçalves, I.L.; Eifler-Lima, V.L. Targeting pteridine reductase 1 and dihydrofolate reductase: The old is a new trend for leishmaniasis drug discovery. Future Med. Chem. 2019, 11, 207–2130. [Google Scholar] [CrossRef]
- Possart, K.; Herrmann, F.C.; Jose, J.; Costi, M.P.; Schmidt, T.J. Sesquiterpene Lactones with Dual Inhibitory Activity against the Trypanosoma brucei Pteridine Reductase 1 and Dihydrofolate Reductase. Molecules 2022, 27, 149. [Google Scholar] [CrossRef]
- Teixeira, B.V.F.; Teles, A.L.B.; da Silva, S.G.; Brito, C.C.B.; de Freitas, H.F.; Pires, A.B.L.; Froes, T.Q.; Castilho, M.S. Dual and selective inhibitors of pteridine reductase 1 (PTR1) and dihydrofolate reductase-thymidylate synthase (DHFR-TS) from Leishmania chagasi. J. Enzym. Inhib. Med. Chem. 2019, 34, 1439–1450. [Google Scholar] [CrossRef] [Green Version]
- Sabt, A.; Eldehna, W.M.; Ibrahim, T.M.; Bekhit, A.A.; Batran, R.Z. New antileishmanial quinoline linked isatin derivatives targeting DHFR-TS and PTR1: Design, synthesis, and molecular modeling studies. Eur. J. Med. Chem. 2023, 246, 114959. [Google Scholar] [CrossRef]
- Nogueira, M.S.; Da Costa, F.B.; Brun, R.; Kaiser, M.; Schmidt, T.J. Ent-pimarane and ent-kaurane diterpenes from Aldama discolor (Asteraceae) and their antiprotozoal activity. Molecules 2016, 21, 1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, M.M.; Panis, C.; Da Silva, S.S.; Macri, J.A.; Kawakami, N.Y.; Hayashida, T.H.; Madeira, T.B.; Acquaro, V.R.; Nixdorf, S.L.; Pizzatti, L.; et al. Kaurenoic acid possesses leishmanicidal activity by triggering a NLRP12/IL-1 β/cNOS/NO Pathway. Mediat. Inflamm. 2015, 2015, 392918. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, A.O.; Izumi, E.; Ueda-Nakamura, T.; Dias-Filho, B.P.; da Veiga-Júnior, V.F.; Vataru Nakamura, C. Antileishmanial activity of diterpene acids in copaiba oil. Mem. Inst. Oswaldo Cruz 2013, 108, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Acevedo, C.; Flores-Gaspar, A.; Scotti, L.; Mendonça-Junior, F.J.B.; Scotti, M.T.; Coy-Barrera, E. Identification of Kaurane-type diterpenes as inhibitors of Leishmania pteridine reductase I. Molecules 2021, 26, 3076. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Prusoff, W.H. the concentration of inhibitor which causes 50 percent inhibition (I) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Scotti, M.T.; Herrera-Acevedo, C.; Barros de Menezes, R.P.; Martin, H.J.; Muratov, E.N.; Ítalo de Souza Silva, Á.; Albuquerque, E.F.; Calado, L.F.; Coy-Barrera, E.; Scotti, L. MolPredictX: Online Biological Activity Predictions by Machine Learning Models. Mol. Inform. 2022, 41, e2200133. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall Iii, W.B.; De Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ, and Cβ deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Vriend, G.; Sander, C. Quality control of protein models: Directional atomic contact analysis. J. Appl. Crystallogr. 1993, 26, 47–60. [Google Scholar] [CrossRef]
- Kosaka, A.; Sakamoto, N.; Hikone, M.; Imai, K.; Ota, M.; Washino, T.; Iwabuchi, S. Failure of liposomal-amphotericin B treatment for new world cutaneous leishmaniasis due to leishmania braziliensis. Intern. Med. 2020, 59, 1227–1230. [Google Scholar] [CrossRef]
- Herrera, G.; Barragán, N.; Luna, N.; Martínez, D.; De Martino, F.; Medina, J.; Niño, S.; Páez, L.; Ramírez, A.; Vega, L.; et al. An interactive database of Leishmania species distribution in the Americas. Sci. Data 2020, 7, 110. [Google Scholar] [CrossRef] [Green Version]
- Davila, M.; Pineda, V.; Calzada, J.E.; Saldaña, A.; Samudio, F. Evaluation of cytochrome b sequence to identify Leishmania species and variants: The case of Panama. Memórias Do Inst. Oswaldo Cruz 2021, 116, e200572. [Google Scholar] [CrossRef]
- Rodrigues, M.P.; Tomaz, D.C.; de Souza, L.A.; Onofre, T.S.; de Menezes, W.A.; Almeida-Silva, J.; Suarez-Fontes, A.M.; de Almeida, M.R.; da Silva, A.M.; Bressan, G.C.; et al. Synthesis of cinnamic acid derivatives and leishmanicidal activity against Leishmania braziliensis. Eur. J. Med. Chem. 2019, 183, 111688. [Google Scholar] [CrossRef]
- Brustolin, A.Á.; Ramos-Milaré, Á.C.F.H.; de Mello, T.F.P.; Aristides, S.M.A.; Lonardoni, M.V.C.; Silveira, T.G.V. In vitro activity of cinnamaldehyde on Leishmania (Leishmania) amazonensis. Exp. Parasitol. 2022, 236, 108244. [Google Scholar] [CrossRef]
- Garcia, L.S.; Nielsen-Saines, K. Leishmaniasis. In Feigin and Cherry’s Textbook of Pediatric Infectious Diseases, 6th ed.; Feigin, R.D., Cherry, J.D., Demmler-Harrison, G.J., Kaplan, S.L., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2009; p. 2920. ISBN 978-1-4160-4044-6. [Google Scholar]
- Rozo-Lugo, C.; Cuca-Suárez, L.E.; Schmidt, T.J.; Coy-Barrera, E. Tetrahydrobenzofuran-6 (2 H)-one neolignans from Ocotea heterochroma: Their platelet activating factor (PAF) antagonistic activity and in silico insights into the PAF receptor binding mode. J. Nat. Prod. 2018, 81, 1968–1975. [Google Scholar] [CrossRef]
- Grumont, R.; Sirawaraporn, W.; Santi, D. V Heterologous Expression of the Bifunctional Thymidylate Synthase-Dihydrofolate Reductase from Leishmania major. Biochemistry 1988, 27, 3776–3784. [Google Scholar] [CrossRef]
- Nare, B.; Hardy, L.W.; Beverley, S.M. The roles of pteridine reductase 1 and dihydrofolate reductase- thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J. Biol. Chem. 1997, 272, 13883–13891. [Google Scholar] [CrossRef] [Green Version]
- Yi-Li, D.; Zhong-Jian, J. Tetracyclic diterpenols from Euphorbia sieboldiana. Phytochemistry 1991, 30, 2413–2415. [Google Scholar] [CrossRef]
- Bon, D.J.-Y.D.; Banwell, M.G.; Willis, A.C. A chemoenzymatic total synthesis of the hirsutene-type sesquiterpene (+)-connatusin B from toluene. Tetrahedron 2010, 66, 7807–7814. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Acevedo, C.H.; Scotti, L.; Scotti, M.T. In silico studies designed to select sesquiterpene lactones with potential antichagasic activity from an in-house asteraceae database. ChemMedChem 2018, 13, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Acevedo, C.; Maia, M.D.S.; Cavalcanti, É.B.V.S.; Coy-Barrera, E.; Scotti, L.; Scotti, M.T. Selection of antileishmanial sesquiterpene lactones from SistematX database using a combined ligand-/structure-based virtual screening approach. Mol. Divers. 2020, 25, 2411–2427. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson− Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 4 | 135 | 302 | 301 | 302a | 301a | MTX |
|---|---|---|---|---|---|---|---|
| IC50 (µM) | 7.6 | 11.2 | 6.3 | 8.8 | 4.5 | 7.9 | 1.4 |
| Confidence Interval (95%) | 6.9–8.1 | 10.2–12.1 | 5.8–6.9 | 8.0–9.9 | 3.9–5.2 | 7.1–8.4 | 1.1–1.8 |
| Kiapp | 0.81 | 1.20 | 0.68 | 0.94 | 0.48 | 0.85 | 0.15 |
| Structure | MolDock Score (kJ/mol) | RMSD (A) | SD |
|---|---|---|---|
| 4 | −70.25 | 0.68 | 5.7 |
| 135 | −62.85 | 1.23 | 8.6 |
| 301 | −73.34 | 1.09 | 10.3 |
| 302 | −76.53 | 1.13 | 4.9 |
| 301a | −72.26 | 0.89 | 9.8 |
| 302a | −81.43 | 1.29 | 6.4 |
| 2,4-diamine | −72.34 | 1.55 | 8.9 |
| MTX | −107.60 | 0.24 | 5.9 |
| L. braziliensis | L. panamensis | L. amazonensis | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Structure | VINA Score (kcal/mol) | SD | RMSD | VINA Score (kcal/mol) | SD | RMSD | VINA Score (kcal/mol) | SD | RMSD |
| 4 | −10.70 | 0.05 | 0.13 | −10.96 | 0.07 | 0.46 | −10.68 | 0.04 | 0.11 |
| 135 | −10.50 | 0 | 0.21 | −10.19 | 0.03 | 0.31 | −10.52 | 0.06 | 0.25 |
| 302 | −10.90 | 0.05 | 0.55 | −10.44 | 0.05 | 2.73 | −10.55 | 0.15 | 0.86 |
| 302a | −11.17 | 0.13 | 0.61 | −12.55 | 0.28 | 0.56 | −10.60 | 0.08 | 0.61 |
| 301 | −10.40 | 0.10 | 0.92 | −10.84 | 0.07 | 1.68 | −10.85 | 0.05 | 0.64 |
| 301a | −10.66 | 0.20 | 0.45 | −12.54 | 0.08 | 0.86 | −11.14 | 0.15 | 0.79 |
| MTX | −9.64 | 0.07 | 1.87 | −9.45 | 0.15 | 1.48 | −9.54 | 0.07 | 1.71 |
| Structure | Van der Waals (kJ/mol) | Electrostatic (kJ/mol) | Polar Solvation (kJ/mol) | SASA (kJ/mol) | Binding Energy (kJ/mol) |
|---|---|---|---|---|---|
| Leishmania major | |||||
| 302 | −122.6 ± 11.8 | −262.3 ± 1.1 | 263.4 ± 23.5 | −16.7 ± 0.7 | −138.2 ± 12.2 |
| 302A | −210.2 ± 10.2 | −127.6 ± 3.0 | 219.5 ± 8.6 | −15.9 ± 1.0 | −134.2 ± 16.8 |
| MTX | −157.5 ± 12.4 | −399.7 ± 10.9 | 436.4 ± 22.4 | −19.4 ± 1.3 | −140.1 ± 18.6 |
| Leishmania braziliensis | |||||
| 302 | −215.4 ± 5.3 | −23.6 ± 2.2 | 124.2 ± 6.8 | −20.0 ± 0.4 | −134.8 ± 9.5 |
| 302A | −199.3 ± 6.0 | −31.4 ± 0.6 | 107.4 ± 7.4 | −21.3 ± 0.6 | −144.5 ± 5.0 |
| MTX | −216.4 ± 5.5 | −51.5 ± 3.5 | 194.6 ± 8.0 | −22.6 ± 0.8 | −95.9 ± 9.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrera-Acevedo, C.; de Menezes, R.P.B.; de Sousa, N.F.; Scotti, L.; Scotti, M.T.; Coy-Barrera, E. Kaurane-Type Diterpenoids as Potential Inhibitors of Dihydrofolate Reductase-Thymidylate Synthase in New World Leishmania Species. Antibiotics 2023, 12, 663. https://doi.org/10.3390/antibiotics12040663
Herrera-Acevedo C, de Menezes RPB, de Sousa NF, Scotti L, Scotti MT, Coy-Barrera E. Kaurane-Type Diterpenoids as Potential Inhibitors of Dihydrofolate Reductase-Thymidylate Synthase in New World Leishmania Species. Antibiotics. 2023; 12(4):663. https://doi.org/10.3390/antibiotics12040663
Chicago/Turabian StyleHerrera-Acevedo, Chonny, Renata Priscila Barros de Menezes, Natália Ferreira de Sousa, Luciana Scotti, Marcus Tullius Scotti, and Ericsson Coy-Barrera. 2023. "Kaurane-Type Diterpenoids as Potential Inhibitors of Dihydrofolate Reductase-Thymidylate Synthase in New World Leishmania Species" Antibiotics 12, no. 4: 663. https://doi.org/10.3390/antibiotics12040663
APA StyleHerrera-Acevedo, C., de Menezes, R. P. B., de Sousa, N. F., Scotti, L., Scotti, M. T., & Coy-Barrera, E. (2023). Kaurane-Type Diterpenoids as Potential Inhibitors of Dihydrofolate Reductase-Thymidylate Synthase in New World Leishmania Species. Antibiotics, 12(4), 663. https://doi.org/10.3390/antibiotics12040663