Complement Evasion in Borrelia spirochetes: Mechanisms and Opportunities for Intervention

Abstract
:1. Introduction
1.1. Borrelia and Lyme Disease
1.1.1. Pathogenesis
1.1.2. Epidemiology
1.1.3. Clinical Presentation, Diagnosis and Treatment
1.1.4. Impact
1.2. Immune System and Treatment Interactions
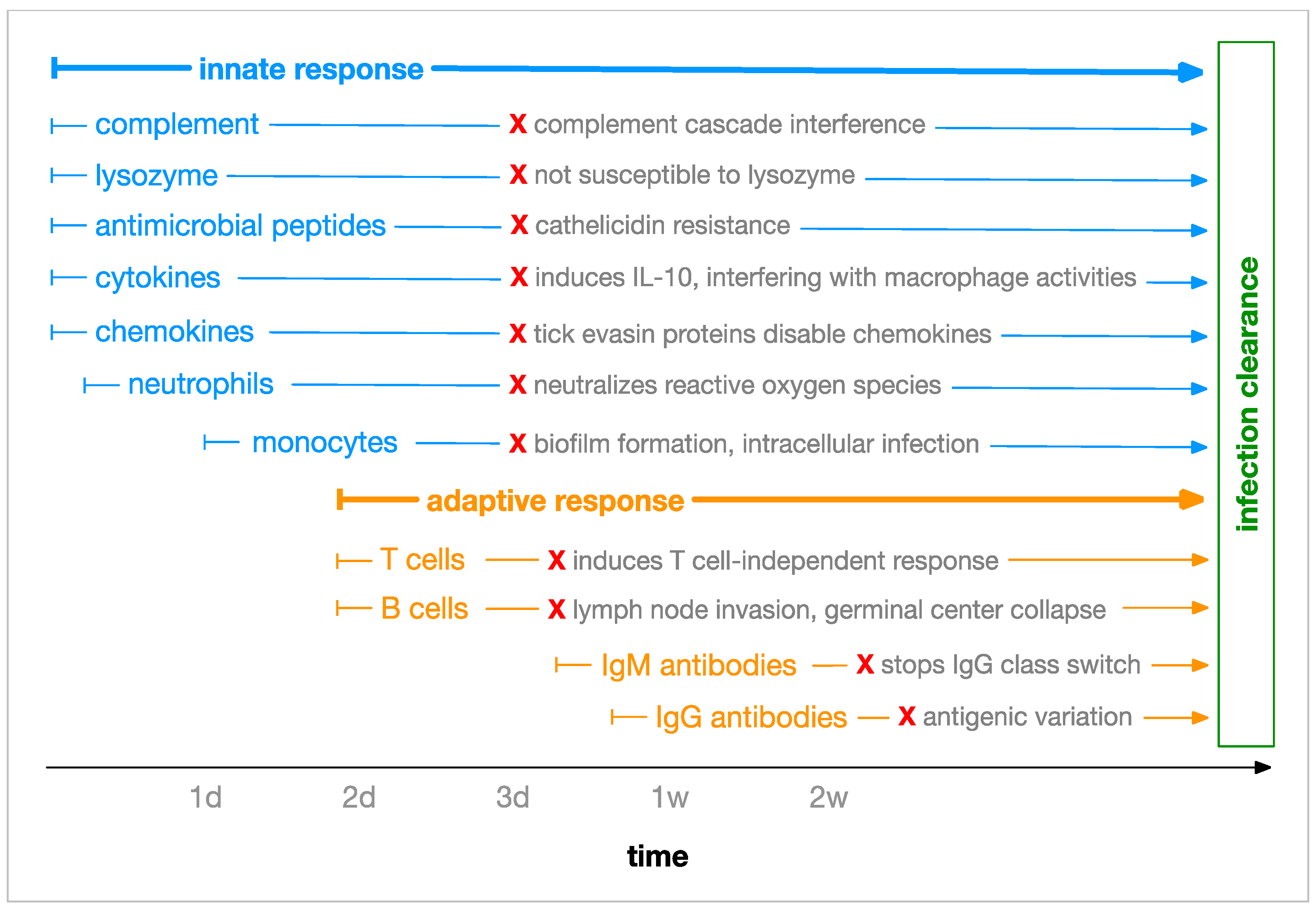
1.2.1. Immune Response
1.2.2. Borrelia Antibiotic Evasion and Tolerance Mechanisms
1.2.3. Borrelia Immune Evasion Mechanisms
2. Results
2.1. Research Overview
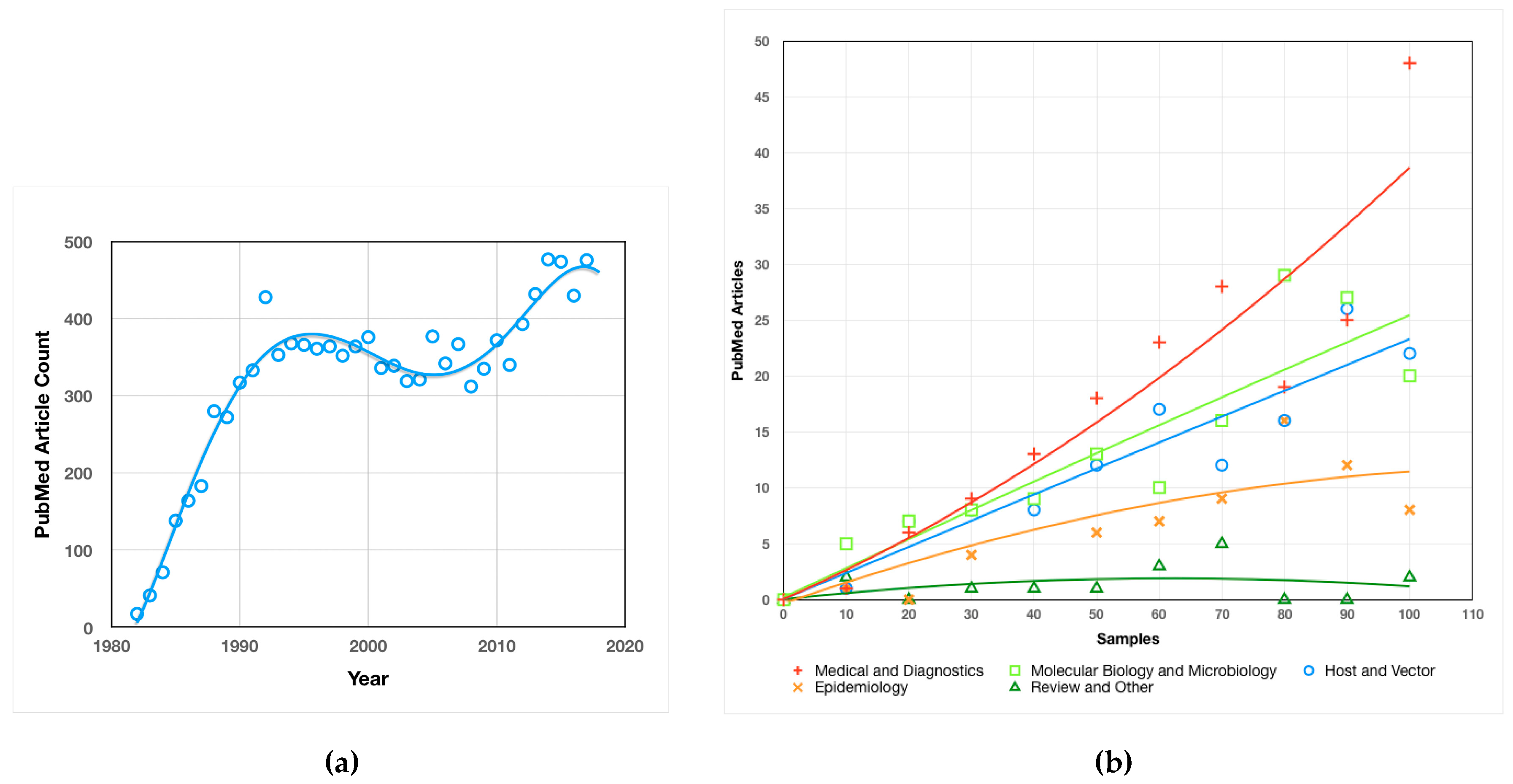
2.1.1. Quantitative Analysis of Borrelia Research
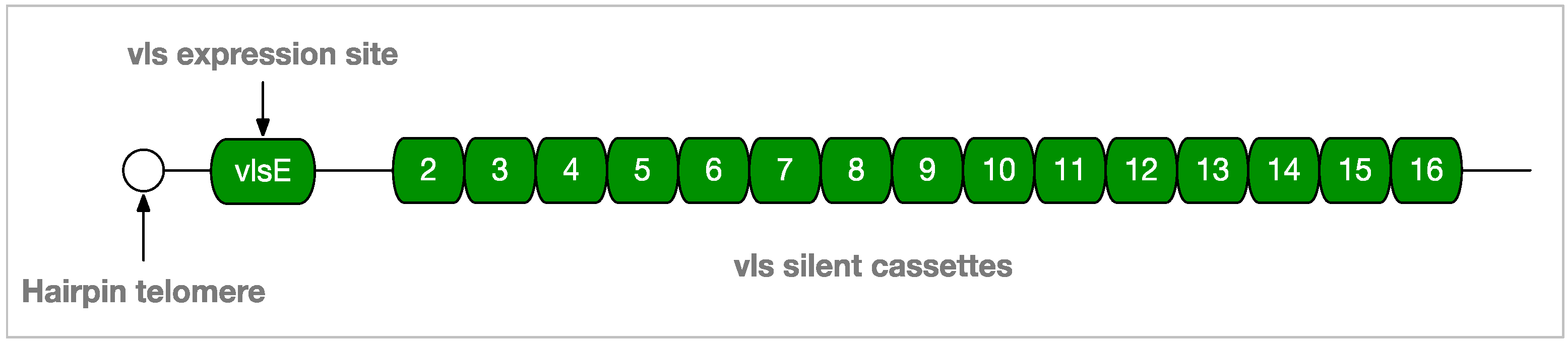
2.1.2. Borrelia Genomics
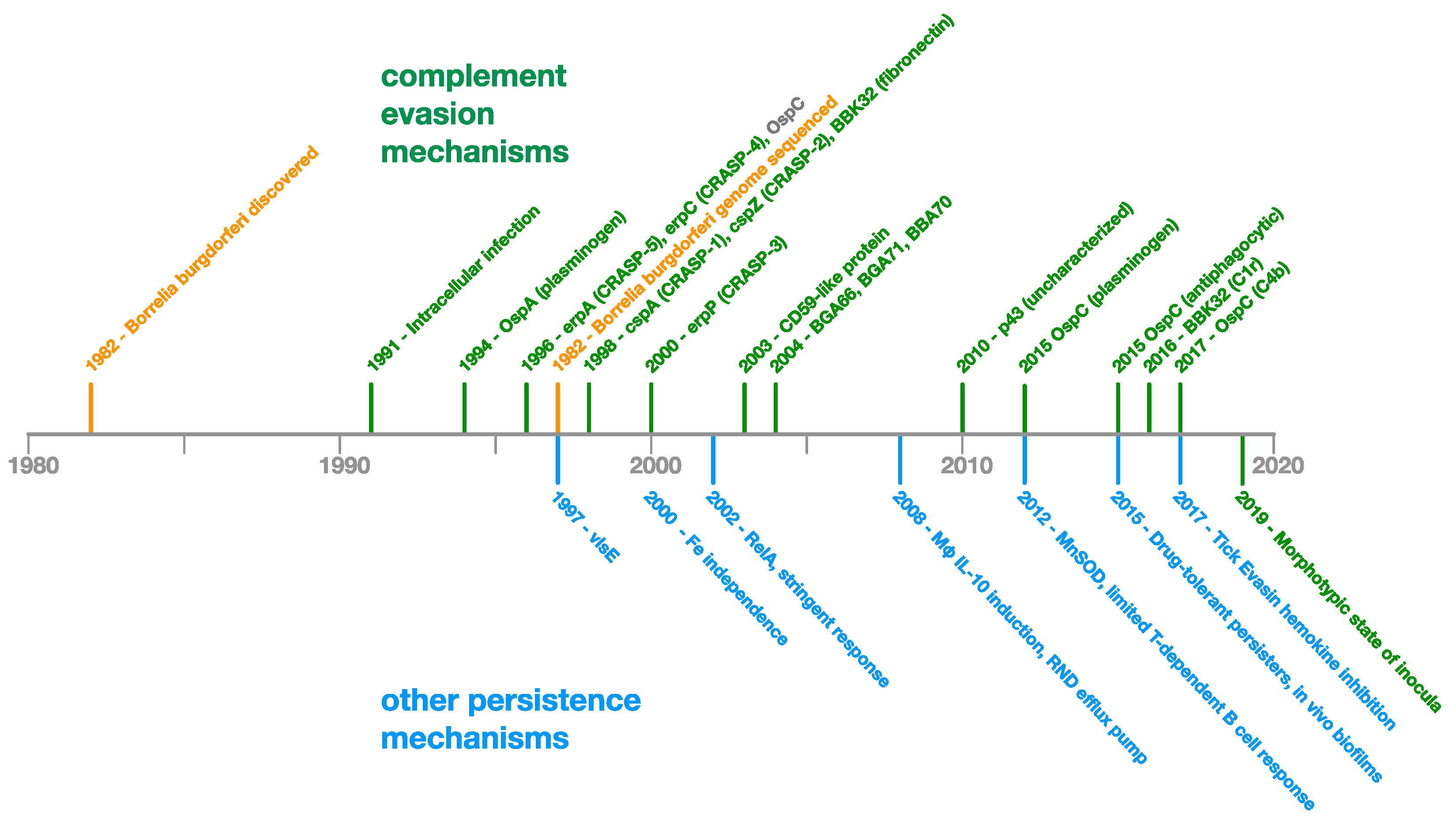
2.1.3. Analysis of Borrelia Immune Evasion and Persistence Research
2.1.4. Relative Importance of Evasion and Persistence Mechanisms
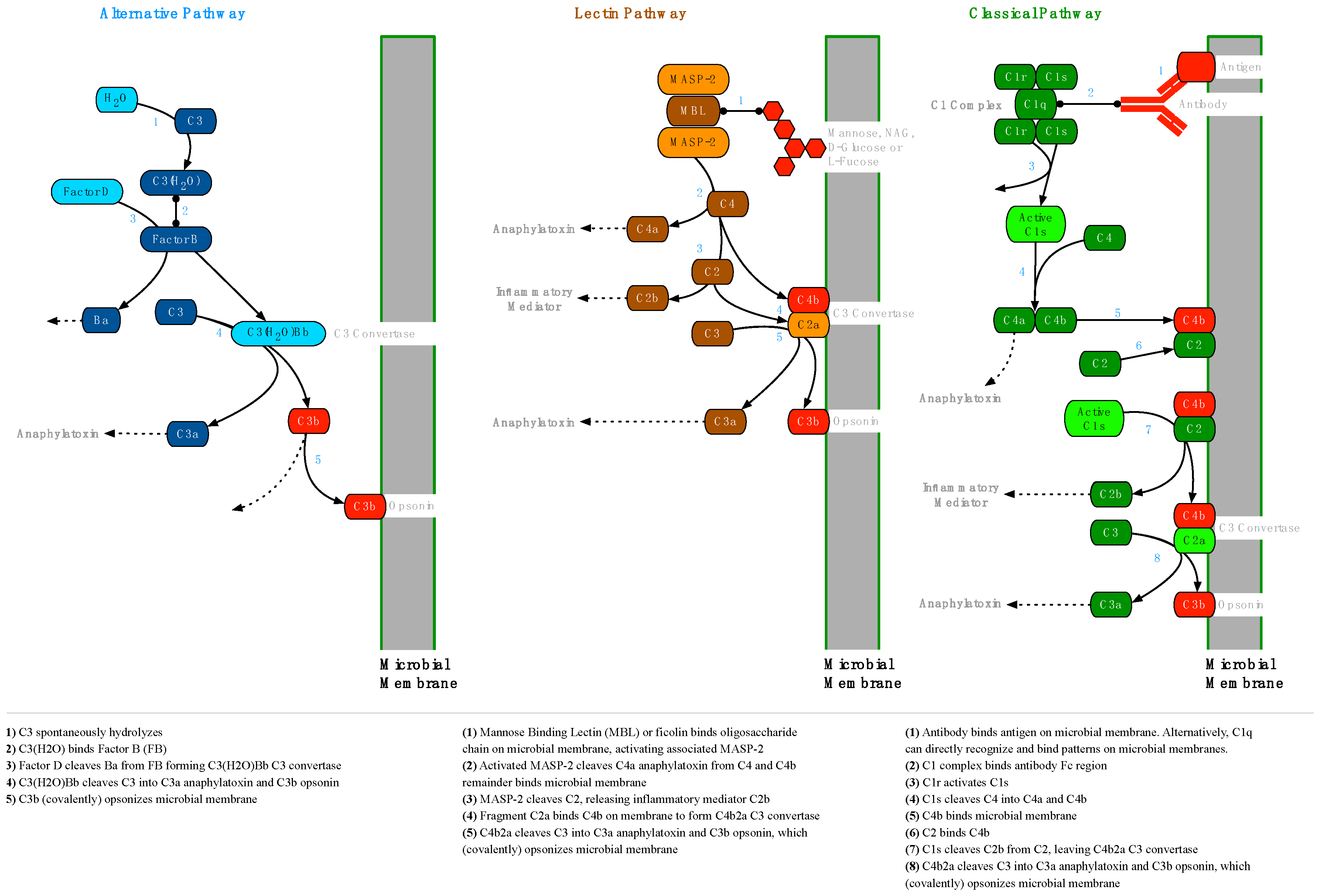
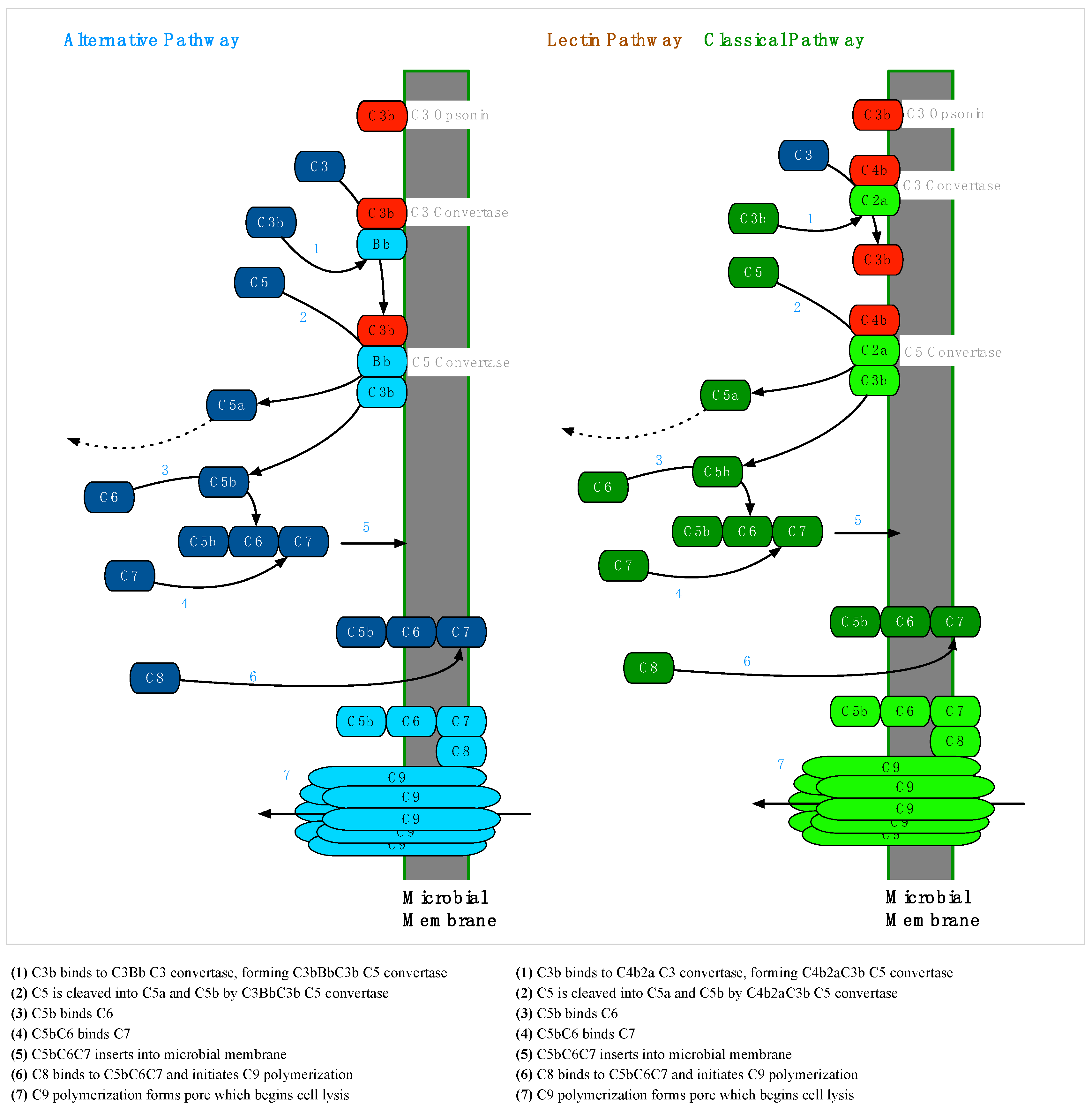
2.2. The Complement System
2.2.1. Opsonization
2.2.2. Membrane Attack Complex Formation
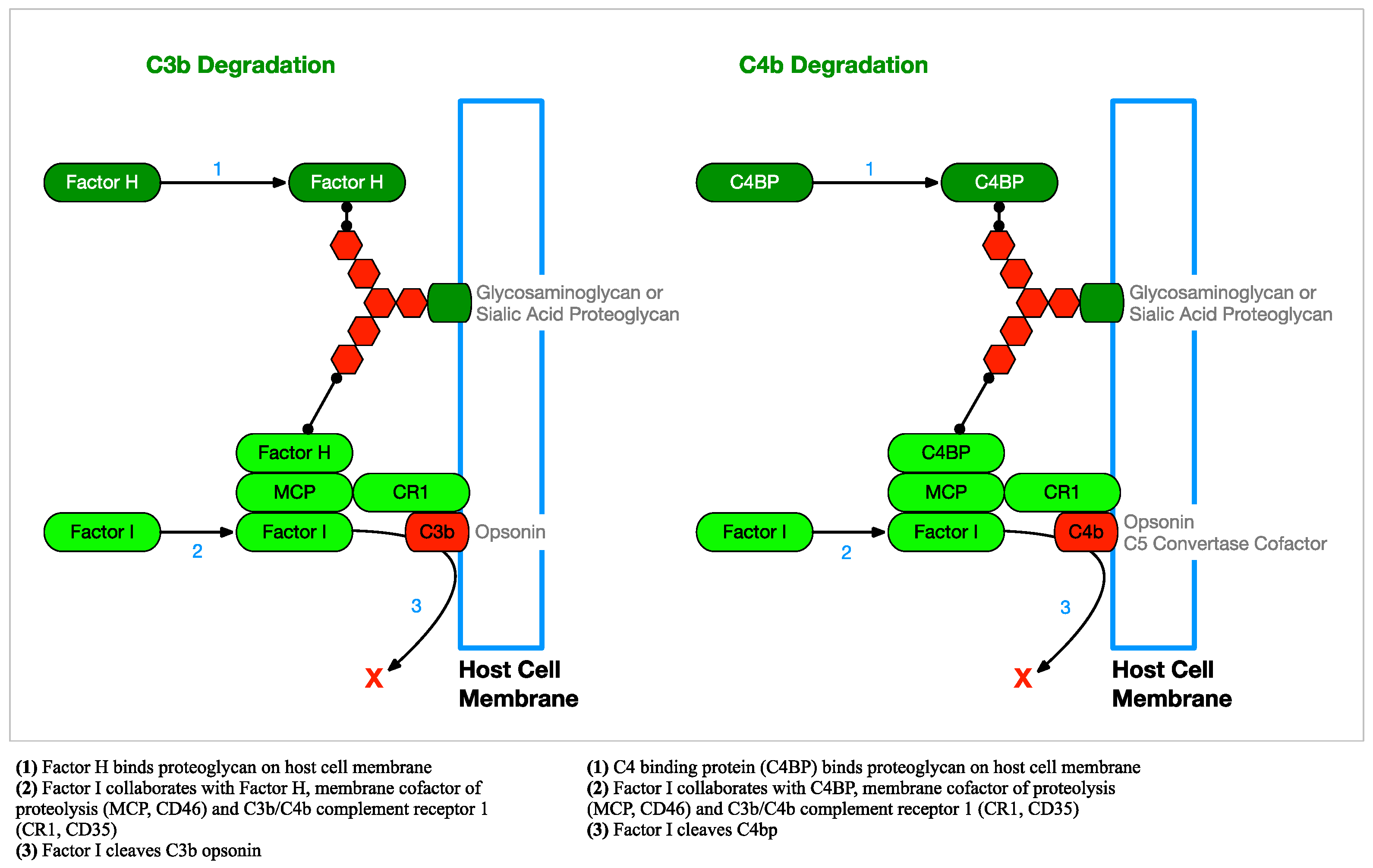
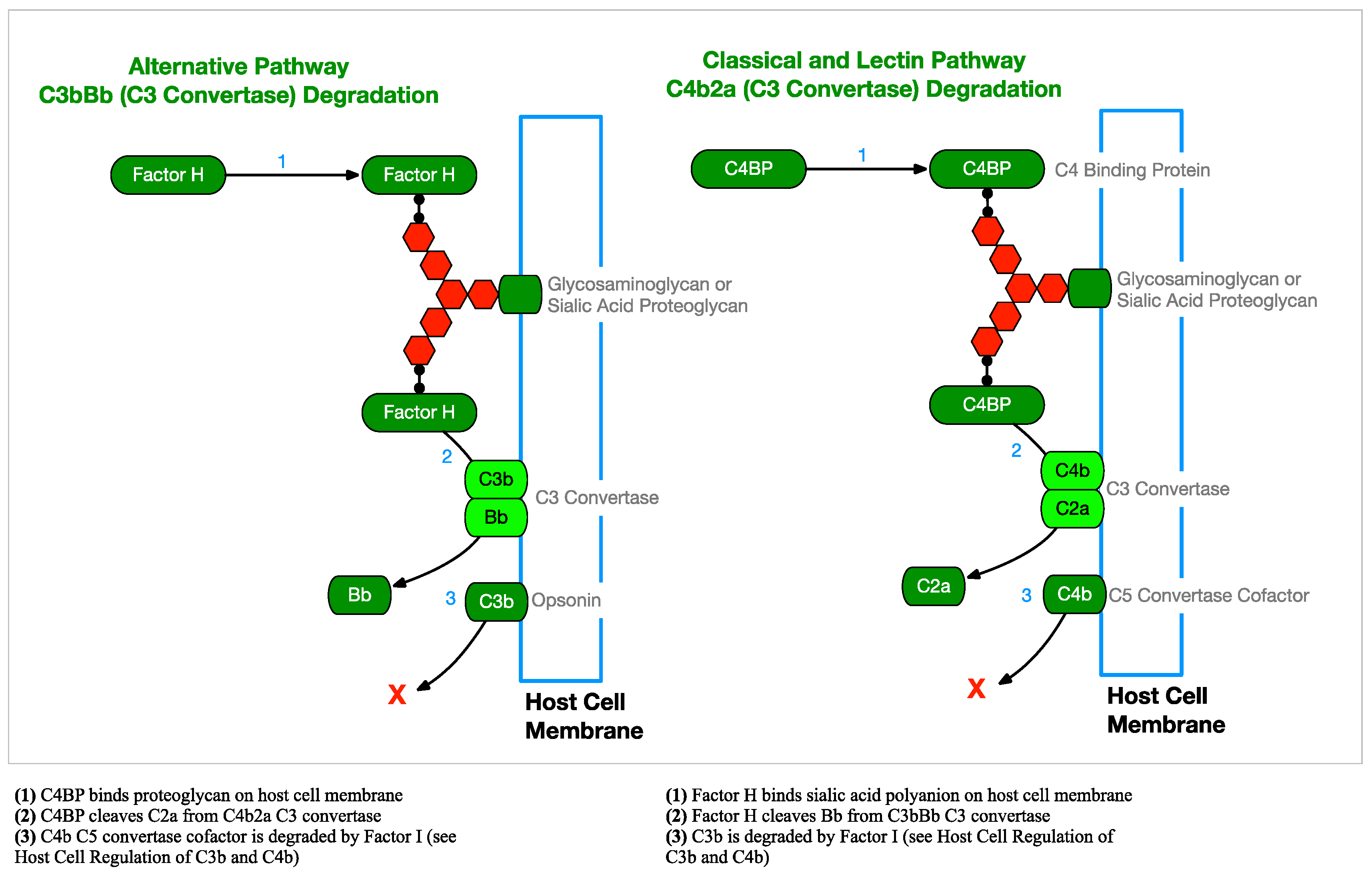
2.2.3. Complement Regulation
2.3. Complement Evasion by Borrelia
2.4. Category I: Direct Complement Interference
2.4.1. Classical Pathway Inhibition by C1r Binding: BBK32
2.4.2. C3b Degradation by Binding Plasminogen
2.4.3. MAC Interference
2.5. Category II: Complement Regulation Interference
2.5.1. Factor H and FHL-1 Binding
2.5.2. C4b Inactivation by Binding C4BP
3. Discussion
3.1. Lyme Disease Impact and Medical Response
3.2. PTLDS or PLD?
3.3. Opportunities for Intervention
3.3.1. Immune System Intervention
3.3.2. Desirable Criteria for Complement Evasion Intervention
- The mechanism must be constitutively expressed, or up-regulated in the vertebrate host at or soon after initial infection;
- The mechanism must be required for infectivity and long-term maintenance of infection;
- The mechanism should be required by all infective morphological forms of Borrelia, including antibiotic-tolerant persister cells and biofilm aggregates;
- The structure of the molecules involved in the mechanism must be known, including ligand binding sites;
- The structure of the molecules involved should be unique so that small molecule inhibition is likely to avoid off-target effects that would impact microbiota or the patient.
3.3.3. BBK32
3.3.4. Erp CRASPs
3.3.5. OmpA-Like OM Proteins
3.3.6. Other Targets
4. Materials and Methods
5. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Brisson, D.; Drecktrah, D.; Eggers, C.H.; Samuels, D.S. Genetics of Borrelia burgdorferi. Annu. Rev. Genet. 2012, 46, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Tilly, K.; Rosa, P.A.; Stewart, P.E. Biology of infection with Borrelia burgdorferi. Infect. Disease Clin. N. Am. 2008, 22, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Steere, A.C.; Coburn, J.; Glickstein, L. The emergence of Lyme disease. J. Clin. Investig. 2004, 113, 1093–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraiczy, P. Hide and Seek: How Lyme Disease Spirochetes Overcome Complement Attack. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Caimano, M.J.; Drecktrah, D.; Kung, F.; Samuels, D.S. Interaction of the Lyme disease spirochete with its tick vector. Cell Microbiol. 2016, 18, 919–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, C.M.; Chao, L.L.; Yu, C.P. Chemotactic migration of the Lyme disease spirochete (Borrelia burgdorferi) to salivary gland extracts of vector ticks. Am. J. Trop. Med. Hyg. 2002, 66, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Francischetti, I.M.; Sa-Nunes, A.; Mans, B.J.; Santos, I.M.; Ribeiro, J.M. The role of saliva in tick feeding. Front. Biosci. Landmark Ed. 2009, 14, 2051–2088. [Google Scholar] [CrossRef] [PubMed]
- Ojaimi, C.; Brooks, C.; Casjens, S.; Rosa, P.; Elias, A.; Barbour, A.; Jasinskas, A.; Benach, J.; Katona, L.; Radolf, J.; et al. Profiling of temperature-induced changes in Borrelia burgdorferi gene expression by using whole genome arrays. Infect. Immun. 2003, 71, 1689–1705. [Google Scholar] [CrossRef] [PubMed]
- Critical Needs and Gaps in Understanding Prevention, Amelioration, and Resolution of Lyme and Other Tick-Borne Diseases: The Short-Term and Long-Term Outcomes: Workshop Report; National Academies Press: Washington, DC, USA, 2011. [CrossRef]
- Sapi, E.K.G.; Wawrzeniak, K.; Gaur, G.; Torres, J.; Filush, K.; Melillo, A.; Zelger, B. Borrelia and Chlamydia Can Form Mixed Biofilms in Infected Human Skin Tissues. Eur. J. Microbiol. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Brown, A.V.; Matluck, N.E.; Hu, L.T.; Lewis, K. Borrelia burgdorferi, the Causative Agent of Lyme Disease, Forms Drug-Tolerant Persister Cells. Antimicrob. Agents Chemother. 2015, 59, 4616–4624. [Google Scholar] [CrossRef] [PubMed]
- Sapi, E.; Balasubramanian, K.; Poruri, A.; Maghsoudlou, J.S.; Socarras, K.M.; Timmaraju, A.V.; Filush, K.R.; Gupta, K.; Shaikh, S.; Theophilus, P.A.; et al. Evidence of In Vivo Existence of Borrelia Biofilm in Borrelial Lymphocytomas. Eur. J. Microbiol. Immunol. (Bp) 2016, 6, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, T.; Yee, R.; Yuan, Y.; Bai, C.; Cai, M.; Shi, W.; Embers, M.; Brayton, C.; Saeki, H.; et al. Stationary phase persister/biofilm microcolony of Borrelia burgdorferi causes more severe disease in a mouse model of Lyme arthritis: Implications for understanding persistence, Post-treatment Lyme Disease Syndrome (PTLDS), and treatment failure. Discov. Med. 2019, 27, 125–138. [Google Scholar] [PubMed]
- Cabello, F.C.; Godfrey, H.P.; Bugrysheva, J.V.; Newman, S.A. Sleeper cells: The stringent response and persistence in the Borreliella (Borrelia) burgdorferi enzootic cycle. Environ. Microbiol. 2017, 19, 3846–3862. [Google Scholar] [CrossRef] [PubMed]
- Hodzic, E.; Imai, D.; Feng, S.; Barthold, S.W. Resurgence of persisting non-cultivable Borrelia burgdorferi following antibiotic treatment in mice. PLoS ONE 2014, 9, e86907. [Google Scholar] [CrossRef] [PubMed]
- Middelveen, M.J.; Sapi, E.; Burke, J.; Filush, K.R.; Franco, A.; Fesler, M.C.; Stricker, R.B. Persistent Borrelia Infection in Patients with Ongoing Symptoms of Lyme Disease. Healthcare 2018, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Dumic, I.; Severnini, E. “Ticking Bomb”: The Impact of Climate Change on the Incidence of Lyme Disease. Can. J. Infect. Dis. Med. Microbiol. 2018, 2018, 5719081. [Google Scholar] [CrossRef] [PubMed]
- Sonenshine, D.E. Range Expansion of Tick Disease Vectors in North America: Implications for Spread of Tick-Borne Disease. Int. J. Environ. Res. Public Health 2018, 15, 478. [Google Scholar] [CrossRef] [PubMed]
- Mead, P.S. Epidemiology of Lyme disease. Infect. Dis. Clin. N. Am. 2015, 29, 187–210. [Google Scholar] [CrossRef]
- Adrion, E.R.; Aucott, J.; Lemke, K.W.; Weiner, J.P. Health care costs, utilization and patterns of care following Lyme disease. PLoS ONE 2015, 10, e0116767. [Google Scholar] [CrossRef]
- Aucott, J.N. Post-treatment Lyme disease syndrome. Infect. Disease Clin. N. Am. 2015, 29, 309–323. [Google Scholar] [CrossRef]
- Waddell, L.A.; Greig, J.; Mascarenhas, M.; Harding, S.; Lindsay, R.; Ogden, N. The Accuracy of Diagnostic Tests for Lyme Disease in Humans, A Systematic Review and Meta-Analysis of North American Research. PLoS ONE 2016, 11, e0168613. [Google Scholar] [CrossRef] [PubMed]
- Rebman, A.W.; Bechtold, K.T.; Yang, T.; Mihm, E.A.; Soloski, M.J.; Novak, C.B.; Aucott, J.N. The Clinical, Symptom, and Quality-of-Life Characterization of a Well-Defined Group of Patients with Posttreatment Lyme Disease Syndrome. Front. Med. Lausanne 2017, 4, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.; Wilcox, S.; Mankoff, J.; Stricker, R.B. Severity of chronic Lyme disease compared to other chronic conditions: A quality of life survey. PeerJ 2014, 2, e322. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease, Control Prevention. Three sudden cardiac deaths associated with Lyme carditis—United States, November 2012-July 2013. MMWR Morb. Mortal. Wkly. Rep. 2013, 62, 993–996. [Google Scholar]
- Bransfield, R.C. Suicide and Lyme and associated diseases. Neuropsychiatr. Disease Treat. 2017, 13, 1575–1587. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Tilly, K.; Stewart, P.; Bestor, A.; Battisti, J.M.; Rosa, P.A. Borrelia burgdorferi resistance to a major skin antimicrobial peptide is independent of outer surface lipoprotein content. Antimicrob. Agents Chemother. 2009, 53, 4490–4494. [Google Scholar] [CrossRef] [PubMed]
- Steere, A.C.; Sikand, V.K.; Schoen, R.T.; Nowakowski, J. Asymptomatic infection with Borrelia burgdorferi. Clin. Infect. Dis. 2003, 37, 528–532. [Google Scholar] [CrossRef]
- Bunikis, I.; Denker, K.; Ostberg, Y.; Andersen, C.; Benz, R.; Bergstrom, S. An RND-type efflux system in Borrelia burgdorferi is involved in virulence and resistance to antimicrobial compounds. PLoS Pathog. 2008, 4, e1000009. [Google Scholar] [CrossRef]
- Lusitani, D.; Malawista, S.E.; Montgomery, R.R. Borrelia burgdorferi are susceptible to killing by a variety of human polymorphonuclear leukocyte components. J. Infect. Dis. 2002, 185, 797–804. [Google Scholar] [CrossRef]
- Hayward, J.; Sanchez, J.; Perry, A.; Huang, C.; Rodriguez Valle, M.; Canals, M.; Payne, R.J.; Stone, M.J. Ticks from diverse genera encode chemokine-inhibitory evasin proteins. J. Biol. Chem. 2017, 292, 15670–15680. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.; Zhang, N.; Wooten, R.M. Borrelia burgdorferi elicited-IL-10 suppresses the production of inflammatory mediators, phagocytosis, and expression of co-stimulatory receptors by murine macrophages and/or dendritic cells. PLoS ONE 2013, 8, e84980. [Google Scholar] [CrossRef] [PubMed]
- Boylan, J.A.; Lawrence, K.A.; Downey, J.S.; Gherardini, F.C. Borrelia burgdorferi membranes are the primary targets of reactive oxygen species. Mol. Microbiol. 2008, 68, 786–799. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive oxygen species in the immune system. Int. Rev. Immunol. 2013, 32, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.D.; Clark, H.M.; McIlvin, M.; Vazquez, C.; Palmere, S.L.; Grab, D.J.; Seshu, J.; Hart, P.J.; Saito, M.; Culotta, V.C. A manganese-rich environment supports superoxide dismutase activity in a Lyme disease pathogen, Borrelia burgdorferi. J. Biol. Chem. 2013, 288, 8468–8478. [Google Scholar] [CrossRef] [PubMed]
- Troxell, B.; Xu, H.; Yang, X.F. Borrelia burgdorferi, a pathogen that lacks iron, encodes manganese-dependent superoxide dismutase essential for resistance to streptonigrin. J. Biol. Chem. 2012, 287, 19284–19293. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.A.; Punt, J.; Stranford, S.A. Kuby Immunology, 7th ed.; W. H. Freeman: New York, NY, USA, 2013. [Google Scholar]
- Ouyang, Z.; He, M.; Oman, T.; Yang, X.F.; Norgard, M.V. A manganese transporter, BB0219 (BmtA), is required for virulence by the Lyme disease spirochete, Borrelia burgdorferi. Proc. Natl. Acad. Sci. USA 2009, 106, 3449–3454. [Google Scholar] [CrossRef] [PubMed]
- Wagh, D.; Pothineni, V.R.; Inayathullah, M.; Liu, S.; Kim, K.M.; Rajadas, J. Borreliacidal activity of Borrelia metal transporter A (BmtA) binding small molecules by manganese transport inhibition. Drug Des. Dev. Ther. 2015, 9, 805–816. [Google Scholar] [CrossRef]
- Sapi, E.; Bastian, S.L.; Mpoy, C.M.; Scott, S.; Rattelle, A.; Pabbati, N.; Poruri, A.; Burugu, D.; Theophilus, P.A.; Pham, T.V.; et al. Characterization of biofilm formation by Borrelia burgdorferi in vitro. PLoS ONE 2012, 7, e48277. [Google Scholar] [CrossRef]
- Wu, J.; Weening, E.H.; Faske, J.B.; Hook, M.; Skare, J.T. Invasion of eukaryotic cells by Borrelia burgdorferi requires beta(1) integrins and Src kinase activity. Infect. Immun. 2011, 79, 1338–1348. [Google Scholar] [CrossRef]
- Tunev, S.S.; Hastey, C.J.; Hodzic, E.; Feng, S.; Barthold, S.W.; Baumgarth, N. Lymphoadenopathy during lyme borreliosis is caused by spirochete migration-induced specific B cell activation. PLoS Pathog. 2011, 7, e1002066. [Google Scholar] [CrossRef]
- Elsner, R.A.; Hastey, C.J.; Olsen, K.J.; Baumgarth, N. Suppression of Long-Lived Humoral Immunity Following Borrelia burgdorferi Infection. PLoS Pathog. 2015, 11, e1004976. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Gao, L.; Edmondson, D.G.; Jacobs, M.B.; Philipp, M.T.; Norris, S.J. Central role of the Holliday junction helicase RuvAB in vlsE recombination and infectivity of Borrelia burgdorferi. PLoS Pathog. 2009, 5, e1000679. [Google Scholar] [CrossRef] [PubMed]
- Norris, S.J. vls Antigenic Variation Systems of Lyme Disease Borrelia: Eluding Host Immunity through both Random, Segmental Gene Conversion and Framework Heterogeneity. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Margos, G.; Hepner, S.; Mang, C.; Marosevic, D.; Reynolds, S.E.; Krebs, S.; Sing, A.; Derdakova, M.; Reiter, M.A.; Fingerle, V. Lost in plasmids: Next generation sequencing and the complex genome of the tick-borne pathogen Borrelia burgdorferi. BMC Genom. 2017, 18, 422. [Google Scholar] [CrossRef] [PubMed]
- Bhide, M.R.; Travnicek, M.; Levkutova, M.; Curlik, J.; Revajova, V.; Levkut, M. Sensitivity of Borrelia genospecies to serum complement from different animals and human: A host-pathogen relationship. FEMS Immunol. Med. Microbiol. 2005, 43, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Šimo, L.; Kazimirova, M.; Richardson, J.; Bonnet, S.I. The Essential Role of Tick Salivary Glands and Saliva in Tick Feeding and Pathogen Transmission. Front. Cell Infect. Microbiol. 2017, 7, 281. [Google Scholar] [CrossRef] [PubMed]
- de Taeye, S.W.; Kreuk, L.; van Dam, A.P.; Hovius, J.W.; Schuijt, T.J. Complement evasion by Borrelia burgdorferi: It takes three to tango. Trends Parasitol. 2013, 29, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Probert, W.S.; Johnson, B.J. Identification of a 47 kDa fibronectin-binding protein expressed by Borrelia burgdorferi isolate B31. Mol. Microbiol. 1998, 30, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.L.; Zhi, H.; Wager, B.; Hook, M.; Skare, J.T. Borrelia burgdorferi BBK32 Inhibits the Classical Pathway by Blocking Activation of the C1 Complement Complex. PLoS Pathog. 2016, 12, e1005404. [Google Scholar] [CrossRef]
- van Dam, A.P.; Oei, A.; Jaspars, R.; Fijen, C.; Wilske, B.; Spanjaard, L.; Dankert, J. Complement-mediated serum sensitivity among spirochetes that cause Lyme disease. Infect. Immun. 1997, 65, 1228–1236. [Google Scholar] [Green Version]
- Seshu, J.; Esteve-Gassent, M.D.; Labandeira-Rey, M.; Kim, J.H.; Trzeciakowski, J.P.; Hook, M.; Skare, J.T. Inactivation of the fibronectin-binding adhesin gene bbk32 significantly attenuates the infectivity potential of Borrelia burgdorferi. Mol. Microbiol. 2006, 59, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Barthel, D.; Schindler, S.; Zipfel, P.F. Plasminogen is a complement inhibitor. J. Biol. Chem. 2012, 287, 18831–18842. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.; Wallich, R.; Simon, M.M.; Kramer, M.D. The outer surface protein A of the spirochete Borrelia burgdorferi is a plasmin(ogen) receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 12594–12598. [Google Scholar] [CrossRef] [PubMed]
- Önder, Ö.; Humphrey, P.T.; McOmber, B.; Korobova, F.; Francella, N.; Greenbaum, D.C.; Brisson, D. OspC is potent plasminogen receptor on surface of Borrelia burgdorferi. J. Biol. Chem. 2012, 287, 16860–16868. [Google Scholar] [CrossRef] [PubMed]
- Koenigs, A.; Hammerschmidt, C.; Jutras, B.L.; Pogoryelov, D.; Barthel, D.; Skerka, C.; Kugelstadt, D.; Wallich, R.; Stevenson, B.; Zipfel, P.F.; et al. BBA70 of Borrelia burgdorferi is a novel plasminogen-binding protein. J. Biol. Chem. 2013, 288, 25229–25243. [Google Scholar] [CrossRef] [PubMed]
- Hallström, T.; Haupt, K.; Kraiczy, P.; Hortschansky, P.; Wallich, R.; Skerka, C.; Zipfel, P.F. Complement regulator-acquiring surface protein 1 of Borrelia burgdorferi binds to human bone morphogenic protein 2, several extracellular matrix proteins, and plasminogen. J. Infect. Dis. 2010, 202, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Brissette, C.A.; Haupt, K.; Barthel, D.; Cooley, A.E.; Bowman, A.; Skerka, C.; Wallich, R.; Zipfel, P.F.; Kraiczy, P.; Stevenson, B. Borrelia burgdorferi infection-associated surface proteins ErpP, ErpA, and ErpC bind human plasminogen. Infect. Immun. 2009, 77, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Kraiczy, P.; Hellwage, J.; Skerka, C.; Becker, H.; Kirschfink, M.; Simon, M.M.; Brade, V.; Zipfel, P.F.; Wallich, R. Complement resistance of Borrelia burgdorferi correlates with the expression of BbCRASP-1, a novel linear plasmid-encoded surface protein that interacts with human factor H and FHL-1 and is unrelated to Erp proteins. J. Biol. Chem. 2004, 279, 2421–2429. [Google Scholar] [CrossRef]
- Kraiczy, P.; Skerka, C.; Brade, V.; Zipfel, P.F. Further characterization of complement regulator-acquiring surface proteins of Borrelia burgdorferi. Infect. Immun. 2001, 69, 7800–7809. [Google Scholar] [CrossRef]
- McDowell, J.V.; Harlin, M.E.; Rogers, E.A.; Marconi, R.T. Putative coiled-coil structural elements of the BBA68 protein of Lyme disease spirochetes are required for formation of its factor H binding site. J. Bacteriol. 2005, 187, 1317–1323. [Google Scholar] [CrossRef]
- Cordes, F.S.; Kraiczy, P.; Roversi, P.; Simon, M.M.; Brade, V.; Jahraus, O.; Wallis, R.; Goodstadt, L.; Ponting, C.P.; Skerka, C.; et al. Structure-function mapping of BbCRASP-1, the key complement factor H and FHL-1 binding protein of Borrelia burgdorferi. Int. J. Med. Microbiol. 2006, 296 (Suppl. 40), 177–184. [Google Scholar] [CrossRef] [PubMed]
- Kenedy, M.R.; Vuppala, S.R.; Siegel, C.; Kraiczy, P.; Akins, D.R. CspA-mediated binding of human factor H inhibits complement deposition and confers serum resistance in Borrelia burgdorferi. Infect. Immun. 2009, 77, 2773–2782. [Google Scholar] [CrossRef] [PubMed]
- Hallström, T.; Siegel, C.; Morgelin, M.; Kraiczy, P.; Skerka, C.; Zipfel, P.F. CspA from Borrelia burgdorferi inhibits the terminal complement pathway. mBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, C.; Klevenhaus, Y.; Koenigs, A.; Hallstrom, T.; Fingerle, V.; Skerka, C.; Pos, K.M.; Zipfel, P.F.; Wallich, R.; Kraiczy, P. BGA66 and BGA71 facilitate complement resistance of Borrelia bavariensis by inhibiting assembly of the membrane attack complex. Mol. Microbiol. 2016, 99, 407–424. [Google Scholar] [CrossRef] [PubMed]
- Pausa, M.; Pellis, V.; Cinco, M.; Giulianini, P.G.; Presani, G.; Perticarari, S.; Murgia, R.; Tedesco, F. Serum-resistant strains of Borrelia burgdorferi evade complement-mediated killing by expressing a CD59-like complement inhibitory molecule. J. Immunol. 2003, 170, 3214–3222. [Google Scholar] [CrossRef] [PubMed]
- Woodman, M.E.; Cooley, A.E.; Miller, J.C.; Lazarus, J.J.; Tucker, K.; Bykowski, T.; Botto, M.; Hellwage, J.; Wooten, R.M.; Stevenson, B. Borrelia burgdorferi binding of host complement regulator factor H is not required for efficient mammalian infection. Infect. Immun. 2007, 75, 3131–3139. [Google Scholar] [CrossRef] [PubMed]
- Kenedy, M.R.; Akins, D.R. The OspE-related proteins inhibit complement deposition and enhance serum resistance of Borrelia burgdorferi, the lyme disease spirochete. Infect. Immun. 2011, 79, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, R.; Mikula, K.M.; Kotila, T.; Postis, V.L.G.; Jokiranta, T.S.; Goldman, A.; Meri, T. Crystal structure of a tripartite complex between C3dg, C-terminal domains of factor H and OspE of Borrelia burgdorferi. PLoS ONE 2017, 12, e0188127. [Google Scholar] [CrossRef]
- Dyer, A.; Brown, G.; Stejskal, L.; Laity, P.R.; Bingham, R.J. The Borrelia afzelii outer membrane protein BAPKO_0422 binds human factor-H and is predicted to form a membrane-spanning beta-barrel. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef]
- Kenedy, M.R.; Scott, E.J., 2nd; Shrestha, B.; Anand, A.; Iqbal, H.; Radolf, J.D.; Dyer, D.W.; Akins, D.R. Consensus computational network analysis for identifying candidate outer membrane proteins from Borrelia spirochetes. BMC Microbiol. 2016, 16, 141. [Google Scholar] [CrossRef]
- Shrestha, B.; Kenedy, M.R.; Akins, D.R. Outer Membrane Proteins BB0405 and BB0406 Are Immunogenic, but Only BB0405 Is Required for Borrelia burgdorferi Infection. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [PubMed]
- Pietikainen, J.; Meri, T.; Blom, A.M.; Meri, S. Binding of the complement inhibitor C4b-binding protein to Lyme disease Borreliae. Mol. Immunol. 2010, 47, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Arvikar, S.L.; Crowley, J.T.; Sulka, K.B.; Steere, A.C. Autoimmune Arthritides, Rheumatoid Arthritis, Psoriatic Arthritis, or Peripheral Spondyloarthritis Following Lyme Disease. Arthritis Rheumatol. 2017, 69, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Maccallini, P.; Bonin, S.; Trevisan, G. Autoimmunity against a glycolytic enzyme as a possible cause for persistent symptoms in Lyme disease. Med. Hypotheses 2018, 110, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bockenstedt, L.K.; Gonzalez, D.G.; Haberman, A.M.; Belperron, A.A. Spirochete antigens persist near cartilage after murine Lyme borreliosis therapy. J. Clin. Investig. 2012, 122, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Socarras, K.M.; Theophilus, P.A.S.; Torres, J.P.; Gupta, K.; Sapi, E. Antimicrobial Activity of Bee Venom and Melittin against Borrelia burgdorferi. Antibiotics 2017, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Theophilus, P.A.; Victoria, M.J.; Socarras, K.M.; Filush, K.R.; Gupta, K.; Luecke, D.F.; Sapi, E. Effectiveness of Stevia Rebaudiana Whole Leaf Extract Against the Various Morphological Forms of Borrelia burgdorferi in Vitro. Eur. J. Microbiol. Immunol. (Bp) 2015, 5, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Yee, R.; Yuan, Y.; Brayton, C.; Leal, A.T.; Feng, J.; Shi, W.; Behrens, A.; Zhang, Y. Biofilm/Persister/Stationary Phase Bacteria Cause More Severe Disease Than Log Phase Bacteria—II Infection with Persister Forms of Staphylococcus aureus Causes a Chronic Persistent Skin Infection with More Severe Lesion that Takes Longer to Heal and is not Eradicated by the Current Recommended Treatment in Mice. bioRxiv Preprint 2018. [Google Scholar] [CrossRef]
- Serdobova, I.; Kieny, M.P. Assembling a global vaccine development pipeline for infectious diseases in the developing world. Am. J. Public Health 2006, 96, 1554–1559. [Google Scholar] [CrossRef]
- El Shikh, M.E.; El Sayed, R.M.; Sukumar, S.; Szakal, A.K.; Tew, J.G. Activation of B cells by antigens on follicular dendritic cells. Trends Immunol. 2010, 31, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Zhi, H.; Garrigues, R.J.; Keightley, A.; Garcia, B.L.; Skare, J.T. Structural determination of the complement inhibitory domain of Borrelia burgdorferi BBK32 provides insight into classical pathway complement evasion by Lyme disease spirochetes. PLoS Pathog. 2019, 15, e1007659. [Google Scholar] [CrossRef] [PubMed]
- Koch, R. Weitere Mittheilungen über ein Heilmittel gegen Tuberculose. Dtsch. Med. Wochenschr. 1890, 16, 1029–1032. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Gene | Plasmid | GenBank Accession | Species |
|---|---|---|---|---|
| BBK32 | bbk32 | lp36 | AE000788.1 | B. burgdorferi, B. afzelii, B. garinii, others |
| OspA | ospA | lp54 | AE000790.2 | B. burgdorferi, B. afzelii, B. garinii, others |
| OspC | ospC | cp26 | AE000792.1 | B. burgdorferi, B. afzelii, B. garinii, others |
| BBA70 | bba70 | lp54 | AY696552.1 | B. burgdorferi 163b, B. bavariensis |
| CRASP | Gene | Plasmid 1 | Interval | GenBank Accession | Species |
|---|---|---|---|---|---|
| CRASP-1 | cspA | lp54 | 46473-47228 2 | AE000790.2 | B. burgdorferi, B. afzelii, B. spielmanii |
| CRASP-2 | cspZ | lp28-3 | 2260-2970 2 | AE000784.1 | B. burgdorferi, B. afzelii |
| CRASP-3 | erpP | cp32-9 | 26210-26770 | AE001581.1 | B. burgdorferi |
| CRASP-4 | erpC | cp32-2 | 26834-27373 | NZ_CP019757.1 | B. burgdorferi |
| CRASP-5 | erpA | cp32-1 | 26235-26768 | AE001575.1 | B. burgdorferi |
| - | BG0407 | - | 417734-418345 | AAU07257.1 | B. garinii, B. bavariensis |
| - | BafPKo_0408 (BAPKO_0422) | - | 419301-419906 | ABH01676.1 (CP000395.1) | B. afzelii |
| Proteins | Identity | Similarity | Gaps |
|---|---|---|---|
| ErpA vs ErpC | 77% | 85% | 5% |
| ErpA vs ErpP | 75% | 83% | 6% |
| ErpC vs ErpP | 70% | 78% | 11% |
| Disease | NIH Funding (Millions, 2018) | US Cases per Year | Funding per Case |
|---|---|---|---|
| Malaria | $202 | 1700 1 | $118,824 |
| HIV/AIDS | $3000 | 38,739 2 | $77,441 |
| West Nile virus | $45 | 2544 3 | $17,689 |
| PTLDS | $23 | 45,000 4 | $511.11 |
| LD | $23 | 300,000 5 | $76.67 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Locke, J.W. Complement Evasion in Borrelia spirochetes: Mechanisms and Opportunities for Intervention. Antibiotics 2019, 8, 80. https://doi.org/10.3390/antibiotics8020080
Locke JW. Complement Evasion in Borrelia spirochetes: Mechanisms and Opportunities for Intervention. Antibiotics. 2019; 8(2):80. https://doi.org/10.3390/antibiotics8020080
Chicago/Turabian StyleLocke, Jonathan W. 2019. "Complement Evasion in Borrelia spirochetes: Mechanisms and Opportunities for Intervention" Antibiotics 8, no. 2: 80. https://doi.org/10.3390/antibiotics8020080
APA StyleLocke, J. W. (2019). Complement Evasion in Borrelia spirochetes: Mechanisms and Opportunities for Intervention. Antibiotics, 8(2), 80. https://doi.org/10.3390/antibiotics8020080