

Clinical Diagnostics of Bacterial Infections and Their Resistance to Antibiotics—Current State and Whole Genome Sequencing Implementation Perspectives


Abstract
:1. Introduction
2. Current Methods for the Clinical Diagnosis of Bacterial Infection
2.1. Pathogen Identification
2.1.1. Culture-Dependent Techniques
2.1.2. Mass Spectroscopy
2.1.3. Nucleic-Acid-Based Techniques
2.2. Antibiotic Susceptibility Profiling
2.2.1. Phenotypic Techniques
2.2.2. Molecular Techniques
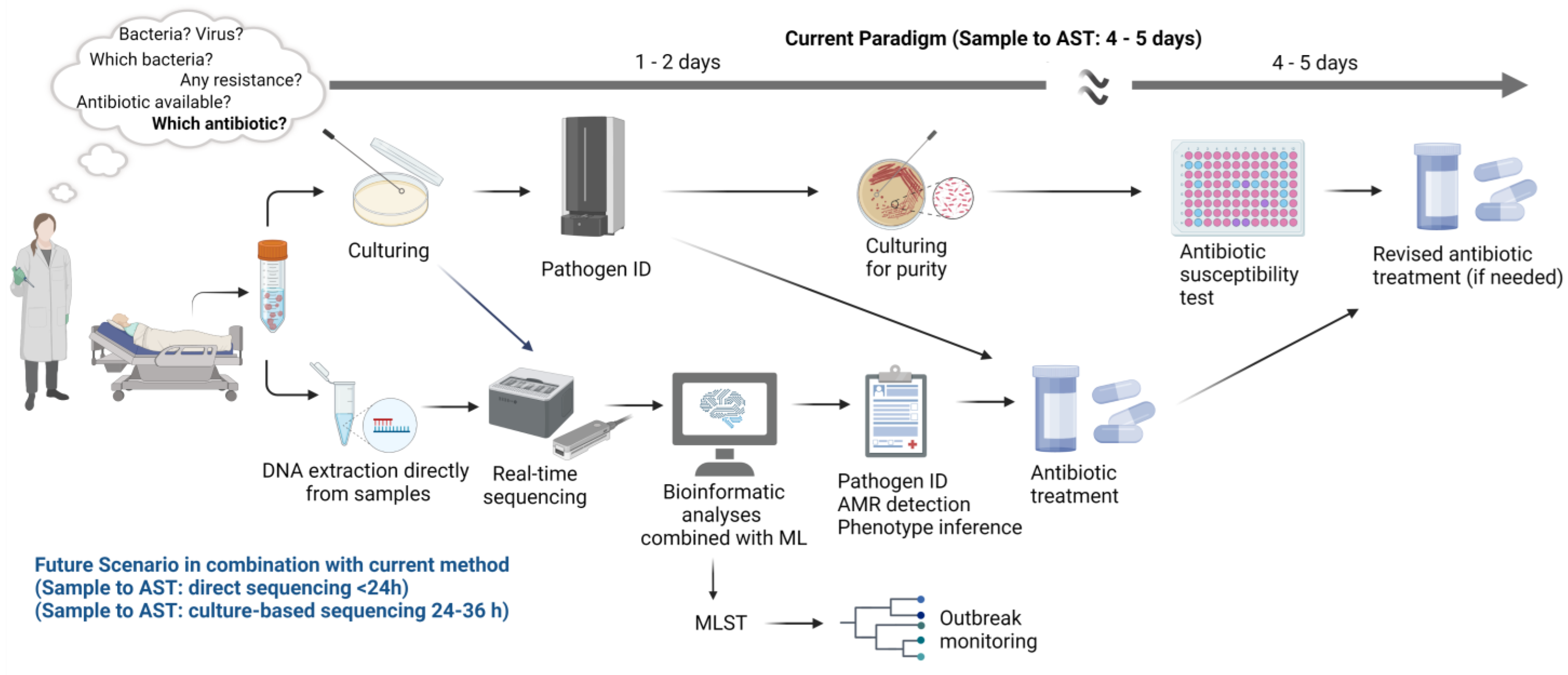
3. The Potential of Whole Genome Sequencing (WGS) for Use in Clinical Routine
3.1. DNA Sequencing Technologies
3.2. WGS Application in Clinical Settings
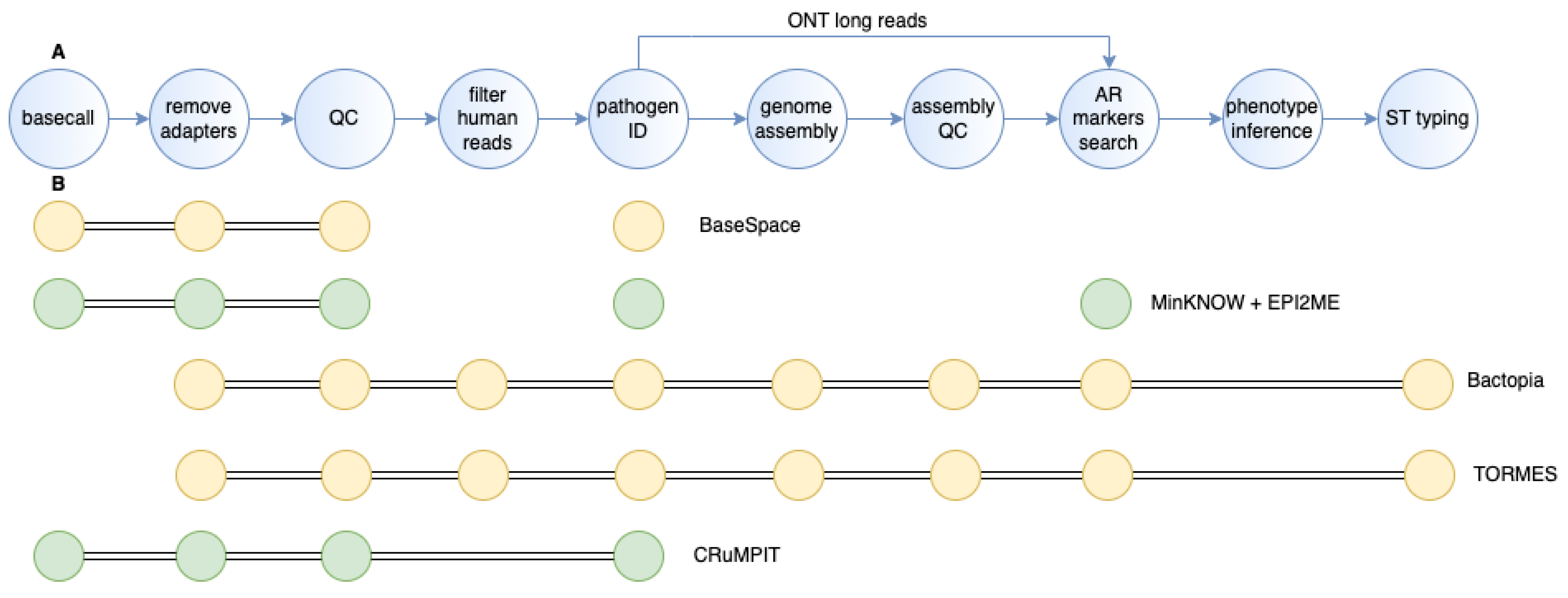
3.3. WGS Data Analysis
3.3.1. Base-Calling and Demultiplexing
3.3.2. Adapter Removal and Quality Filtering
3.3.3. Filtering of Human Reads
3.3.4. Pathogen Identification
3.3.5. Genome Assembly and Assembly Quality Control
3.3.6. Resistome Identification and Phenotype Inference
4. Machine Learning in Data Analysis for Tackling Antibiotic Resistance
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dadgostar, P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef]
- Reygaert, W.C. An Overview of the Antimicrobial Resistance Mechanisms of Bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.; Martins, A.; Spengler, G.; Molnar, J. Efflux Pumps of Gram-Negative Bacteria: What They Do, How They Do It, with What and How to Deal with Them. Front. Pharmacol. 2014, 4, 168. [Google Scholar] [CrossRef] [PubMed]
- Lambert, P.A. Bacterial Resistance to Antibiotics: Modified Target Sites. Adv. Drug Deliv. Rev. 2005, 57, 1471–1485. [Google Scholar] [CrossRef]
- Peterson, E.; Kaur, P. Antibiotic Resistance Mechanisms in Bacteria: Relationships between Resistance Determinants of Antibiotic Producers, Environmental Bacteria, and Clinical Pathogens. Front. Microbiol. 2018, 9, 2928. [Google Scholar] [CrossRef] [PubMed]
- Egorov, A.M.; Ulyashova, M.M.; Rubtsova, M.Y. Bacterial Enzymes and Antibiotic Resistance. Acta Nat. 2018, 10, 33–48. [Google Scholar] [CrossRef]
- Arora, G.; Bothra, A.; Prosser, G.; Arora, K.; Sajid, A. Role of Post-Translational Modifications in the Acquisition of Drug Resistance in Mycobacterium tuberculosis. FEBS J. 2021, 288, 3375–3393. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the Wild: Antibiotic Resistance Genes in Natural Environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef]
- Kadri, S.S. Key Takeaways from the U.S. Cdc’s 2019 Antibiotic Resistance Threats Report for Frontline Providers. Crit. Care Med. 2020, 48, 939–945. [Google Scholar] [CrossRef]
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Rosca, A.; Balcaen, T.; Lanoix, J.P.; Michaud, A.; Moyet, J.; Marcq, I.; Schmit, J.L.; Bloch, F.; Deschasse, G. Mortality Risk and Antibiotic Use for Covid-19 in Hospitalized Patients over 80. Biomed. Pharmacother. 2022, 146, 112481. [Google Scholar] [CrossRef] [PubMed]
- Sokolović, D.; Drakul, D.; Joksimović, B.; Lalović, N.; Avram, N.; Milić, M.; Nogo-Živanović, D.; Mijović, B. Consumption of Antibiotics in Primary Care Setting before and During Covid-19 Pandemic in Republic of Srpska, Bosnia and Herzegovina. Antibiotics 2022, 11, 1319. [Google Scholar] [CrossRef]
- Munk, P.; Brinch, C.; Moller, F.D.; Petersen, T.N.; Hendriksen, R.S.; Seyfarth, A.M.; Kjeldgaard, J.S.; Svendsen, C.A.; van Bunnik, B.; Berglund, F.; et al. Genomic Analysis of Sewage from 101 Countries Reveals Global Landscape of Antimicrobial Resistance. Nat. Commun. 2022, 13, 7251. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016.
- Pereira, B.; Kulkarni, S. Antibiotic Misuse and Improper Practices in India: Identifying the Scope to Improve through a Narrative Review. Int. J. Risk Saf. Med. 2021, 33, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.; Sarkar, A. Review on Multiple Facets of Drug Resistance: A Rising Challenge in the 21st Century. J. Xenobiot 2021, 11, 197–214. [Google Scholar] [CrossRef]
- Chokshi, A.; Sifri, Z.; Cennimo, D.; Horng, H. Global Contributors to Antibiotic Resistance. J. Glob. Infect. Dis. 2019, 11, 36–42. [Google Scholar]
- Anderson, M.; Clift, C.; Schulze, K.; Sagan, A.; Nahrgang, S.; Ouakrim, D.A.; Mossialos, E. European Observatory Policy Briefs. In Averting the Amr Crisis: What Are the Avenues for Policy Action for Countries in Europe? European Observatory on Health Systems and Policies: Copenhagen, Denmark, 2019. [Google Scholar]
- Santoro-Lopes, G.; de Gouvêa, E.F. Multidrug-Resistant Bacterial Infections after Liver Transplantation: An Ever-Growing Challenge. World J. Gastroenterol. 2014, 20, 6201–6210. [Google Scholar] [CrossRef]
- Van Boeckel, T.P.; Brower, C.; Gilbert, M.; Grenfell, B.T.; Levin, S.A.; Robinson, T.P.; Teillant, A.; Laxminarayan, R. Global Trends in Antimicrobial Use in Food Animals. Proc. Natl. Acad. Sci. USA 2015, 112, 5649–5654. [Google Scholar] [CrossRef] [PubMed]
- Jonas, O.B.; Irwin, A.; Berthe; Jean, F.C.; Gall, L.; Francois, G.; Marquez, P.V. Drug-Resistant Infections a Threat to Our Economic Future; World Bank Group: Washington, DC, USA, 2017. [Google Scholar]
- Naylor, N.R.; Atun, R.; Zhu, N.; Kulasabanathan, K.; Silva, S.; Chatterjee, A.; Knight, G.M.; Robotham, J.V. Estimating the Burden of Antimicrobial Resistance: A Systematic Literature Review. Antimicrob. Resist. Infect. Control. 2018, 7, 58. [Google Scholar] [CrossRef]
- Lekagul, A.; Tangcharoensathien, V.; Yeung, S. Patterns of Antibiotic Use in Global Pig Production: A Systematic Review. Vet. Anim. Sci. 2019, 7, 100058. [Google Scholar] [CrossRef] [PubMed]
- van Belkum, A.; Durand, G.; Peyret, M.; Chatellier, S.; Zambardi, G.; Schrenzel, J.; Shortridge, D.; Engelhardt, A.; Dunne, W.M., Jr. Rapid Clinical Bacteriology and Its Future Impact. Ann. Lab. Med. 2013, 33, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Bradley, P.; Gordon, N.C.; Walker, T.M.; Dunn, L.; Heys, S.; Huang, B.; Earle, S.; Pankhurst, L.J.; Anson, L.; De Cesare, M.; et al. Rapid Antibiotic-Resistance Predictions from Genome Sequence Data for Staphylococcus Aureus and Mycobacterium Tuberculosis. Nat. Commun. 2015, 6, 10063. [Google Scholar] [CrossRef]
- Lievens, B.; Thomma, B.P. Recent Developments in Pathogen Detection Arrays: Implications for Fungal Plant Pathogens and Use in Practice. Phytopathology 2005, 95, 1374–1380. [Google Scholar] [CrossRef]
- Mohamed Salleh, N.A.B.; Tanaka, Y.; Sutarlie, L.; Su, X. Detecting Bacterial Infections in Wounds: A Review of Biosensors and Wearable Sensors in Comparison with Conventional Laboratory Methods. Analyst 2022, 147, 1756–1776. [Google Scholar] [CrossRef]
- Książczyk, M.; Kuczkowski, M.; Dudek, B.; Korzekwa, K.; Tobiasz, A.; Korzeniowska-Kowal, A.; Paluch, E.; Wieliczko, A.; Bugla-Płoskońska, G. Application of Routine Diagnostic Procedure, Vitek 2 Compact, Maldi-Tof Ms, and Pcr Assays in Identification Procedure of Bacterial Strain with Ambiguous Phenotype. Curr. Microbiol. 2016, 72, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Verma, J.; Saxena, S.; Babu, S.G. Elisa-Based Identification and Detection of Microbes. In Analyzing Microbes: Manual of Molecular Biology Techniques; Springer: Berlin, Heidelberg, 2013. [Google Scholar]
- Torres-Sangiao, E.; Rodriguez, C.L.; García-Riestra, C. Application and Perspectives of Maldi–Tof Mass Spectrometry in Clinical Microbiology Laboratories. Microorganisms 2021, 9, 1539. [Google Scholar] [CrossRef]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. Maldi-Tof Mass Spectrometry: An Emerging Technology for Microbial Identification and Diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef]
- Rychert, J. Benefits and Limitations of Maldi-Tof Mass Spectrometry for the Identification of Microorganisms. J. Infect. Epidemiol. 2019, 2, 1–5. [Google Scholar] [CrossRef]
- Wieser, A.; Schneider, L.; Jung, J.; Schubert, S. Maldi-Tof Ms in Microbiological Diagnostics-Identification of Microorganisms and Beyond (Mini Review). Appl. Microbiol. Biotechnol. 2012, 93, 965–974. [Google Scholar] [CrossRef]
- Florio, W.; Baldeschi, L.; Rizzato, C.; Tavanti, A.; Ghelardi, E.; Lupetti, A. Detection of Antibiotic-Resistance by Maldi-Tof Mass Spectrometry: An Expanding Area. Front. Cell. Infect. Microbiol. 2020, 10, 572909. [Google Scholar] [CrossRef] [PubMed]
- Weis, C.; Rieck, B.; Balzer, S.; Cuénod, A.; Egli, A.; Borgwardt, K. Improved Maldi-Tof Ms Based Antimicrobial Resistance Prediction through Hierarchical Stratification. bioRxiv 2022. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16s Rrna Gene Sequencing for Species and Strain-Level Microbiome Analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Karaoz, U.; Volegova, M.; MacKichan, J.; Kato-Maeda, M.; Miller, S.; Nadarajan, R.; Brodie, E.L.; Lynch, S.V. Use of 16s Rrna Gene for Identification of a Broad Range of Clinically Relevant Bacterial Pathogens. PLoS ONE 2015, 10, e0117617. [Google Scholar] [CrossRef]
- Rogers, G.B.; Carroll, M.P.; Bruce, K.D. Studying Bacterial Infections through Culture-Independent Approaches. J. Med. Microbiol. 2009, 58, 1401–1418. [Google Scholar] [CrossRef]
- Pilecky, M.; Schildberger, A.; Orth-Höller, D.; Weber, V. Pathogen Enrichment from Human Whole Blood for the Diagnosis of Bloodstream Infection: Prospects and Limitations. Diagn. Microbiol. Infect. Dis. 2019, 94, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, P.; Bryson, A.L.; Binnicker, M.J.; Pritt, B.S.; Patel, R. Syndromic Panel-Based Testing in Clinical Microbiology. Clin. Microbiol. Rev. 2018, 31, e00024-17. [Google Scholar] [CrossRef]
- Bard, J.D.; McElvania, E. Panels and Syndromic Testing in Clinical Microbiology. Clin. Lab. Med. 2020, 40, 393–420. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and Broth Dilution Methods to Determine the Minimal Inhibitory Concentration (Mic) of Antimicrobial Substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- Marroki, A.; Bousmaha-Marroki, L. Antibiotic Resistance Diagnostic Methods for Pathogenic Bacteria. In Encyclopedia of Infection and Immunity; Rezaei, N., Ed.; Elsevier: Oxford, UK, 2022; pp. 320–341. [Google Scholar]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in Vitro Evaluating Antimicrobial Activity: A Review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef]
- Gajic, I.; Kabic, J.; Kekic, D.; Jovicevic, M.; Milenkovic, M.; Culafic, D.M.; Trudic, A.; Ranin, L.; Opavski, N. Antimicrobial Susceptibility Testing: A Comprehensive Review of Currently Used Methods. Antibiotics 2022, 11, 427. [Google Scholar] [CrossRef] [PubMed]
- Reller, L.B.; Weinstein, M.; Jorgensen, J.H.; Ferraro, M.J. Antimicrobial Susceptibility Testing: A Review of General Principles and Contemporary Practices. Clinical. Infect. Dis. 2009, 49, 1749–1755. [Google Scholar]
- Chapot, V.; Effenberg, L.; Dohmen-Ruetten, J.; Buer, J.; Kehrmann, J. Evaluation of the Accelerate Pheno System for Rapid Identification and Antimicrobial Susceptibility Testing of Positive Blood Culture Bottles Inoculated with Primary Sterile Specimens from Patients with Suspected Severe Infections. J. Clin. Microbiol. 2021, 59, e02637-20. [Google Scholar] [CrossRef] [PubMed]
- Inglis, T.J.J.; Paton, T.F.; Kopczyk, M.K.; Mulroney, K.T.; Carson, C.F. Same-day antimicrobial susceptibility test using acoustic-enhanced flow cytometry visualized with supervised machine learning. J. Med. Microbiol. 2020, 69, 657–669. [Google Scholar] [CrossRef]
- Weile, J.; Knabbe, C. Current Applications and Future Trends of Molecular Diagnostics in Clinical Bacteriology. Anal. Bioanal. Chem. 2009, 394, 731–742. [Google Scholar] [CrossRef]
- Vasudevan, H.N.; Xu, P.; Servellita, V.; Miller, S.; Liu, L.; Gopez, A.; Chiu, C.Y.; Abate, A.R. Digital Droplet Pcr Accurately Quantifies Sars-Cov-2 Viral Load from Crude Lysate without Nucleic Acid Purification. Sci. Rep. 2021, 11, 780. [Google Scholar] [CrossRef]
- Varlamov, D.A.; Blagodatskikh, K.A.; Smirnova, E.V.; Kramarov, V.M.; Ignatov, K.B. Combinations of Pcr and Isothermal Amplification Techniques Are Suitable for Fast and Sensitive Detection of Sars-Cov-2 Viral Rna. Front. Bioeng. Biotechnol. 2020, 8, 604793. [Google Scholar] [CrossRef]
- Yee, R.; Bard, J.D.; Simner, P.J. The Genotype-to-Phenotype Dilemma: How Should Laboratories Approach Discordant Susceptibility Results? J. Clin. Microbiol. 2021, 59, e00138-20. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, M100. In Appendix H. Using Molecular Assays for Resistance Detection; CLSI: Wayne, PA, USA, 2020. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Ludwig, W.; Schleifer, K.H. Bacterial Phylogeny Based on 16s and 23s Rrna Sequence Analysis. FEMS Microbiol. Rev. 1994, 15, 155–173. [Google Scholar] [CrossRef]
- Athanasopoulou, K.; Boti, M.A.; Adamopoulos, P.G.; Skourou, P.C.; Scorilas, A. Third-Generation Sequencing: The Spearhead Towards the Radical Transformation of Modern Genomics. Life 2021, 12, 30. [Google Scholar] [CrossRef] [PubMed]
- Besser, J.; Carleton, H.A.; Gerner-Smidt, P.; Lindsey, R.L.; Trees, E. Next-Generation Sequencing Technologies and Their Application to the Study and Control of Bacterial Infections. Clin. Microbiol. Infect. 2018, 24, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, B.; Tekin, M.; Mahdieh, N. The Promise of Whole-Exome Sequencing in Medical Genetics. J. Hum. Genet. 2014, 59, 5–15. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of Age: Ten Years of Next-Generation Sequencing Technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Hufnagel, D.E.; Hufford, M.B.; Seetharam, A.S. Sequeltools: A Suite of Tools for Working with Pacbio Sequel Raw Sequence Data. BMC Bioinform. 2020, 21, 429. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From Squiggle to Basepair: Computational Approaches for Improving Nanopore Sequencing Read Accuracy. Genome. Biol. 2018, 19, 90. [Google Scholar] [CrossRef]
- Petersen, L.M.; Martin, I.W.; Moschetti, W.E.; Kershaw, C.M.; Tsongalis, G.J. Third-Generation Sequencing in the Clinical Laboratory: Exploring the Advantages and Challenges of Nanopore Sequencing. J. Clin. Microbiol. 2019, 58, e01315-19. [Google Scholar] [CrossRef]
- Vaca, D.J.; Dobler, G.; Fischer, S.F.; Keller, C.; Konrad, M.; von Loewenich, F.D.; Orenga, S.; Sapre, S.U.; van Belkum, A.; Kempf, V.A.J. Contemporary Diagnostics for Medically Relevant Fastidious Microorganisms Belonging to the Genera Anaplasma, Bartonella, Coxiella, Orientia and Rickettsia. FEMS Microbiol. Rev. 2022, 46, fuac013. [Google Scholar] [CrossRef]
- Taxt, A.M.; Avershina, E.; Frye, S.A.; Naseer, U.; Ahmad, R. Rapid Identification of Pathogens, Antibiotic Resistance Genes and Plasmids in Blood Cultures by Nanopore Sequencing. Sci. Rep. 2020, 10, 7622. [Google Scholar] [CrossRef]
- Bourke, T.W.; McKenna, J.P.; Coyle, P.V.; Shields, M.D.; Fairley, D.J. Diagnostic Accuracy of Loop-Mediated Isothermal Amplification as a near-Patient Test for Meningococcal Disease in Children: An Observational Cohort Study. Lancet Infect. Dis. 2015, 15, 552–558. [Google Scholar] [CrossRef]
- Stoler, N.; Nekrutenko, A. Sequencing Error Profiles of Illumina Sequencing Instruments. NAR Genom. Bioinform. 2021, 3, lqab019. [Google Scholar] [CrossRef]
- Charalampous, T.; Kay, G.L.; Richardson, H.; Aydin, A.; Baldan, R.; Jeanes, C.; Rae, D.; Grundy, S.; Turner, D.J.; Wain, J.; et al. Nanopore Metagenomics Enables Rapid Clinical Diagnosis of Bacterial Lower Respiratory Infection. Nat. Biotechnol. 2019, 37, 783–792. [Google Scholar] [CrossRef]
- Janes, V.A.; Matamoros, S.; Munk, P.; Clausen, P.T.L.C.; Koekkoek, S.M.; Koster, L.A.M.; Jakobs, M.E.; de Wever, B.; Visser, C.E.; Aarestrup, F.M.; et al. Metagenomic DNA Sequencing for Semi-Quantitative Pathogen Detection from Urine: A Prospective, Laboratory-Based, Proof-of-Concept Study. Lancet Microbe 2022, 3, e588–e597. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, W.; Zhang, S.; Li, Q.; Wang, Y.; Chen, T.; Jiang, H.; Kong, D.; Lv, Q.; Zheng, Y.; et al. Rapid Detection of Bacterial Pathogens and Antimicrobial Resistance Genes in Clinical Urine Samples with Urinary Tract Infection by Metagenomic Nanopore Sequencing. Front. Microbiol. 2022, 13, 858777. [Google Scholar] [CrossRef]
- Morsli, M.; Bechah, Y.; Coulibaly, O.; Toro, A.; Fournier, P.E.; Houhamdi, L.; Drancourt, M. Direct Diagnosis of Pasteurella Multocida Meningitis Using Next-Generation Sequencing. Lancet Microbe 2022, 3, e6. [Google Scholar] [CrossRef]
- Morsli, M.; Kerharo, Q.; Amrane, S.; Parola, P.; Fournier, P.E.; Drancourt, M. Real-Time Whole Genome Sequencing Direct Diagnosis of Streptococcus pneumoniae Meningitis: A Case Report. J. Infect. 2021, 83, 709–737. [Google Scholar] [CrossRef]
- Morsli, M.; Kerharo, Q.; Delerce, J.; Roche, P.-H.; Troude, L.; Drancourt, M. Haemophilus Influenzae Meningitis Direct Diagnosis by Metagenomic Next-Generation Sequencing: A Case Report. Pathogens 2021, 10, 461. [Google Scholar] [CrossRef]
- Jang, Y.; Kim, S.; Kim, N.; Son, H.; Ha, E.J.; Koh, E.J.; Phi, J.H.; Park, C.-K.; Kim, J.E.; Kim, S.-K.; et al. Nanopore 16s Sequencing Enhances the Detection of Bacterial Meningitis after Neurosurgery. Ann. Clin. Transl. Neurol. 2022, 9, 312–325. [Google Scholar] [CrossRef]
- Avershina, E.; Frye, S.A.; Ali, J.; Taxt, A.M.; Ahmad, R. Ultrafast and Cost-Effective Pathogen Identification and Resistance Gene Detection in a Clinical Setting Using Nanopore Flongle Sequencing. Front. Microbiol. 2022, 13, 822402. [Google Scholar] [CrossRef] [PubMed]
- Whittle, E.; Yonkus, J.A.; Jeraldo, P.; Alva-Ruiz, R.; Nelson, H.; Kendrick, M.L.; Grys, T.E.; Patel, R.; Truty, M.J.; Chia, N. Optimizing Nanopore Sequencing for Rapid Detection of Microbial Species and Antimicrobial Resistance in Patients at Risk of Surgical Site Infections. Msphere 2022, 7, e00964-21. [Google Scholar] [CrossRef] [PubMed]
- Yonkus, J.A.; Whittle, E.; Alva-Ruiz, R.; Abdelrahman, A.M.; Horsman, S.E.; Suh, G.A.; Cunningham, S.A.; Nelson, H.; Grotz, T.E.; Smoot, R.L.; et al. Answers in Hours: A Prospective Clinical Study Using Nanopore Sequencing for Bile Duct Cultures. Surgery 2022, 171, 693–702. [Google Scholar] [CrossRef]
- Ahmadi, A.; Khezri, A.; Nørstebø, H.; Ahmad, R. A Culture-, Amplification-Independent, and Rapid Method for Identification of Pathogens and Antibiotic Resistance Profile in Bovine Mastitis Milk. Front. Microbiol. 2022, 13, 1104701. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, N.D.; Street, T.L.; Foster, D.; Swann, J.; Atkins, B.L.; Brent, A.J.; McNally, M.A.; Oakley, S.; Taylor, A.; Peto, T.E.A.; et al. Real-Time Analysis of Nanopore-Based Metagenomic Sequencing from Infected Orthopaedic Devices. BMC Genom. 2018, 19, 714. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-X.; Huang, Z.; Fang, W.; Zhang, Z.; Fang, X.; Li, W.; Yang, B.; Chen, L.; Liang, X.; Hu, H.; et al. Preliminary Assessment of Nanopore-Based Metagenomic Sequencing for the Diagnosis of Prosthetic Joint Infection. Int. J. Infect. Dis. 2020, 97, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Cacho, A.; Smirnova, E.; Huzurbazar, S.; Cui, X. A Comparison of Base-Calling Algorithms for Illumina Sequencing Technology. Brief. Bioinform. 2015, 17, 786–795. [Google Scholar] [CrossRef]
- Wilkins, O.G.; Capitanchik, C.; Luscombe, N.M.; Ule, J. Ultraplex: A Rapid, Flexible, All-in-One Fastq Demultiplexer. Wellcome Open Res. 2021, 6, 141. [Google Scholar] [CrossRef]
- Perešíni, P.; Boža, V.; Brejová, B.; Vinař, T. Nanopore Base Calling on the Edge. Bioinformatics 2021, 37, 4661–4667. [Google Scholar] [CrossRef]
- Andrews, S. Fastqc: A Quality Control Tool for High Throughput Sequence Data. 2015. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 9 February 2023).
- Fukasawa, Y.; Ermini, L.; Wang, H.; Carty, K.; Cheung, M.S. Longqc: A Quality Control Tool for Third Generation Sequencing Long Read Data. G3 Genes Genom. Genet. 2020, 10, 1193–1196. [Google Scholar] [CrossRef]
- Leger, A.; Leonardi, T. Pycoqc, Interactive Quality Control for Oxford Nanopore Sequencing. J. Open Source Softw. 2019, 4, 1236. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing Bacterial Genome Assemblies with Multiplex Minion Sequencing. Microbial. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and Sensitive Classification of Metagenomic Sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Spades: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly Via Adaptive K-Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Li, H. Minimap and Miniasm: Fast Mapping and De Novo Assembly for Noisy Long Sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Šikić, M. Time- and Memory-Efficient Genome Assembly with Raven. Nat. Comput. Sci. 2021, 1, 332–336. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Cerdeira, L.T.; Hawkey, J.; Méric, G.; Vezina, B.; Wyres, K.L.; Holt, K.E. Trycycler: Consensus Long-Read Assemblies for Bacterial Genomes. Genome Biol. 2021, 22, 266. [Google Scholar] [CrossRef] [PubMed]
- Khezri, A.; Avershina, E.; Ahmad, R. Hybrid Assembly Provides Improved Resolution of Plasmids, Antimicrobial Resistance Genes, and Virulence Factors in Escherichia Coli and Klebsiella Pneumoniae Clinical Isolates. Microorganisms 2021, 9, 2560. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. Quast: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. Busco: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. Checkm: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Manchanda, N.; Portwood, J.L.; Woodhouse, M.R.; Seetharam, A.S.; Lawrence-Dill, C.J.; Andorf, C.M.; Hufford, M.B. Genomeqc: A Quality Assessment Tool for Genome Assemblies and Gene Structure Annotations. BMC Genom. 2020, 21, 193. [Google Scholar] [CrossRef]
- Seemann, T. ABRicate: Mass screening of contigs for antimicrobial resistance or virulence genes. Available online: https://github.com/tseemann/abricate (accessed on 9 February 2023).
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. Amrfinderplus and the Reference Gene Catalog Facilitate Examination of the Genomic Links among Antimicrobial Resistance, Stress Response, and Virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. Resfinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.V.; Cheng, A.A.; Liu, S.; et al. Card 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic. Acids. Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.; Mather, A.E.; Sánchez-Busó, L.; Page, A.J.; Parkhill, J.; Keane, J.A.; Harris, S.R. Ariba: Rapid Antimicrobial Resistance Genotyping Directly from Sequencing Reads. Microbial. Genom. 2017, 3, e000131. [Google Scholar] [CrossRef] [PubMed]
- Khezri, A.; Avershina, E.; Ahmad, R. Plasmid Identification and Plasmid-Mediated Antimicrobial Gene Detection in Norwegian Isolates. Microorganisms 2021, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Petit, R.A.; Read, T.D. Bactopia: A Flexible Pipeline for Complete Analysis of Bacterial Genomes. mSystems 2020, 5, e00190-20. [Google Scholar] [CrossRef]
- Quijada, N.M.; Rodríguez-Lázaro, D.; Eiros, J.M.; Hernández, M. Tormes: An Automated Pipeline for Whole Bacterial Genome Analysis. Bioinformatics 2019, 35, 4207–4212. [Google Scholar] [CrossRef]
- Seemann, T.; da Silva, A.G.; Bulach, D.M.; Schultz, M.B.; Kwong, J.C.; Howden, B.P. Nullarbor: Pipeline to generate complete public health microbiology reports from sequenced isolates. Available online: https://github.com/tseemann/nullarbor (accessed on 9 February 2023).
- Schürch, A.C.; van Schaik, W. Challenges and Opportunities for Whole-Genome Sequencing–Based Surveillance of Antibiotic Resistance. Ann. N. Y. Acad. Sci. 2017, 1388, 108–120. [Google Scholar] [CrossRef]
- Dahl, L.G.; Joensen, K.G.; Østerlund, M.T.; Kiil, K.; Nielsen, E.M. Prediction of Antimicrobial Resistance in Clinical Campylobacter Jejuni Isolates from Whole-Genome Sequencing Data. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 673–682. [Google Scholar] [CrossRef]
- Rebelo, A.R.; Bortolaia, V.; Leekitcharoenphon, P.; Hansen, D.S.; Nielsen, H.L.; Ellermann-Eriksen, S.; Kemp, M.; Røder, B.L.; Frimodt-Møller, N.; Søndergaard, T.S.; et al. One Day in Denmark: Comparison of Phenotypic and Genotypic Antimicrobial Susceptibility Testing in Bacterial Isolates from Clinical Settings. Front. Microbiol. 2022, 13, 804627. [Google Scholar] [CrossRef]
- Qi, L.; Li, H.; Zhang, C.; Liang, B.; Li, J.; Wang, L.; Du, X.; Liu, X.; Qiu, S.; Song, H. Relationship between Antibiotic Resistance, Biofilm Formation, and Biofilm-Specific Resistance in Acinetobacter Baumannii. Front. Microbiol. 2016, 7, 483. [Google Scholar] [CrossRef]
- Moran, R.A.; Anantham, S.; Holt, K.E.; Hall, R.M. Prediction of Antibiotic Resistance from Antibiotic Resistance Genes Detected in Antibiotic-Resistant Commensal Escherichia Coli Using Pcr or Wgs. J. Antimicrob. Chemother. 2016, 72, 700–704. [Google Scholar]
- Kumburu, H.H.; Sonda, T.; van Zwetselaar, M.; Leekitcharoenphon, P.; Lukjancenko, O.; Mmbaga, B.T.; Alifrangis, M.; Lund, O.; Aarestrup, F.M.; Kibiki, G.S. Using Wgs to Identify Antibiotic Resistance Genes and Predict Antimicrobial Resistance Phenotypes in Mdr Acinetobacter Baumannii in Tanzania. J. Antimicrob. Chemother. 2019, 74, 1484–1493. [Google Scholar] [CrossRef] [PubMed]
- Dey, A. Machine Learning Algorithms: A Review. Int. J. Comput. Sci. Inf. Technol. 2016, 7, 1174–1179. [Google Scholar]
- Sarker, I.H. Machine Learning: Algorithms, Real-World Applications and Research Directions. SN Comput. Sci. 2021, 2, 160. [Google Scholar] [CrossRef] [PubMed]
- Feretzakis, G.; Loupelis, E.; Sakagianni, A.; Kalles, D.; Martsoukou, M.; Lada, M.; Skarmoutsou, N.; Christopoulos, C.; Valakis, K.; Velentza, A.; et al. Using Machine Learning Techniques to Aid Empirical Antibiotic Therapy Decisions in the Intensive Care Unit of a General Hospital in Greece. Antibiotics 2020, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Wright, S.N.; Hamblin, M.; McCloskey, D.; Alcantar, M.A.; Schrübbers, L.; Lopatkin, A.J.; Satish, S.; Nili, A.; Palsson, B.O.; et al. A White-Box Machine Learning Approach for Revealing Antibiotic Mechanisms of Action. Cell 2019, 177, 1649–1661.e9. [Google Scholar] [CrossRef]
- Ferreira, I.; Beisken, S.; Lueftinger, L.; Weinmaier, T.; Klein, M.; Bacher, J.; Patel, R.; von Haeseler, A.; Posch, A.E. Species Identification and Antibiotic Resistance Prediction by Analysis of Whole-Genome Sequence Data by Use of Aresdb: An Analysis of Isolates from the Unyvero Lower Respiratory Tract Infection Trial. J. Clin. Microbiol. 2020, 58, e00273-20. [Google Scholar] [CrossRef]
- Sundermann, A.J.; Chen, J.; Kumar, P.; Ayres, A.M.; Cho, S.-T.; Ezeonwuka, C.; Griffith, M.P.; Miller, J.K.; Mustapha, M.M.; Pasculle, A.W.; et al. Whole-Genome Sequencing Surveillance and Machine Learning of the Electronic Health Record for Enhanced Healthcare Outbreak Detection. Clin. Infect. Dis. 2022, 75, 476–482. [Google Scholar] [CrossRef]
- McCoubrey, L.E.; Gaisford, S.; Orlu, M.; Basit, A.W. Predicting Drug-Microbiome Interactions with Machine Learning. Biotechnol. Adv. 2022, 54, 107797. [Google Scholar] [CrossRef]
- Nguyen, M.; Brettin, T.; Long, S.W.; Musser, J.M.; Olsen, R.J.; Olson, R.; Shukla, M.; Stevens, R.L.; Xia, F.; Yoo, H.; et al. Developing an in Silico Minimum Inhibitory Concentration Panel Test for Klebsiella Pneumoniae. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Kim, J.; Greenberg, D.E.; Pifer, R.; Jiang, S.; Xiao, G.; Shelburne, S.A.; Koh, A.; Xie, Y.; Zhan, X. Vampr: Variant Mapping and Prediction of Antibiotic Resistance Via Explainable Features and Machine Learning. PLOS Comput. Biol. 2020, 16, e1007511. [Google Scholar] [CrossRef]
- Avershina, E.; Sharma, P.; Taxt, A.M.; Singh, H.; Frye, S.A.; Paul, K.; Kapil, A.; Naseer, U.; Kaur, P.; Ahmad, R. Amr-Diag: Neural Network Based Genotype-to-Phenotype Prediction of Resistance Towards Β-Lactams in Escherichia Coli and Klebsiella Pneumoniae. Comput. Struct. Biotechnol. J. 2021, 19, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Sunuwar, J.; Azad, R.K. A Machine Learning Framework to Predict Antibiotic Resistance Traits and yet Unknown Genes Underlying Resistance to Specific Antibiotics in Bacterial Strains. Brief Bioinform. 2021, 22, bbab179. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Chakraborty, T.; Doijad, S.; Falgenhauer, L.; Falgenhauer, J.; Goesmann, A.; Hauschild, A.C.; Schwengers, O.; Heider, D. Prediction of Antimicrobial Resistance Based on Whole-Genome Sequencing and Machine Learning. Bioinformatics 2022, 38, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Ellington, M.J.; Ekelund, O.; Aarestrup, F.M.; Canton, R.; Doumith, M.; Giske, C.; Grundman, H.; Hasman, H.; Holden, M.T.G.; Hopkins, K.L.; et al. The Role of Whole Genome Sequencing in Antimicrobial Susceptibility Testing of Bacteria: Report from the Eucast Subcommittee. Clin. Microbiol. Infect. 2017, 23, 2–22. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Antimicrobial Resistance and Use Surveillance System (Glass). Available online: https://www.who.int/initiatives/glass (accessed on 24 October 2022).
- Li, Y.; Yang, X.; Zhao, W. Emerging Microtechnologies and Automated Systems for Rapid Bacterial Identification and Antibiotic Susceptibility Testing. SLAS Technol. 2017, 22, 585–608. [Google Scholar] [CrossRef]
- Ahmad, A.; Hettiarachchi, R.; Khezri, A.; Singh Ahluwalia, B.; Wadduwage, D.N.; Ahmad, R. Highly sensitive quantitative phase microscopy and deep learning aided with whole genome sequencing for rapid detection of infection and antimicrobial resistance. Front. Microbiol. 2023, 14, 1154620. [Google Scholar] [CrossRef]
- Lister, A.; Avershina, E.; Ali, J.; Devitt, G.; Hanrahan, N.; Highmore, C.; Webb, J.; Muller, F.; Mahajan, S.; Ahmad, R. Multi-Excitation Raman Spectroscopy Complements Whole Genome Sequencing for Rapid Detection of Bacterial Infection and Resistance in Who Priority Pathogens. bioRxiv 2022. [Google Scholar] [CrossRef]
- Gieroba, B.; Krysa, M.; Wojtowicz, K.; Wiater, A.; Pleszczyńska, M.; Tomczyk, M.; Sroka-Bartnicka, A. The Ft-Ir and Raman Spectroscopies as Tools for Biofilm Characterization Created by Cariogenic Streptococci. Int. J. Mol. Sci. 2020, 21, 3811. [Google Scholar] [CrossRef]
- Tewes, T.J.; Centeleghe, I.; Maillard, J.Y.; Platte, F.; Bockmühl, D.P. Raman Microscopic Analysis of Dry-Surface Biofilms on Clinically Relevant Materials. Microorganisms 2022, 10, 1369. [Google Scholar] [CrossRef]
- Afolayan, A.O.; Bernal, J.F.; Gayeta, J.M.; Masim, M.L.; Shamanna, V.; Abrudan, M.; Abudahab, K.; Argimón, S.; Carlos, C.C.; Sia, S.; et al. Overcoming Data Bottlenecks in Genomic Pathogen Surveillance. Clin. Infect. Dis. 2021, 73, S267–S274. [Google Scholar] [CrossRef]
- EU. Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions Building Trust in Human-Centric Artificial Intelligence; EU: Brussels, Belgium, 2019. [Google Scholar]
- EU. Draft Ethics Guidelines for Trustworthy AI; EU: Brussels, Belgium, 2021. [Google Scholar]



{kind=link}
{kind=link}
| Disease | Sample Type | Number of samples | Culture Independent | Turnaround Time (h) | Sensitivity (%) | Specificity (%) | ARG Detection | Main Outcome | Reference |
|---|---|---|---|---|---|---|---|---|---|
| LRTI | Sputum | 81 | yes | 6 | 96.6 | 41.7 | yes | mONS can rapidly and accurately characterize bacterial LRIs and might contribute to a reduction in broad-spectrum antibiotic use | [70] |
| UTI | Urine | 86 | yes | NR * | 71–100 | - | yes | mONS pathogen and resistome detection for the diagnosis of UTIs warrants prospective clinical validation | [71] |
| 76 | yes | 6 | 86.7 | 96.8 | yes | mONS is a promising clinical diagnostic tool for infectious diseases | [72] | ||
| Meningitis | Cerebral spinal fluid | 1 | yes | ~5 | - | - | yes | mONS enabled the direct diagnosis of meningitis caused by Pasteurella multocida, which is not routinely detected by current point-of-care assays | [73] |
| 1 | yes | 2 | - | - | yes | mONS enabled the diagnosis of community-acquired Streptococcus pneumoniae meningitis, providing a less than two-hour workflow including only a 20 min sequencing time, detection, identification, typing, and an in silico antibiogram | [74] | ||
| 1 | yes | 6 | - | - | yes | mONS could be considered for the point-of-care diagnosis of infectious meningitis, by direct identification of pathogenic genomes and their genotypes/serotypes | [75] | ||
| 285 | yes | NR | 100 | 94.4 | no | 16S rDNA mONS was more effective than conventional culture in postoperative bacterial meningitis and may contribute to evidence-based decisions for antibiotic maintenance and discontinuation | [76] | ||
| BSI | Blood | 8 | no | 3.5–6 | 100 ** | 83.1 ** | yes | Identification of pathogens was possible after 10 min of sequencing and all predefined AMR-encoding genes and plasmids from monoculture experiments were detected within one hour using raw mONS data | [67] |
| 2 | no | 6.5 | 100 ** | - | yes | Flongle data allowed for rapid bacterial ID and resistome detection based on the first 1000–3000 generated sequences (10 min to 3 h from the sequencing start) | [77] | ||
| SSI | IBS | 42 | yes | 6–14 | 100 | - | yes | The results generated using mONS were similar to the current methods of detection but were obtained in a significantly shorter amount of time. mONS could be used to tailor antibiotics in surgical patients and reduce the use of broad-spectrum antibiotics | [78] |
| 42 | yes | 8 | 100 | 100 | yes | Rapid microbial profiling with mONS is feasible with broader organism and resistance profiling compared to standard cultures. mONS has perfect negative predictive values and can potentially improve antibiotic stewardship | [79] | ||
| Mastitis | Milk | 24 | yes | 9 | 100 | 92.3 | yes | A proof-of-concept study that established an effective method for host removal and direct MinION sequencing from mastitis milk | [80] |
| PJI | Sonication fluids | 7 | yes | NR | 100 ** | 100 ** | no | A novel, scalable pipeline for the real-time analysis of MinION sequence data and the use of this pipeline to show initial the proof of concept that mONS can provide rapid and accurate diagnosis for prosthetic joint infections | [81] |
| JF and PT | 9 | yes | 14–22 | 100 ** | 80 ** | yes | A proof-of-concept study that established that mONS can function as a rapid and accurate tool in PJI diagnostic microbiology | [82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avershina, E.; Khezri, A.; Ahmad, R. Clinical Diagnostics of Bacterial Infections and Their Resistance to Antibiotics—Current State and Whole Genome Sequencing Implementation Perspectives. Antibiotics 2023, 12, 781. https://doi.org/10.3390/antibiotics12040781
Avershina E, Khezri A, Ahmad R. Clinical Diagnostics of Bacterial Infections and Their Resistance to Antibiotics—Current State and Whole Genome Sequencing Implementation Perspectives. Antibiotics. 2023; 12(4):781. https://doi.org/10.3390/antibiotics12040781
Chicago/Turabian StyleAvershina, Ekaterina, Abdolrahman Khezri, and Rafi Ahmad. 2023. "Clinical Diagnostics of Bacterial Infections and Their Resistance to Antibiotics—Current State and Whole Genome Sequencing Implementation Perspectives" Antibiotics 12, no. 4: 781. https://doi.org/10.3390/antibiotics12040781
APA StyleAvershina, E., Khezri, A., & Ahmad, R. (2023). Clinical Diagnostics of Bacterial Infections and Their Resistance to Antibiotics—Current State and Whole Genome Sequencing Implementation Perspectives. Antibiotics, 12(4), 781. https://doi.org/10.3390/antibiotics12040781