
Horizontal Gene Transfer and Drug Resistance Involving Mycobacterium tuberculosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
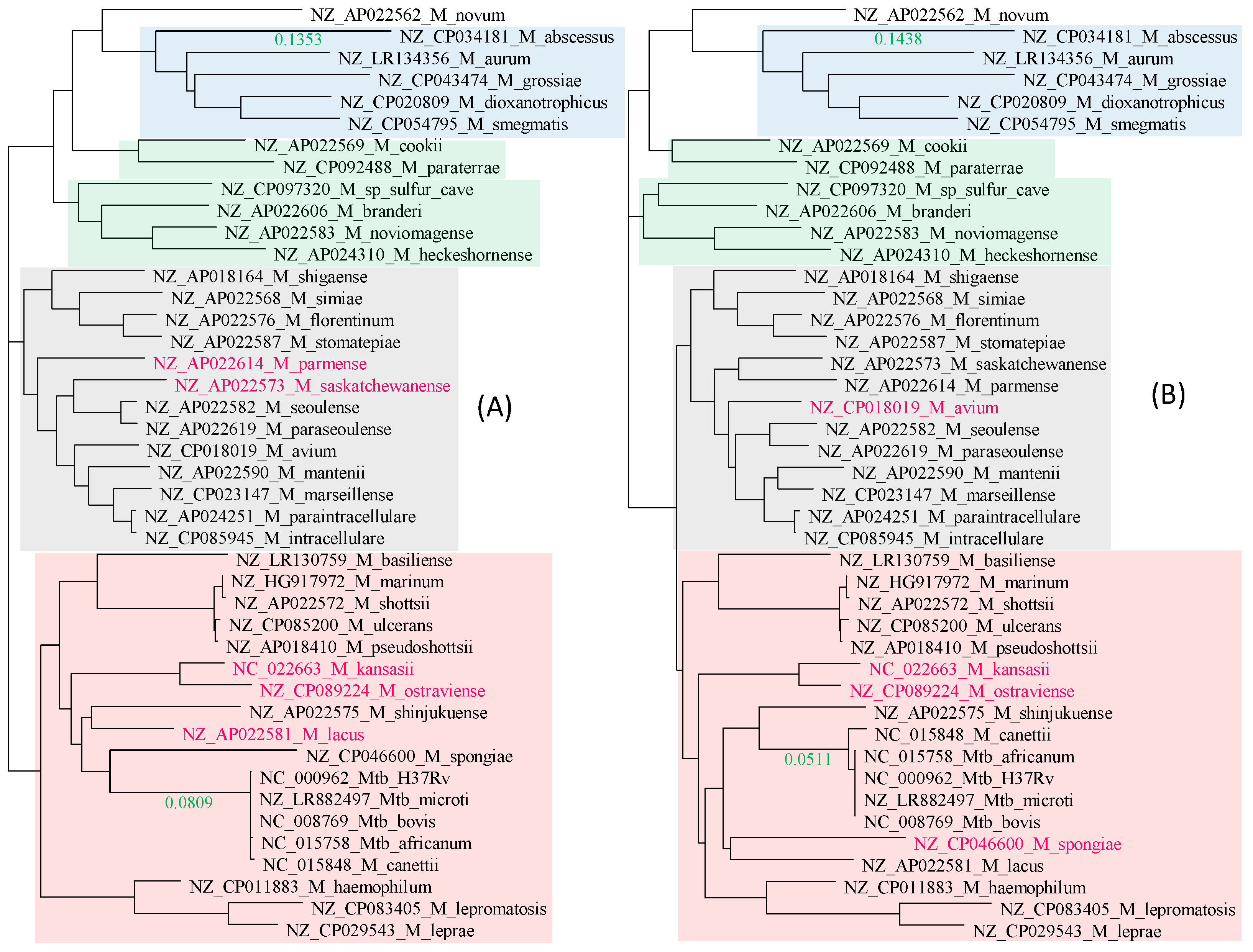
2.1. Species Tree: Approximation from Ribosomal Protein Genes
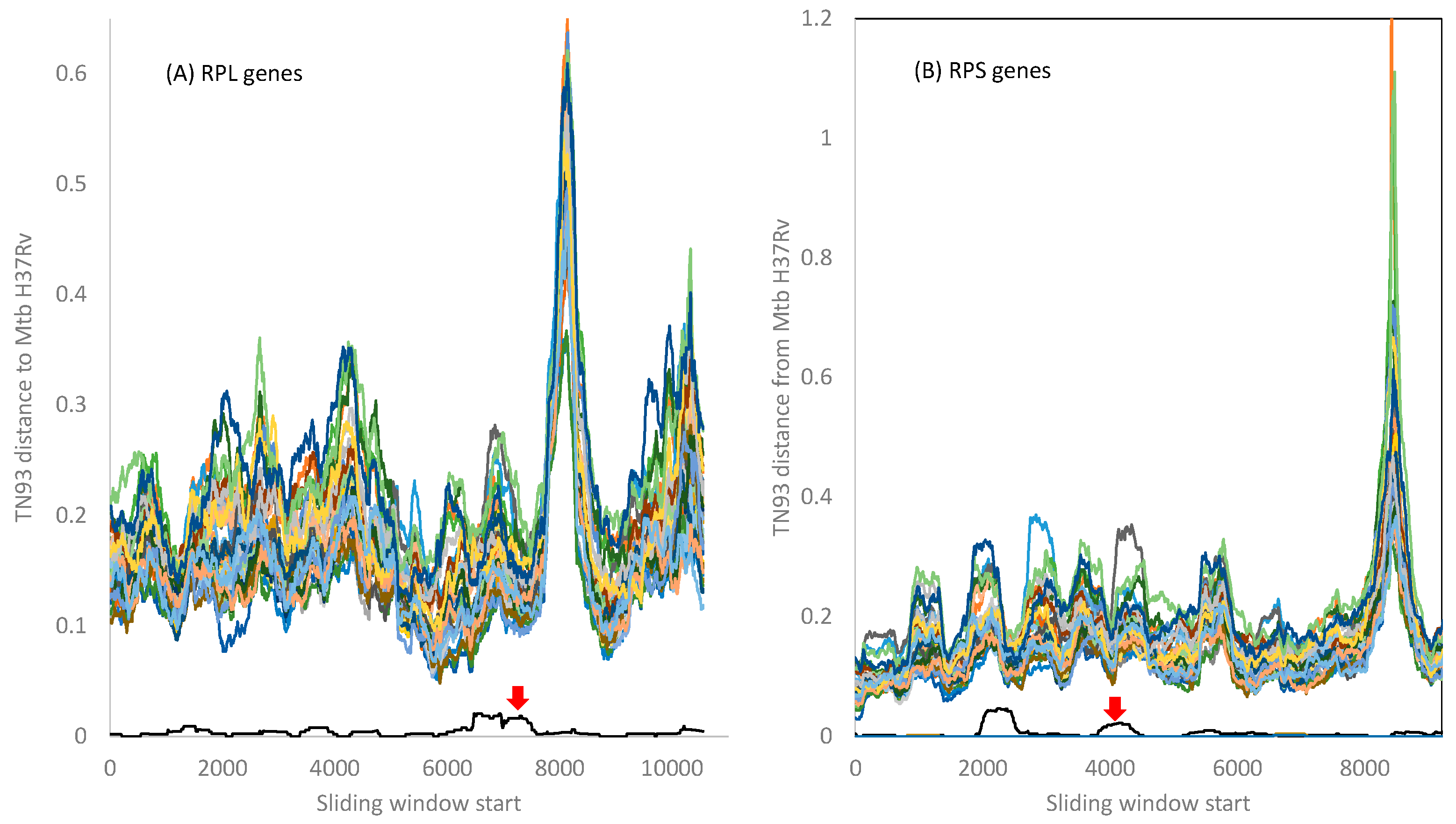
2.2. Genomic Integrity in the Mtb Complex as Revealed by Ribosomal Proteins
2.3. Genomic Integrity in the Mtb Complex as Revealed by Ribosomal RNA Genes
2.4. RNA Polymerase β and β′ Subunit (rpoB and rpoC)
2.5. Genomic Integrity in the Mtb Complex as Revealed by inhA and katG Genes
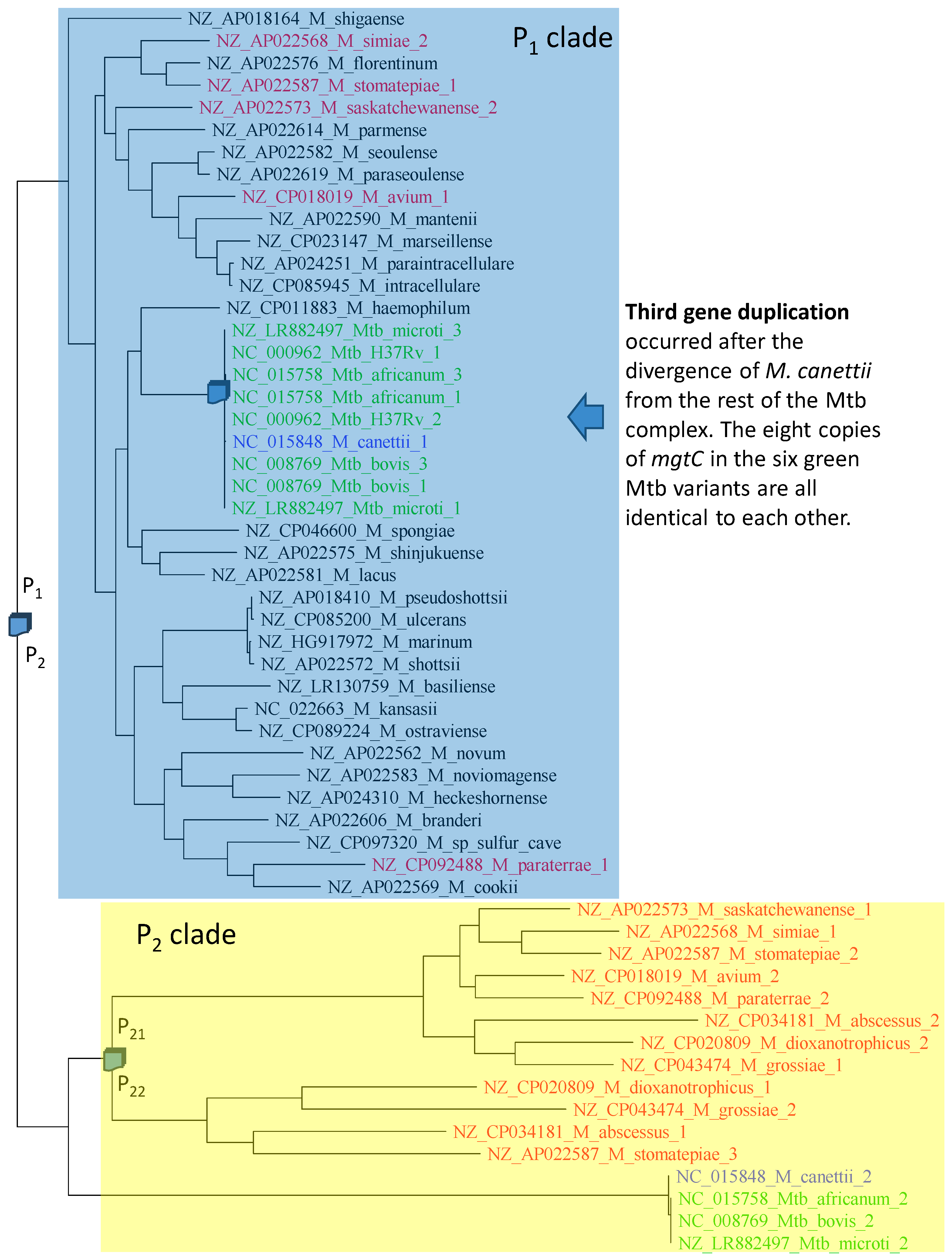
2.6. Is mgtC Involved in HGT?
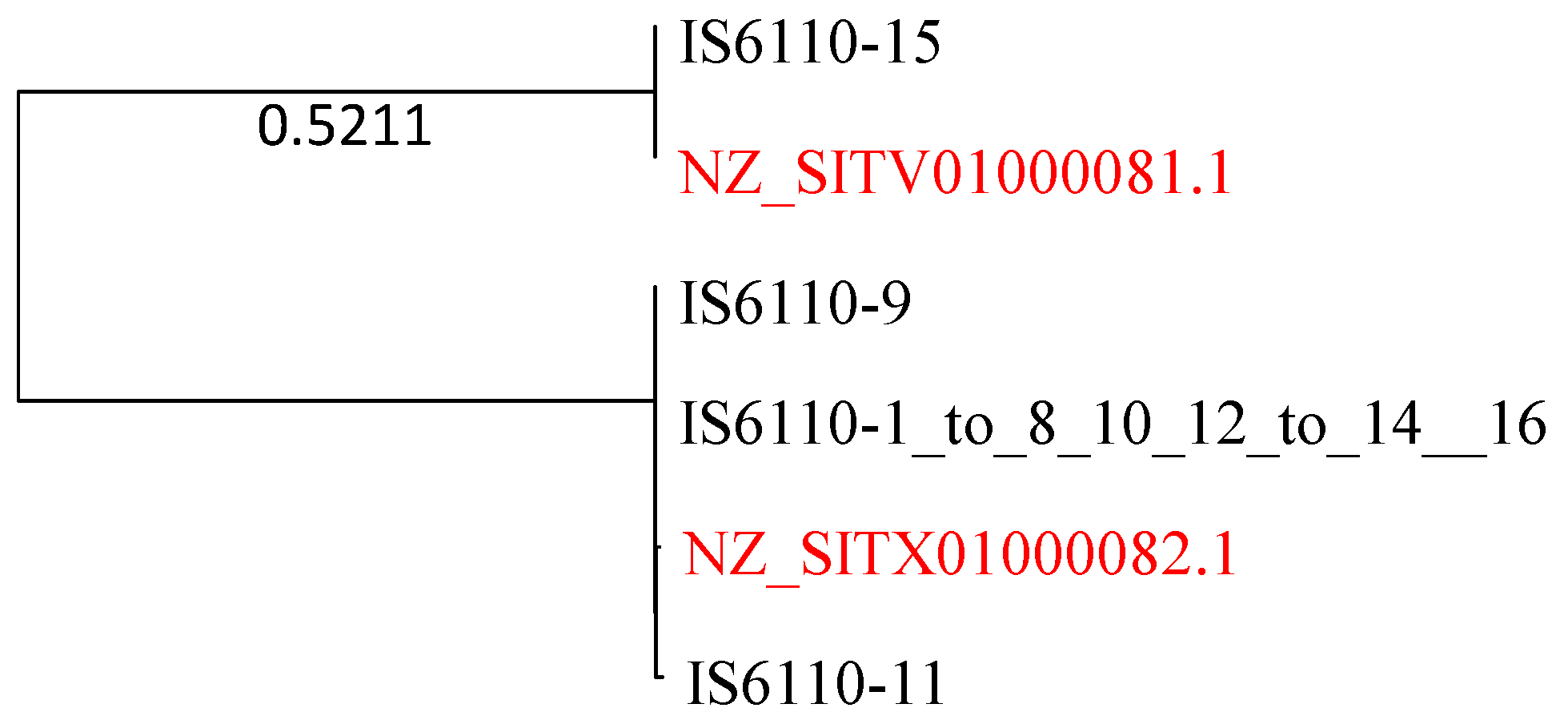
2.7. Strong Evidence of HGT Involving Insertion Sequence IS6110
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cole, S.T. Comparative and functional genomics of the Mycobacterium tuberculosis complex. Microbiology 2002, 148, 2919–2928. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Dai, X. On the intrinsic constraint of bacterial growth rate: M. tuberculosis’s view of the protein translation capacity. Crit. Rev. Microbiol. 2018, 44, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Gengenbacher, M.; Kaufmann, S.H. Mycobacterium tuberculosis: Success through dormancy. FEMS Microbiol. Rev. 2012, 36, 514–532. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.B.; Lin, P.L.; Chase, M.R.; Shah, R.R.; Iartchouk, O.; Galagan, J.; Mohaideen, N.; Ioerger, T.R.; Sacchettini, J.C.; Lipsitch, M.; et al. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat. Genet. 2011, 43, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Emergence of Mycobacterium tuberculosis with extensive resistance to second-line drugs–worldwide, 2000–2004. MMWR Morb. Mortal. Wkly. Rep. 2006, 55, 301–305. [Google Scholar]
- Gandhi, N.R.; Moll, A.; Sturm, A.W.; Pawinski, R.; Govender, T.; Lalloo, U.; Zeller, K.; Andrews, J.; Friedland, G. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet 2006, 368, 1575–1580. [Google Scholar] [CrossRef] [PubMed]
- Oppong, Y.E.A.; Phelan, J.; Perdigão, J.; Machado, D.; Miranda, A.; Portugal, I.; Viveiros, M.; Clark, T.G.; Hibberd, M.L. Genome-wide analysis of Mycobacterium tuberculosis polymorphisms reveals lineage-specific associations with drug resistance. BMC Genom. 2019, 20, 252. [Google Scholar] [CrossRef]
- Udwadia, Z.F.; Amale, R.A.; Ajbani, K.K.; Rodrigues, C. Totally drug-resistant tuberculosis in India. Clin. Infect. Dis. 2012, 54, 579–581. [Google Scholar] [CrossRef]
- Velayati, A.A.; Masjedi, M.R.; Farnia, P.; Tabarsi, P.; Ghanavi, J.; ZiaZarifi, A.H.; Hoffner, S.E. Emergence of new forms of totally drug-resistant tuberculosis bacilli: Super extensively drug-resistant tuberculosis or totally drug-resistant strains in iran. Chest 2009, 136, 420–425. [Google Scholar] [CrossRef]
- Kempker, R.R.; Kipiani, M.; Mirtskhulava, V.; Tukvadze, N.; Magee, M.J.; Blumberg, H.M. Acquired Drug Resistance in Mycobacterium tuberculosis and Poor Outcomes among Patients with Multidrug-Resistant Tuberculosis. Emerg. Infect. Dis. 2015, 21, 992–1001. [Google Scholar] [CrossRef]
- Cillóniz, C.; Garcia-Vidal, C.; Ceccato, A.; Torres, A. Antimicrobial Resistance among Streptococcus pneumoniae. In Antimicrobial Resistance in the 21st Century; Fong, I.W., Shlaes, D., Drlica, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 13–38. [Google Scholar]
- Ercoli, G.; Fernandes, V.E.; Chung, W.Y.; Wanford, J.J.; Thomson, S.; Bayliss, C.D.; Straatman, K.; Crocker, P.R.; Dennison, A.; Martinez-Pomares, L.; et al. Intracellular replication of Streptococcus pneumoniae inside splenic macrophages serves as a reservoir for septicaemia. Nat. Microbiol. 2018, 3, 600–610. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; Ling, C.; Chaguza, C.; Salter, S.J.; Hinfonthong, P.; Nikolaou, E.; Tate, N.; Pastusiak, A.; Turner, C.; Chewapreecha, C.; et al. Pneumococcal within-host diversity during colonization, transmission and treatment. Nat. Microbiol. 2022, 7, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.E.; Sebert, M.E. Frequent beneficial mutations during single-colony serial transfer of Streptococcus pneumoniae. PLoS Genet. 2011, 7, e1002232. [Google Scholar] [CrossRef] [PubMed]
- Eldholm, V.; Balloux, F. Antimicrobial Resistance in Mycobacterium tuberculosis: The Odd One Out. Trends Microbiol. 2016, 24, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.; Mouton, J.; Ummels, R.; Ten Hagen-Jongman, C.; Kriel, N.; Pain, A.; Warren, R.M.; Bitter, W.; Heunis, T.; Sampson, S.L. Identification of gene fusion events in Mycobacterium tuberculosis that encode chimeric proteins. NAR Genom. Bioinform. 2020, 2, lqaa033. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, A.E.; Tsolaki, A.G.; DeRiemer, K.; Feldman, M.W.; Small, P.M. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc. Natl. Acad. Sci. USA 2004, 101, 4871–4876. [Google Scholar] [CrossRef] [PubMed]
- Warner, D.F.; Rock, J.M.; Fortune, S.M.; Mizrahi, V. DNA Replication Fidelity in the Mycobacterium tuberculosis Complex. Adv. Exp. Med. Biol. 2017, 1019, 247–262. [Google Scholar] [PubMed]
- Coros, A.; DeConno, E.; Derbyshire, K.M. IS6110, a Mycobacterium tuberculosis complex-specific insertion sequence, is also present in the genome of Mycobacterium smegmatis, suggestive of lateral gene transfer among mycobacterial species. J. Bacteriol. 2008, 190, 3408–3410. [Google Scholar] [CrossRef]
- Mariani, F.; Piccolella, E.; Colizzi, V.; Rappuoli, R.; Gross, R. Characterization of an IS-like element from Mycobacterium tuberculosis. J. Gen. Microbiol. 1993, 139, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Gaur, M.; Sharma, V.; Khanna, P.; Bothra, A.; Bhaduri, A.; Mondal, A.K.; Dash, D.; Singh, Y.; Misra, R. Comparative Genomic Analysis of Mycobacteriaceae Reveals Horizontal Gene Transfer-Mediated Evolution of the CRISPR-Cas System in the Mycobacterium tuberculosis Complex. mSystems 2021, 6, e00934-20. [Google Scholar] [CrossRef]
- Razavi, M.; Kristiansson, E.; Flach, C.F.; Larsson, D.G.J. The Association between Insertion Sequences and Antibiotic Resistance Genes. mSphere 2020, 5, e00418-20. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. Comparative Genomics; Springer: Berlin/Heidelberg, Germany, 2013; p. VIII. 67p. [Google Scholar]
- Rivera, M.C.; Jain, R.; Moore, J.E.; Lake, J.A. Genomic evidence for two functionally distinct gene classes. Proc. Natl. Acad. Sci. USA 1998, 95, 6239–6244. [Google Scholar] [CrossRef]
- Sorek, R.; Zhu, Y.; Creevey, C.J.; Francino, M.P.; Bork, P.; Rubin, E.M. Genome-wide experimental determination of barriers to horizontal gene transfer. Science 2007, 318, 1449–1452. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.J.; Springman, R.; Molineux, I.J. Compensatory evolution in response to a novel RNA polymerase: Orthologous replacement of a central network gene. Mol. Biol. Evol. 2007, 24, 900–908. [Google Scholar] [CrossRef]
- Hudson, R.R. Gene trees, species trees and the segregation of ancestral alleles. Genetics 1992, 131, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Page, R.D.M. Introduction. In Tangled Trees: Phylogeny, Cospeciation and Coevolution; Page, R.D.M., Ed.; University of Chicago Press: Chicago, IL, USA, 2003; pp. 1–21. [Google Scholar]
- Xia, X. A Mathematical Primer of Molecular Phylogenetics; CRC Press: New York, NY, USA, 2020; p. 380. [Google Scholar]
- Kinsella, R.J.; Fitzpatrick, D.A.; Creevey, C.J.; McInerney, J.O. Fatty acid biosynthesis in Mycobacterium tuberculosis: Lateral gene transfer, adaptive evolution, and gene duplication. Proc. Natl. Acad. Sci. USA 2003, 100, 10320–10325. [Google Scholar] [CrossRef]
- Gupta, R.S.; Lo, B.; Son, J. Phylogenomics and Comparative Genomic Studies Robustly Support Division of the Genus Mycobacterium into an Emended Genus Mycobacterium and Four Novel Genera. Front. Microbiol. 2018, 9, 67. [Google Scholar] [CrossRef]
- Yamada, H.; Chikamatsu, K.; Aono, A.; Murata, K.; Miyazaki, N.; Kayama, Y.; Bhatt, A.; Fujiwara, N.; Maeda, S.; Mitarai, S. Fundamental Cell Morphologies Examined With Cryo-TEM of the Species in the Novel Five Genera Robustly Correlate with New Classification in Family Mycobacteriaceae. Front. Microbiol. 2020, 11, 562395. [Google Scholar] [CrossRef]
- Yamada, H.; Yamaguchi, M.; Igarashi, Y.; Chikamatsu, K.; Aono, A.; Murase, Y.; Morishige, Y.; Takaki, A.; Chibana, H.; Mitarai, S. Mycolicibacterium smegmatis, Basonym Mycobacterium smegmatis, Expresses Morphological Phenotypes Much more Similar to Escherichia coli than Mycobacterium tuberculosis in Quantitative Structome Analysis and CryoTEM Examination. Front. Microbiol. 2018, 9, 1992. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Kinjo, T.; Motooka, D.; Nabeya, D.; Jung, N.; Uechi, K.; Horii, T.; Iida, T.; Fujita, J.; Nakamura, S. Comprehensive subspecies identification of 175 nontuberculous mycobacteria species based on 7547 genomic profiles. Emerg. Microbes Infect. 2019, 8, 1043–1053. [Google Scholar] [CrossRef]
- Nouioui, I.; Carro, L.; García-López, M.; Meier-Kolthoff, J.P.; Woyke, T.; Kyrpides, N.C.; Pukall, R.; Klenk, H.-P.; Goodfellow, M.; Göker, M. Genome-Based Taxonomic Classification of the Phylum Actinobacteria. Front. Microbiol. 2018, 9, 2007. [Google Scholar] [CrossRef]
- He, Y.; Wei, K.; Si, K.; Mathieu, J.; Li, M.; Alvarez, P.J.J. Whole-Genome Sequence of the 1,4-Dioxane-Degrading Bacterium Mycobacterium dioxanotrophicus PH-06. Genome Announc. 2017, 5, e00625-17. [Google Scholar] [CrossRef] [PubMed]
- Cech, J.S.; Hartman, P.; Slosárek, M.; Chudoba, J. Isolation and identification of a morpholine-degrading bacterium. Appl. Environ. Microbiol. 1988, 54, 619–621. [Google Scholar] [CrossRef]
- Poupin, P.; Mazure, N.; Truffaut, N. Morpholine degradation by strain Mycobacterium aurum MOI: Improvement of cells growth and morpholine degradation rate by cells immobilization. In Progress in Biotechnology; Wijffels, R.H., Buitelaar, R.M., Bucke, C., Tramper, J., Eds.; Elsevier: Amsterdam, The Netherlands, 1996; Volume 11, pp. 770–776. [Google Scholar]
- Lelovic, N.; Mitachi, K.; Yang, J.; Lemieux, M.R.; Ji, Y.; Kurosu, M. Application of Mycobacterium smegmatis as a surrogate to evaluate drug leads against Mycobacterium tuberculosis. J. Antibiot. 2020, 73, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Reyrat, J.M.; Kahn, D. Mycobacterium smegmatis: An absurd model for tuberculosis? Trends Microbiol. 2001, 9, 472–474. [Google Scholar] [CrossRef]
- Paniz-Mondolfi, A.E.; Greninger, A.L.; Ladutko, L.; Brown-Elliott, B.A.; Vasireddy, R.; Jakubiec, W.; Vasireddy, S.; Wallace Jr, R.J.; Simmon, K.E.; Dunn, B.E.; et al. Mycobacterium grossiae sp. nov., a rapidly growing, scotochromogenic species isolated from human clinical respiratory and blood culture specimens. Int. J. Syst. Evol. Microbiol. 2017, 67, 4345–4351. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Xia, X. Optimizing Protein Production in Therapeutic Phages against a Bacterial Pathogen, Mycobacterium abscessus. Drugs Drug Candidates 2023, 2, 189–209. [Google Scholar] [CrossRef]
- Change, Y.T.; Andersen, R.N.; Vaituzis, Z. Growth of Mycobacterium lepraemurium in cultures of mouse peritoneal macrophages. J. Bacteriol. 1967, 93, 1119–1131. [Google Scholar] [CrossRef]
- van Spanning, R.J.M.; Guan, Q.; Melkonian, C.; Gallant, J.; Polerecky, L.; Flot, J.-F.; Brandt, B.W.; Braster, M.; Iturbe Espinoza, P.; Aerts, J.W.; et al. Methanotrophy by a Mycobacterium species that dominates a cave microbial ecosystem. Nat. Microbiol. 2022, 7, 2089–2100. [Google Scholar] [CrossRef]
- Kazda, J.; Stackebrandt, E.; Smida, J.; Minnikin, D.E.; Daffe, M.; Parlett, J.H.; Pitulle, C. Mycobacterium cookii sp. nov. Int. J. Syst. Bacteriol. 1990, 40, 217–223. [Google Scholar] [CrossRef]
- Lee, H.; Lee, S.A.; Lee, I.K.; Yu, H.K.; Park, Y.G.; Jeong, J.; Lee, S.H.; Kim, S.R.; Hyun, J.W.; Kim, K.; et al. Mycobacterium paraterrae sp. nov. recovered from a clinical specimen: Novel chromogenic slow growing mycobacteria related to Mycobacterium terrae complex. Microbiol. Immunol. 2010, 54, 46–53. [Google Scholar] [CrossRef] [PubMed]
- van Ingen, J.; Turenne, C.Y.; Tortoli, E.; Wallace, R.J., Jr.; Brown-Elliott, B.A. A definition of the Mycobacterium avium complex for taxonomical and clinical purposes, a review. Int. J. Syst. Evol. Microbiol. 2018, 68, 3666–3677. [Google Scholar] [CrossRef] [PubMed]
- Goring, S.M.; Wilson, J.B.; Risebrough, N.R.; Gallagher, J.; Carroll, S.; Heap, K.J.; Obradovic, M.; Loebinger, M.R.; Diel, R. The cost of Mycobacterium avium complex lung disease in Canada, France, Germany, and the United Kingdom: A nationally representative observational study. BMC Health Serv. Res. 2018, 18, 700. [Google Scholar] [CrossRef] [PubMed]
- Taoka, T.; Shinohara, T.; Hatakeyama, N.; Iwamura, S.; Murase, Y.; Mitarai, S.; Ogushi, F. Mycobacterium Shinjukuense Pulmonary Disease Progressed to Pleuritis after Iatrogenic Pneumothorax: A Case Report. J. Clin. Tuberc. Other Mycobact. Dis. 2020, 19, 100160. [Google Scholar] [CrossRef]
- Shinabarger, D. Mechanism of action of the oxazolidinone antibacterial agents. Expert Opin. Investig. Drugs 1999, 8, 1195–1202. [Google Scholar] [CrossRef]
- Foti, C.; Piperno, A.; Scala, A.; Giuffrè, O. Oxazolidinone Antibiotics: Chemical, Biological and Analytical Aspects. Molecules 2021, 26, 4280. [Google Scholar] [CrossRef]
- Ma, K.C.; Mortimer, T.D.; Duckett, M.A.; Hicks, A.L.; Wheeler, N.E.; Sánchez-Busó, L.; Grad, Y.H. Increased power from conditional bacterial genome-wide association identifies macrolide resistance mutations in Neisseria gonorrhoeae. Nat. Commun. 2020, 11, 5374. [Google Scholar] [CrossRef]
- Gregory, S.T.; Dahlberg, A.E. Erythromycin resistance mutations in ribosomal proteins L22 and L4 perturb the higher order structure of 23 S ribosomal RNA. J. Mol. Biol. 1999, 289, 827–834. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Shine, J.; Dalgarno, L. The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: Complementarity to nonsense triplets and ribosome binding sites. Proc. Natl. Acad. Sci. USA 1974, 71, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Shine, J.; Dalgarno, L. Determinant of cistron specificity in bacterial ribosomes. Nature 1975, 254, 34–38. [Google Scholar] [CrossRef]
- Hui, A.; de Boer, H.A. Specialized ribosome system: Preferential translation of a single mRNA species by a subpopulation of mutated ribosomes in Escherichia coli. Proc. Natl. Acad. Sci. USA 1987, 84, 4762–4766. [Google Scholar] [CrossRef] [PubMed]
- Steitz, J.A.; Jakes, K. How ribosomes select initiator regions in mRNA: Base pair formation between the 3′ terminus of 16S rRNA and the mRNA during initiation of protein synthesis in Escherichia coli. Proc. Natl. Acad. Sci. USA 1975, 72, 4734–4738. [Google Scholar] [CrossRef]
- Taniguchi, T.; Weissmann, C. Inhibition of Qbeta RNA 70S ribosome initiation complex formation by an oligonucleotide complementary to the 3′ terminal region of E. coli 16S ribosomal RNA. Nature 1978, 275, 770–772. [Google Scholar] [CrossRef] [PubMed]
- Ross, W.; Gosink, K.K.; Salomon, J.; Igarashi, K.; Zou, C.; Ishihama, A.; Severinov, K.; Gourse, R.L. A Third Recognition Element in Bacterial Promoters: DNA Binding by the α Subunit of RNA Polymerase. Science 1993, 262, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, S.; Scott, M.; Pedersen, S.; Hwa, T. Molecular crowding limits translation and cell growth. Proc. Natl. Acad. Sci. USA 2013, 110, 16754–16759. [Google Scholar] [CrossRef] [PubMed]
- Karpinets, T.V.; Greenwood, D.J.; Sams, C.E.; Ammons, J.T. RNA: Protein ratio of the unicellular organism as a characteristic of phosphorous and nitrogen stoichiometry and of the cellular requirement of ribosomes for protein synthesis. BMC Biol. 2006, 4, 30. [Google Scholar] [CrossRef]
- Chambers, H.F.; Sande, M.A. Antimicrobial agents—The aminoglycosides. In Goodman and Gilman’s the Pharmacological Basis of Therapeutics, 9th ed.; Goodmann, L.S., Limbird, L.E., Milinoff, P.B., Gilman, A.G., Hardmann, J.G., Eds.; McGraw-Hill: New York, NY, USA, 1996; pp. 1103–1121. [Google Scholar]
- Shcherbakov, D.; Akbergenov, R.; Matt, T.; Sander, P.; Andersson, D.I.; Böttger, E.C. Directed mutagenesis of Mycobacterium smegmatis 16S rRNA to reconstruct the in vivo evolution of aminoglycoside resistance in Mycobacterium tuberculosis. Mol. Microbiol. 2010, 77, 830–840. [Google Scholar] [CrossRef]
- Chisholm, S.A.; Dave, J.; Ison, C.A. High-level azithromycin resistance occurs in Neisseria gonorrhoeae as a result of a single point mutation in the 23S rRNA genes. Antimicrob. Agents Chemother. 2010, 54, 3812–3816. [Google Scholar] [CrossRef]
- Pham, C.D.; Nash, E.; Liu, H.; Schmerer, M.W.; Sharpe, S.; Woods, G.; Roland, B.; Schlanger, K.; St Cyr, S.B.; Carlson, J.; et al. Atypical Mutation in Neisseria gonorrhoeae 23S rRNA Associated with High-Level Azithromycin Resistance. Antimicrob. Agents Chemother. 2021, 65, e00885-20. [Google Scholar] [CrossRef] [PubMed]
- Ummels, R.; Abdallah, A.M.; Kuiper, V.; Aâjoud, A.; Sparrius, M.; Naeem, R.; Spaink, H.P.; Soolingen, D.V.; Pain, A.; Bitter, W. Identification of a Novel Conjugative Plasmid in Mycobacteria That Requires Both Type IV and Type VII Secretion. mBio 2014, 5, e01744-14. [Google Scholar] [CrossRef] [PubMed]
- Heil, A.; Zillig, W. Reconstitution of bacterial DNA-dependent RNA-polymerase from isolated subunits as a tool for the elucidation of the role of the subunits in transcription. FEBS Lett. 1970, 11, 165–168. [Google Scholar] [CrossRef]
- Rabussay, D.; Zillig, W. A rifampicin resistent rna-polymerase from E. coli altered in the β-subunit. FEBS Lett. 1969, 5, 104–106. [Google Scholar] [CrossRef]
- Wehrli, W.; Knüsel, F.; Schmid, K.; Staehelin, M. Interaction of rifamycin with bacterial RNA polymerase. Proc. Natl. Acad. Sci. USA 1968, 61, 667–673. [Google Scholar] [CrossRef]
- Boyd, D.H.; Zillig, W.; Scaife, F.J.G. Reference mutations for the β subunit of RNA polymerase. Mol. Gen. Genet. MGG 1974, 130, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.P.; Crawford, J.T.; Shinnick, T.M. The rpoB gene of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1994, 38, 805–811. [Google Scholar] [CrossRef]
- Madacki, J.; Orgeur, M.; Mas Fiol, G.; Frigui, W.; Ma, L.; Brosch, R. ESX-1-Independent Horizontal Gene Transfer by Mycobacterium tuberculosis Complex Strains. mBio 2021, 12, e00965-21. [Google Scholar] [CrossRef]
- Nataraj, V.; Varela, C.; Javid, A.; Singh, A.; Besra, G.S.; Bhatt, A. Mycolic acids: Deciphering and targeting the Achilles’ heel of the tubercle bacillus. Mol. Microbiol. 2015, 98, 7–16. [Google Scholar] [CrossRef]
- Lee, W.; Engels, B. The protonation state of catalytic residues in the resting state of KasA revisited: Detailed mechanism for the activation of KasA by its own substrate. Biochemistry 2014, 53, 919–931. [Google Scholar] [CrossRef]
- Zhao, J.; Siddiqui, S.; Shang, S.; Bian, Y.; Bagchi, S.; He, Y.; Wang, C.R. Mycolic acid-specific T cells protect against Mycobacterium tuberculosis infection in a humanized transgenic mouse model. eLife 2015, 4, e08525. [Google Scholar] [CrossRef]
- Khan, S.R.; Manialawy, Y.; Siraki, A.G. Isoniazid and host immune system interactions: A proposal for a novel comprehensive mode of action. Br. J. Pharmacol. 2019, 176, 4599–4608. [Google Scholar] [CrossRef]
- Asselineau, J.; Lederer, E. Structure of the mycolic acids of Mycobacteria. Nature 1950, 166, 782–783. [Google Scholar] [CrossRef]
- Quémard, A.; Sacchettini, J.C.; Dessen, A.; Vilcheze, C.; Bittman, R.; Jacobs, W.R., Jr.; Blanchard, J.S. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry 1995, 34, 8235–8241. [Google Scholar] [CrossRef]
- Iwao, Y.; Nakata, N. Roles of the three Mycobacterium smegmatis katG genes for peroxide detoxification and isoniazid susceptibility. Microbiol. Immunol. 2018, 62, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Timmins, G.S.; Deretic, V. Mechanisms of action of isoniazid. Mol. Microbiol. 2006, 62, 1220–1227. [Google Scholar] [CrossRef]
- Parikh, S.L.; Xiao, G.; Tonge, P.J. Inhibition of InhA, the Enoyl Reductase from Mycobacterium tuberculosis, by Triclosan and Isoniazid. Biochemistry 2000, 39, 7645–7650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Garbe, T.; Young, D. Transformation with katG restores isoniazid-sensitivity in Mycobacterium tuberculosis isolates resistant to a range of drug concentrations. Mol. Microbiol. 1993, 8, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, M.; Marostenmaki, J.; Wong, A.; FitzGerald, M.; Black, W.A.; Smith, J.A. Mutations in the catalase-peroxidase gene from isoniazid-resistant Mycobacterium tuberculosis isolates. J. Infect. Dis. 1994, 169, 1162–1165. [Google Scholar] [CrossRef]
- Heym, B.; Alzari, P.M.; Honoré, N.; Cole, S.T. Missense mutations in the catalase-peroxidase gene, katG, are associated with isoniazid resistance in Mycobacterium tuberculosis. Mol. Microbiol. 1995, 15, 235–245. [Google Scholar] [CrossRef]
- Alix, E.; Blanc-Potard, A.B. MgtC: A key player in intramacrophage survival. Trends Microbiol. 2007, 15, 252–256. [Google Scholar] [CrossRef]
- Choi, S.; Choi, E.; Cho, Y.J.; Nam, D.; Lee, J.; Lee, E.J. The Salmonella virulence protein MgtC promotes phosphate uptake inside macrophages. Nat. Commun. 2019, 10, 3326. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Lee, E.J. Regulation and function of the Salmonella MgtC virulence protein. J. Microbiol. 2015, 53, 667–672. [Google Scholar] [CrossRef]
- Alix, E.; Godreuil, S.; Blanc-Potard, A.B. Identification of a Haarlem genotype-specific single nucleotide polymorphism in the mgtC virulence gene of Mycobacterium tuberculosis. J. Clin. Microbiol. 2006, 44, 2093–2098. [Google Scholar] [CrossRef]
- Guilhot, C.; Gicquel, B.; Davies, J.; Martín, C. Isolation and analysis of IS6120, a new insertion sequence from Mycobacterium smegmatis. Mol. Microbiol. 1992, 6, 107–113. [Google Scholar] [CrossRef]
- Ichikawa, K.; Yagi, T.; Moriyama, M.; Inagaki, T.; Nakagawa, T.; Uchiya, K.I.; Nikai, T.; Ogawa, K. Characterization of Mycobacterium avium clinical isolates in Japan using subspecies-specific insertion sequences, and identification of a new insertion sequence, ISMav6. J. Med. Microbiol 2009, 58, 945–950. [Google Scholar] [CrossRef]
- Park, H.T.; Park, H.E.; Jung, Y.H.; Yoo, H.S. An ISMap02-like insertion sequence in Mycobacterium spp. interferes with specific detection of Mycobacterium avium subsp. paratuberculosis. Vet. Microbiol. 2018, 216, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Thierry, D.; Brisson-Noël, A.; Vincent-Lévy-Frébault, V.; Nguyen, S.; Guesdon, J.L.; Gicquel, B. Characterization of a Mycobacterium tuberculosis insertion sequence, IS6110, and its application in diagnosis. J. Clin. Microbiol. 1990, 28, 2668–2673. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, C.R.; Falmer, A.A.; Gey van Pittius, N.C.; Victor, T.C.; van Helden, P.D.; Warren, R.M. The role of IS6110 in the evolution of Mycobacterium tuberculosis. Tuberculosis 2007, 87, 393–404. [Google Scholar] [CrossRef]
- Thierry, D.; Cave, M.D.; Eisenach, K.D.; Crawford, J.T.; Bates, J.H.; Gicquel, B.; Guesdon, J.L. IS6110, an IS-like element of Mycobacterium tuberculosis complex. Nucleic Acids Res. 1990, 18, 188. [Google Scholar] [CrossRef]
- van Embden, J.D.; Cave, M.D.; Crawford, J.T.; Dale, J.W.; Eisenach, K.D.; Gicquel, B.; Hermans, P.; Martin, C.; McAdam, R.; Shinnick, T.M.; et al. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: Recommendations for a standardized methodology. J. Clin. Microbiol. 1993, 31, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Itoh, T.; Matsuda, H.; Gojobori, T. Biased biological functions of horizontally transferred genes in prokaryotic genomes. Nat. Genet. 2004, 36, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Creevey, C.J.; Doerks, T.; Fitzpatrick, D.A.; Raes, J.; Bork, P. Universally distributed single-copy genes indicate a constant rate of horizontal transfer. PLoS ONE 2011, 6, e22099. [Google Scholar] [CrossRef]
- Smith, H.O.; Tomb, J.F.; Dougherty, B.A.; Fleischmann, R.D.; Venter, J.C. Frequency and distribution of DNA uptake signal sequences in the Haemophilus influenzae Rd genome. Science 1995, 269, 538–540. [Google Scholar] [CrossRef]
- Maughan, H.; Wilson, L.A.; Redfield, R.J. Bacterial DNA uptake sequences can accumulate by molecular drive alone. Genetics 2010, 186, 613–627. [Google Scholar] [CrossRef]
- Frye, S.A.; Nilsen, M.; Tønjum, T.; Ambur, O.H. Dialects of the DNA uptake sequence in Neisseriaceae. PLoS Genet. 2013, 9, e1003458. [Google Scholar] [CrossRef] [PubMed]
- Bishai, W. The Mycobacterium tuberculosis genomic sequence: Anatomy of a master adaptor. Trends Microbiol. 1998, 6, 464–465. [Google Scholar] [CrossRef]
- Mukherjee, T.; Goswami, A.; Chakraborty, U.; Majumdar, M.; Sinha, S.; Pal, N.K. A Study on Generation Time of Sensitive and Resistant Mycobacterium tuberculosis Isolates. J. Evol. Med. Dent. Sci. 2019, 8, 2489–2494. [Google Scholar] [CrossRef]
- Wang, J.; Behr, M.A. Building a better bacillus: The emergence of Mycobacterium tuberculosis. Front. Microbiol. 2014, 5, 139. [Google Scholar] [CrossRef]
- Mi, Y.; Bao, L.; Gu, D.; Luo, T.; Sun, C.; Yang, G. Mycobacterium tuberculosis PPE25 and PPE26 proteins expressed in Mycobacterium smegmatis modulate cytokine secretion in mouse macrophages and enhance mycobacterial survival. Res. Microbiol. 2017, 168, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Batoni, G.; Bottai, D.; Maisetta, G.; Pardini, M.; Boschi, A.; Florio, W.; Esin, S.; Campa, M. Involvement of the Mycobacterium tuberculosis secreted antigen SA-5K in intracellular survival of recombinant Mycobacterium smegmatis. FEMS Microbiol. Lett. 2001, 205, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, H.; Li, H.; Dang, G.; Cui, Z.; Song, N.; Wang, Q.; Liu, S.; Chen, L. PE17 protein from Mycobacterium tuberculosis enhances Mycobacterium smegmatis survival in macrophages and pathogenicity in mice. Microb. Pathog. 2019, 126, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Ganaie, A.A.; Trivedi, G.; Kaur, A.; Jha, S.S.; Anand, S.; Rana, V.; Singh, A.; Kumar, S.; Sharma, C. Interaction of Erp Protein of Mycobacterium tuberculosis with Rv2212 Enhances Intracellular Survival of Mycobacterium smegmatis. J. Bacteriol. 2016, 198, 2841–2852. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Zhang, F.; Yang, S.; Kang, J.; Sha, S.; Ma, Y. Mycobacterium tuberculosis Rv0431 expressed in Mycobacterium smegmatis, a potentially mannosylated protein, mediated the immune evasion of RAW 264.7 macrophages. Microb. Pathog. 2016, 100, 285–292. [Google Scholar] [CrossRef]
- Li, J.M.; Li, N.; Zhu, D.Y.; Wan, L.G.; He, Y.L.; Yang, C. Isocitrate lyase from Mycobacterium tuberculosis promotes survival of Mycobacterium smegmatis within macrophage by suppressing cell apoptosis. Chin. Med. J. 2008, 121, 1114–1119. [Google Scholar] [CrossRef]
- Xia, X. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Xia, X. Nucleotide Substitution Models and Evolutionary Distances. In Bioinformatics and the Cell: Modern Computational Approaches in Genomics, Proteomics and Transcriptomics; Springer: Cham, Switzerland, 2018; pp. 269–314. [Google Scholar]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, X. Horizontal Gene Transfer and Drug Resistance Involving Mycobacterium tuberculosis. Antibiotics 2023, 12, 1367. https://doi.org/10.3390/antibiotics12091367
Xia X. Horizontal Gene Transfer and Drug Resistance Involving Mycobacterium tuberculosis. Antibiotics. 2023; 12(9):1367. https://doi.org/10.3390/antibiotics12091367
Chicago/Turabian StyleXia, Xuhua. 2023. "Horizontal Gene Transfer and Drug Resistance Involving Mycobacterium tuberculosis" Antibiotics 12, no. 9: 1367. https://doi.org/10.3390/antibiotics12091367
APA StyleXia, X. (2023). Horizontal Gene Transfer and Drug Resistance Involving Mycobacterium tuberculosis. Antibiotics, 12(9), 1367. https://doi.org/10.3390/antibiotics12091367