Re-Understanding the Mechanisms of Action of the Anti-Mycobacterial Drug Bedaquiline



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mechanisms of Action of BDQ—Before TBAJ-876′s Discovery
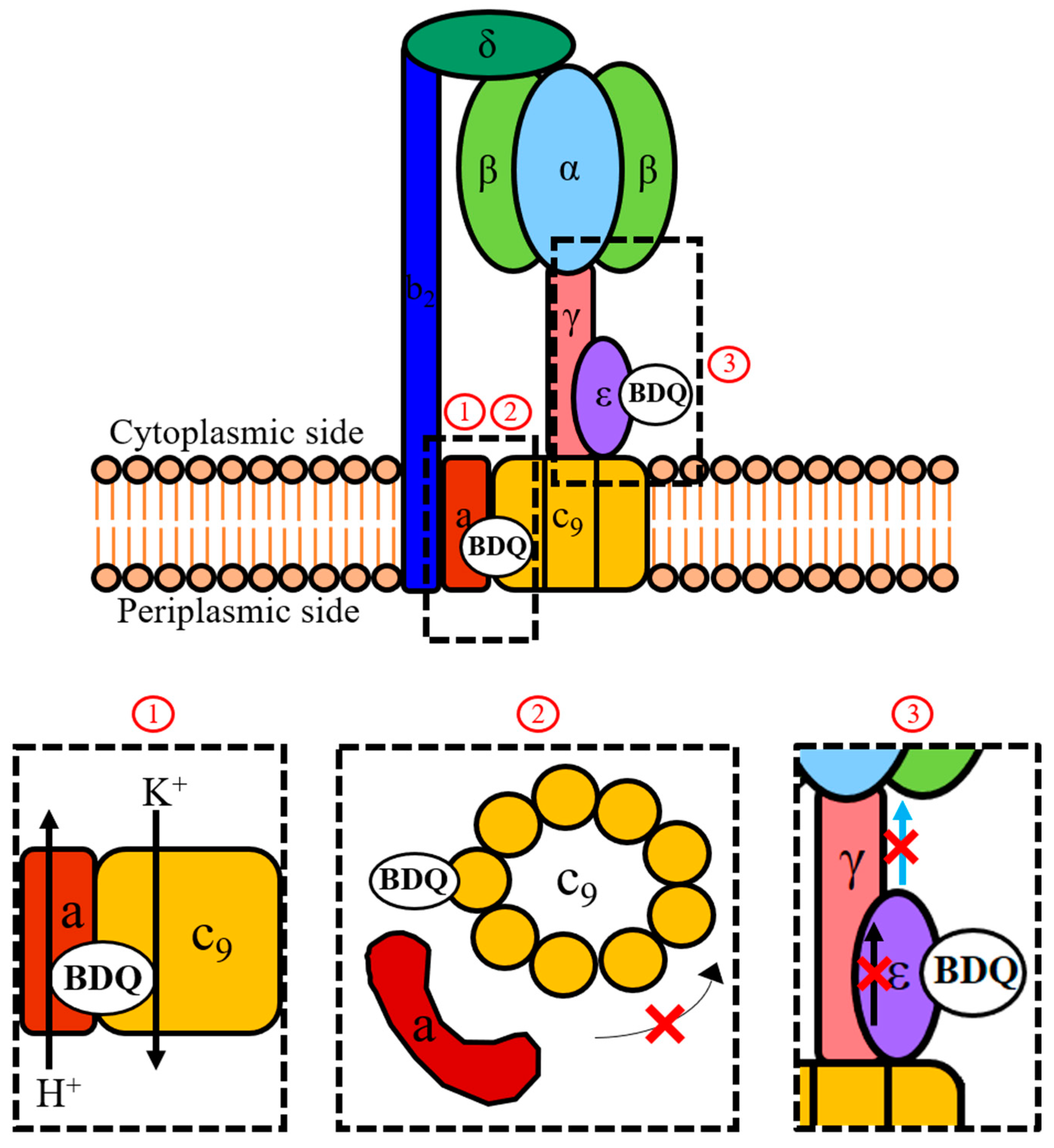
2.1. Stalling of Rotation of the Mycobacterial F-ATP Synthase c-Ring
2.2. Uncoupling Electron Transport from ATP Synthesis
2.3. Disrupting the Mycobacterial F-ATP Synthase ε-Subunit’s Function of Linking c-Ring Rotation to ATP Synthesis
3. Insights into BDQ’s Mechanisms of Action Derived from TBAJ-876 Studies
3.1. TBAJ-876 Retained BDQ’s Targeting of Both the c-Ring and ε-Subunit
3.2. TBAJ-876 Did Not Retain BDQ’s Uncoupler Activity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lawn, S.D.; Zumla, A.I. Tuberculosis. Lancet 2011, 378, 57–72. [Google Scholar] [CrossRef]
- Gradmann, C. Die Ätiologie Der Tuberkulose (1882). In Robert Koch: Zentrale Texte, 1st ed.; Springer: Berlin, Germany, 2018; pp. 113–131. [Google Scholar]
- WHO. Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Sharma, A.; Hill, A.; Kurbatova, E.; van der Walt, M.; Kvasnovsky, C.; Tupasi, T.E.; Caoili, J.C.; Gler, M.T.; Volchenkov, G.V.; Kazennyy, B.Y. Estimating the future burden of multidrug-resistant and extensively drug-resistant tuberculosis in India, the Philippines, Russia, and South Africa: A mathematical modelling study. Lancet Infect. Dis. 2017, 17, 707–715. [Google Scholar] [CrossRef]
- Mullard, A. 2012 FDA Drug Approvals. Nat. Rev. Drug Discov. 2013, 12, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, M.; Xavier, A.S. Bedaquiline–The first ATP synthase inhibitor against multi drug resistant tuberculosis. J. Young Pharm. 2013, 5, 112–115. [Google Scholar] [CrossRef] [Green Version]
- Mingote, L.R.; Namutamba, D.; Apina, F.; Barnabas, N.; Contreras, C.; Elnour, T.; Frick, M.W.; Lee, C.; Seaworth, B.; Shelly, D. The use of bedaquiline in regimens to treat drug-resistant and drug-susceptible tuberculosis: A perspective from tuberculosis-affected communities. Lancet 2015, 385, 477–479. [Google Scholar] [CrossRef]
- SIRTURO (Bedaquiline): Medication Guide; Janssen Pharmaceuticals, Inc.: Titusville, NJ, USA, 2012.
- WHO. Consolidated Guidelines on Drug-Resistant Tuberculosis Treatment; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.; Neefs, J.-M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef]
- Huitric, E.; Verhasselt, P.; Andries, K.; Hoffner, S.E. In vitro antimycobacterial spectrum of a diarylquinoline ATP synthase inhibitor. Antimicrob. Agents Chemother. 2007, 51, 4202–4204. [Google Scholar] [CrossRef] [Green Version]
- Tasneen, R.; Li, S.-Y.; Peloquin, C.A.; Taylor, D.; Williams, K.N.; Andries, K.; Mdluli, K.E.; Nuermberger, E.L. Sterilizing activity of novel TMC207-and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 5485–5492. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.; Andries, K.; Lounis, N.; Chauffour, A.; Truffot-Pernot, C.; Jarlier, V.; Veziris, N. Synergistic activity of R207910 combined with pyrazinamide against murine tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1011–1015. [Google Scholar] [CrossRef] [Green Version]
- Vocat, A.; Hartkoorn, R.C.; Lechartier, B.; Zhang, M.; Dhar, N.; Cole, S.T.; Sala, C. Bioluminescence for assessing drug potency against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 4012–4019. [Google Scholar] [CrossRef] [Green Version]
- Koul, A.; Vranckx, L.; Dendouga, N.; Balemans, W.; Van den Wyngaert, I.; Vergauwen, K.; Göhlmann, H.W.; Willebrords, R.; Poncelet, A.; Guillemont, J. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 2008, 283, 25273–25280. [Google Scholar] [CrossRef] [Green Version]
- Udwadia, Z.; Amale, R.; Mullerpattan, J. Initial experience of bedaquiline use in a series of drug-resistant tuberculosis patients from India. Int. J. Tuberc. Lung Dis. 2014, 18, 1315–1318. [Google Scholar] [CrossRef]
- Pym, A.S.; Diacon, A.H.; Tang, S.-J.; Conradie, F.; Danilovits, M.; Chuchottaworn, C.; Vasilyeva, I.; Andries, K.; Bakare, N.; De Marez, T. Bedaquiline in the treatment of multidrug-and extensively drug-resistant tuberculosis. Eur. Respir. J. 2016, 47, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Diacon, A.H.; Pym, A.; Grobusch, M.; Patientia, R.; Rustomjee, R.; Page-Shipp, L.; Pistorius, C.; Krause, R.; Bogoshi, M.; Churchyard, G. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Engl. J. Med. 2009, 360, 2397–2405. [Google Scholar] [CrossRef] [Green Version]
- Diacon, A.H.; Pym, A.; Grobusch, M.P.; de los Rios, J.M.; Gotuzzo, E.; Vasilyeva, I.; Leimane, V.; Andries, K.; Bakare, N.; De Marez, T. Multidrug-resistant tuberculosis and culture conversion with bedaquiline. N. Engl. J. Med. 2014, 371, 723–732. [Google Scholar] [CrossRef] [Green Version]
- Libardo, M.D.J.; Boshoff, H.I.; Barry, C.E., III. The present state of the tuberculosis drug development pipeline. Curr. Opin. Pharmacol. 2018, 42, 81–94. [Google Scholar] [CrossRef]
- FDA Approves New Drug for Treatment-Resistant Forms of Tuberculosis that Affects the Lungs; U.S. Food and Drug Administration (FDA): Silver Spring, MD, USA, 2019.
- FDA Approves New Treatment for Highly Drug-Resistant Forms of Tuberculosis; PR Newswire: New York, NY, USA, 2019.
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef]
- Tran, S.L.; Cook, G.M. The F1Fo-ATP synthase of Mycobacterium smegmatis is essential for growth. J. Bacteriol. 2005, 187, 5023–5028. [Google Scholar] [CrossRef] [Green Version]
- Koul, A.; Dendouga, N.; Vergauwen, K.; Molenberghs, B.; Vranckx, L.; Willebrords, R.; Ristic, Z.; Lill, H.; Dorange, I.; Guillemont, J. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 2007, 3, 323–324. [Google Scholar] [CrossRef]
- Haagsma, A.C.; Podasca, I.; Koul, A.; Andries, K.; Guillemont, J.; Lill, H.; Bald, D. Probing the interaction of the diarylquinoline TMC207 with its target mycobacterial ATP synthase. PLoS ONE 2011, 6, e23575. [Google Scholar] [CrossRef] [Green Version]
- Koul, A.; Vranckx, L.; Dhar, N.; Göhlmann, H.W.; Özdemir, E.; Neefs, J.-M.; Schulz, M.; Lu, P.; Mørtz, E.; McKinney, J.D. Delayed bactericidal response of Mycobacterium tuberculosis to bedaquiline involves remodelling of bacterial metabolism. Nat. Commun. 2014, 5, 3369. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Soni, V.; Marriner, G.; Kaneko, T.; Boshoff, H.I.; Barry, C.E.; Rhee, K.Y. Mode-of-action profiling reveals glutamine synthetase as a collateral metabolic vulnerability of M. tuberculosis to bedaquiline. Proc. Natl. Acad. Sci. USA 2019, 116, 19646–19651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, T.; Young, D. How antibacterials really work: Impact on drug discovery. Future Microbiol. 2011, 6, 603–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diacon, A.; Donald, P.; Pym, A.; Grobusch, M.; Patientia, R.; Mahanyele, R.; Bantubani, N.; Narasimooloo, R.; De Marez, T.; Van Heeswijk, R. Randomized pilot trial of eight weeks of bedaquiline (TMC207) treatment for multidrug-resistant tuberculosis: Long-term outcome, tolerability, and effect on emergence of drug resistance. Antimicrob. Agents Chemother. 2012, 56, 3271–3276. [Google Scholar] [CrossRef] [Green Version]
- Anti-Infective Drugs Advisory Committee Meeting Briefing Document TMC207 (Bedaquiline). Treatment of Patients with MDR-TB, NDA 204-384; U.S. Food and Drug Administration (FDA): Silver Spring, MD, USA, 2012. [Google Scholar]
- Kakkar, A.K.; Dahiya, N. Bedaquiline for the treatment of resistant tuberculosis: Promises and pitfalls. Tuberculosis 2014, 94, 357–362. [Google Scholar] [CrossRef]
- Tong, A.S.T.; Choi, P.J.; Blaser, A.; Sutherland, H.S.; Tsang, S.K.Y.; Guillemont, J.; Motte, M.; Cooper, C.B.; Andries, K.; Van den Broeck, W.; et al. 6-Cyano Analogues of Bedaquiline as Less Lipophilic and Potentially Safer Diarylquinolines for Tuberculosis. ACS Med. Chem. Lett. 2017, 8, 1019–1024. [Google Scholar] [CrossRef] [Green Version]
- Choi, P.J.; Sutherland, H.S.; Tong, A.S.T.; Blaser, A.; Franzblau, S.G.; Cooper, C.B.; Lotlikar, M.U.; Upton, A.M.; Guillemont, J.; Motte, M.; et al. Synthesis and evaluation of analogues of the tuberculosis drug bedaquiline containing heterocyclic B-ring units. Bioorganic Med. Chem. Lett. 2017, 27, 5190–5196. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Conole, D.; Blaser, A.; Franzblau, S.G.; Cooper, C.B.; Upton, A.M.; Lotlikar, M.U.; Denny, W.A.; et al. Structure-activity relationships for analogs of the tuberculosis drug bedaquiline with the naphthalene unit replaced by bicyclic heterocycles. Bioorganic Med. Chem. 2018, 26, 1797–1809. [Google Scholar] [CrossRef]
- Blaser, A.; Sutherland, H.S.; Tong, A.S.; Choi, P.J.; Conole, D.; Franzblau, S.G.; Cooper, C.B.; Upton, A.M.; Lotlikar, M.; Denny, W.A.; et al. Structure-activity relationships for unit C pyridyl analogues of the tuberculosis drug bedaquiline. Bioorganic Med. Chem. 2019, 27, 1283–1291. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Tong, A.S.; Choi, P.J.; Blaser, A.; Conole, D.; Franzblau, S.G.; Lotlikar, M.U.; Cooper, C.B.; Upton, A.M.; Denny, W.A.; et al. 3, 5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorganic Med. Chem. 2019, 27, 1292–1307. [Google Scholar] [CrossRef]
- Bald, D.; Villellas, C.; Lu, P.; Koul, A. Targeting energy metabolism in Mycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. Microbiology 2017, 8, e00272-17. [Google Scholar] [CrossRef] [Green Version]
- Mitome, N.; Ono, S.; Sato, H.; Suzuki, T.; Sone, N.; Yoshida, M. Essential arginine residue of the Fo-a subunit in FoF1-ATP synthase has a role to prevent the proton shortcut without c-ring rotation in the Fo proton channel. Biochem. J. 2010, 430, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Diez, M.; Zimmermann, B.; Börsch, M.; König, M.; Schweinberger, E.; Steigmiller, S.; Reuter, R.; Felekyan, S.; Kudryavtsev, V.; Seidel, C.A. Proton-powered subunit rotation in single membrane-bound FoF1-ATP synthase. Nat. Struct. Mol. Biol. 2004, 11, 135–141. [Google Scholar] [CrossRef]
- Kamariah, N.; Huber, R.G.; Nartey, W.; Bhushan, S.; Bond, P.J.; Grüber, G. Structure and subunit arrangement of Mycobacterial F1Fo ATP synthase and novel features of the unique mycobacterial subunit δ. J. Struct. Biol. 2019, 207, 199–208. [Google Scholar] [CrossRef]
- Preiss, L.; Langer, J.D.; Yildiz, Ö.; Eckhardt-Strelau, L.; Guillemont, J.E.; Koul, A.; Meier, T. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci. Adv. 2015, 1, e1500106. [Google Scholar] [CrossRef] [Green Version]
- Huitric, E.; Verhasselt, P.; Koul, A.; Andries, K.; Hoffner, S.; Andersson, D.I. Rates and mechanisms of resistance development in Mycobacterium tuberculosis to a novel diarylquinoline ATP synthase inhibitor. Antimicrob. Agents Chemother 2010, 54, 1022–1028. [Google Scholar] [CrossRef] [Green Version]
- Segala, E.; Sougakoff, W.; Nevejans-Chauffour, A.; Jarlier, V.; Petrella, S. New mutations in the mycobacterial ATP synthase: New insights into the binding of the diarylquinoline TMC207 to the ATP synthase C-ring structure. Antimicrob. Agents Chemother. 2012, 56, 2326–2334. [Google Scholar] [CrossRef] [Green Version]
- Salifu, E.Y.; Agoni, C.; Olotu, F.A.; Dokurugu, Y.M.; Soliman, M.E. Halting ionic shuttle to disrupt the synthetic machinery—Structural and molecular insights into the inhibitory roles of Bedaquiline towards Mycobacterium tuberculosis ATP synthase in the treatment of tuberculosis. J. Cell. Biochem. 2019, 120, 16108–16119. [Google Scholar] [CrossRef]
- Zimenkov, D.V.; Nosova, E.Y.; Kulagina, E.V.; Antonova, O.V.; Arslanbaeva, L.R.; Isakova, A.I.; Krylova, L.Y.; Peretokina, I.V.; Makarova, M.V.; Safonova, S.G. Examination of bedaquiline-and linezolid-resistant Mycobacterium tuberculosis isolates from the Moscow region. J. Antimicrob. Chemother. 2017, 72, 1901–1906. [Google Scholar] [CrossRef] [Green Version]
- Hards, K.; Robson, J.R.; Berney, M.; Shaw, L.; Bald, D.; Koul, A.; Andries, K.; Cook, G.M. Bactericidal mode of action of bedaquiline. J. Antimicrob. Chemother. 2015, 70, 2028–2037. [Google Scholar] [CrossRef] [Green Version]
- Hards, K.; McMillan, D.G.; Schurig-Briccio, L.A.; Gennis, R.B.; Lill, H.; Bald, D.; Cook, G.M. Ionophoric effects of the antitubercular drug bedaquiline. Proc. Natl. Acad. Sci. USA 2018, 115, 7326–7331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Zhu, W.; Schurig-Briccio, L.A.; Lindert, S.; Shoen, C.; Hitchings, R.; Li, J.; Wang, Y.; Baig, N.; Zhou, T. Antiinfectives targeting enzymes and the proton motive force. Proc. Natl. Acad. Sci. USA 2015, 112, E7073–E7082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, S. Interpretation of the mechanism of action of antituberculosis drug bedaquiline based on a novel two-ion theory of energy coupling in ATP synthesis. Bioeng. Transl. Med. 2019, 4, 164–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, S. Two-ion theory of energy coupling in ATP synthesis rectifies a fundamental flaw in the governing equations of the chemiosmotic theory. Biophys. Chem. 2017, 230, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.P.; Alonso, S.; Rand, L.; Dick, T.; Pethe, K. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2008, 105, 11945–11950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.; Streur, T.L.; Aldwell, F.E.; Cook, G.M. Intracellular pH regulation by Mycobacterium smegmatis and Mycobacterium bovis BCG. Microbiology 2001, 147, 1017–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biuković, G.; Basak, S.; Manimekalai, M.S.S.; Rishikesan, S.; Roessle, M.; Dick, T.; Rao, S.P.; Hunke, C.; Grüber, G. Variations of subunit ε of the Mycobacterium tuberculosis F1Fo ATP synthase and a novel model for mechanism of action of the tuberculosis drug TMC207. Antimicrob. Agents Chemother. 2013, 57, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Kundu, S.; Biukovic, G.; Grüber, G.; Dick, T. Bedaquiline targets the ε subunit of mycobacterial F-ATP synthase. Antimicrob. Agents Chemother. 2016, 60, 6977–6979. [Google Scholar] [CrossRef] [Green Version]
- Joon, S.; Ragunathan, P.; Sundararaman, L.; Nartey, W.; Kundu, S.; Manimekalai, M.S.; Bogdanović, N.; Dick, T.; Grüber, G. The NMR solution structure of Mycobacterium tuberculosis F-ATP synthase subunit ε provides new insight into energy coupling inside the rotary engine. FEBS J. 2018, 285, 1111–1128. [Google Scholar] [CrossRef] [Green Version]
- Bogdanović, N.; Sundararaman, L.; Kamariah, N.; Tyagi, A.; Bhushan, S.; Ragunathan, P.; Shin, J.; Dick, T.; Grüber, G. Structure and function of Mycobacterium-specific components of F-ATP synthase subunits α and ε. J. Struct. Biol. 2018, 204, 420–434. [Google Scholar] [CrossRef]
- Saw, W.-G.; Wu, M.-L.; Ragunathan, P.; Biuković, G.; Lau, A.-M.; Shin, J.; Harikishore, A.; Cheung, C.-Y.; Hards, K.; Sarathy, J.P.; et al. Disrupting coupling within mycobacterial F-ATP synthase subunit ε causes dysregulated energy production and cell wall biosynthesis. Sci. Rep. 2019, 9, 16759. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, J.P.; Ragunathan, P.; Shin, J.; Cooper, C.B.; Upton, A.M.; Grüber, G.; Dick, T. TBAJ-876 retains bedaquiline’s activity against subunit c and ϵ of Mycobacterium tuberculosis F-ATP synthase. Antimicrob. Agents Chemother. 2019, 63, e01191-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarathy, J.P.; Ragunathan, P.; Cooper, C.B.; Upton, A.M.; Grüber, G.; Dick, T. TBAJ-876 displays Bedaquiline-like mycobactericidal potency without retaining the parental drug’s uncoupler activity. Antimicrob. Agents Chemother. 2019. [Google Scholar] [CrossRef] [Green Version]
- Toei, M.; Noji, H. Single-molecule analysis of FoF1-ATP synthase inhibited by N, N-dicyclohexylcarbodiimide. J. Biol. Chem. 2013, 288, 25717–25726. [Google Scholar] [CrossRef] [Green Version]
- Abrams, A.; Smith, J.B.; Baron, C. Carbodiimide-resistant membrane adenosine triphosphatase in mutants of Streptococcus faecalis. I. Studies of the mechanism of resistance. J. Biol. Chem. 1972, 247, 1484–1488. [Google Scholar]
- Cohen, N.S.; Lee, S.H.; Brodie, A.F. Purification and characteristics of hydrophobic membrane protein(s) required for DCCD sensitivity of ATPase in Mycobacterium phlei. J. Supramol. Struct. 1978, 8, 111–117. [Google Scholar] [CrossRef]
- Fillingame, R.H. Identification of the dicyclohexylcarbodiimide-reactive protein component of the adenosine 5’-triphosphate energy-transducing system of Escherichia coli. J. Bacteriol. 1975, 124, 870–883. [Google Scholar]
- Yoshida, M.; Poser, J.W.; Allison, W.S.; Esch, F.S. Identification of an essential glutamic acid residue in the beta subunit of the adenosine triphosphatase from the thermophilic bacterium PS3. J. Biol. Chem. 1981, 256, 148–153. [Google Scholar]
- Yoshida, M.; Allison, W.; Esch, F.; Futai, M. The specificity of carboxyl group modification during the inactivation of the Escherichia coli F1-ATPase with dicyclohexyl [14C] carbodiimide. J. Biol. Chem. 1982, 257, 10033–10037. [Google Scholar]
- Parsonage, D.; Wilke-Mounts, S.; Senior, A.E.E. E. coli F1-ATPase: Site-directed mutagenesis of the β-subunit. FEBS Lett. 1988, 232, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Naven, R.T.; Swiss, R.; Klug-Mcleod, J.; Will, Y.; Greene, N. The development of structure-activity relationships for mitochondrial dysfunction: Uncoupling of oxidative phosphorylation. Toxicol. Sci. 2012, 131, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarathy, J.P.; Gruber, G.; Dick, T. Re-Understanding the Mechanisms of Action of the Anti-Mycobacterial Drug Bedaquiline. Antibiotics 2019, 8, 261. https://doi.org/10.3390/antibiotics8040261
Sarathy JP, Gruber G, Dick T. Re-Understanding the Mechanisms of Action of the Anti-Mycobacterial Drug Bedaquiline. Antibiotics. 2019; 8(4):261. https://doi.org/10.3390/antibiotics8040261
Chicago/Turabian StyleSarathy, Jickky Palmae, Gerhard Gruber, and Thomas Dick. 2019. "Re-Understanding the Mechanisms of Action of the Anti-Mycobacterial Drug Bedaquiline" Antibiotics 8, no. 4: 261. https://doi.org/10.3390/antibiotics8040261
APA StyleSarathy, J. P., Gruber, G., & Dick, T. (2019). Re-Understanding the Mechanisms of Action of the Anti-Mycobacterial Drug Bedaquiline. Antibiotics, 8(4), 261. https://doi.org/10.3390/antibiotics8040261