
Analysis of Influence of Insulating Resin Paint Film on Enameled Wire Properties Based on Molecular Simulation

Abstract
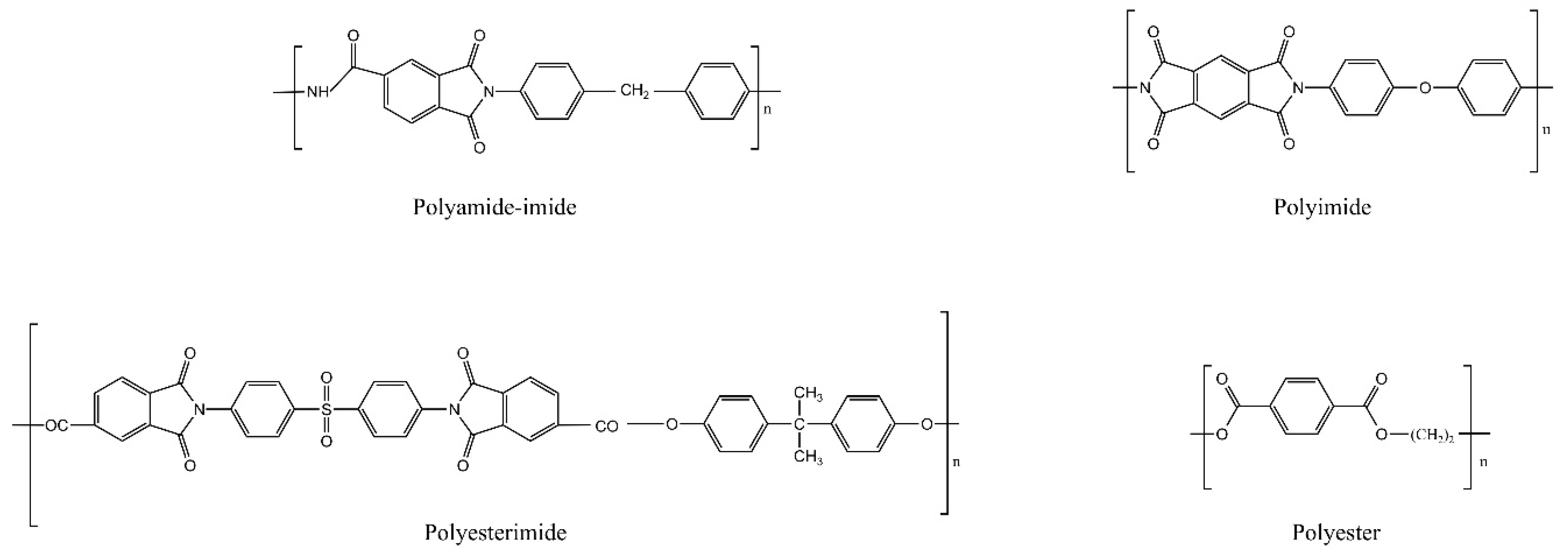
:1. Introduction
2. Computational Methods
2.1. DMol3
2.2. Forcite
3. Results and Discussions
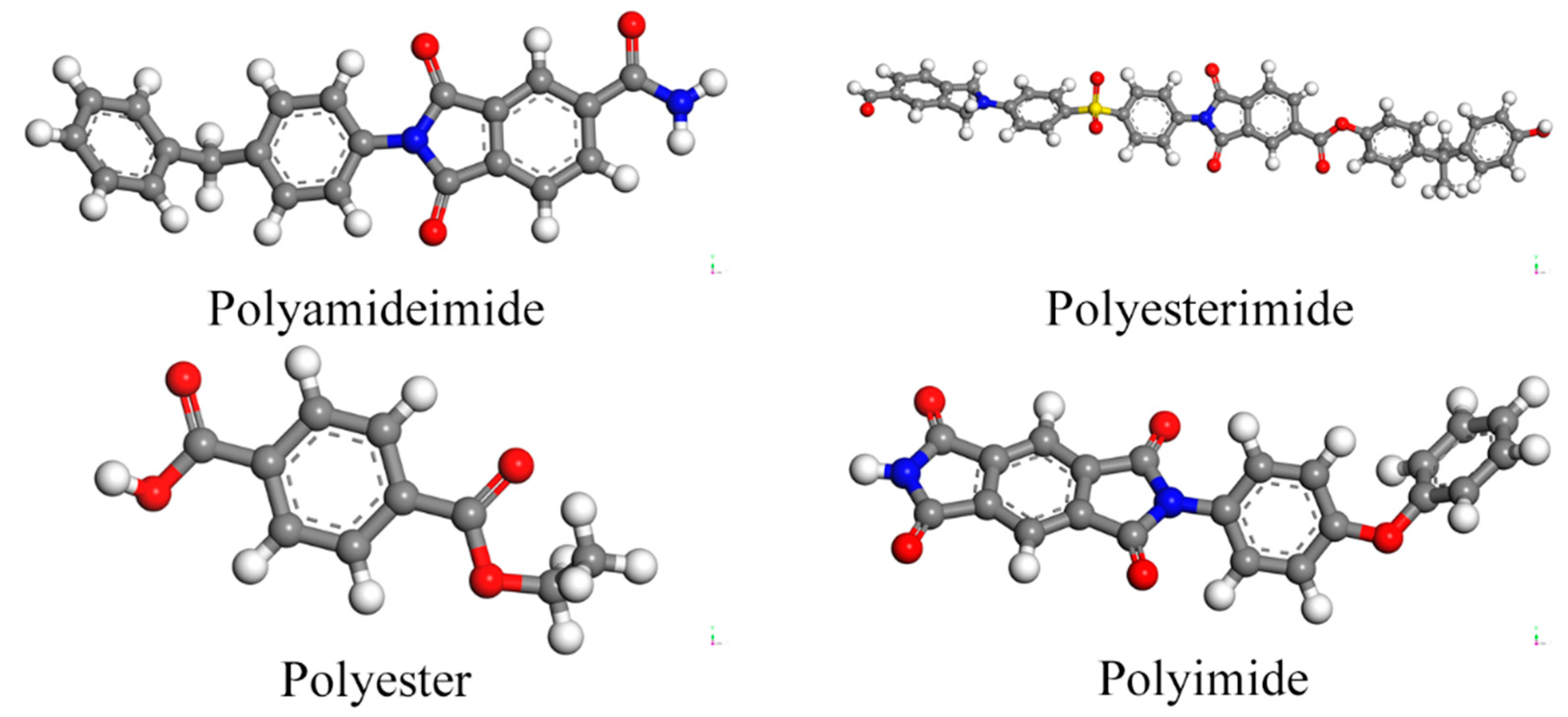
3.1. Adsorption Energy of Physical Adsorption
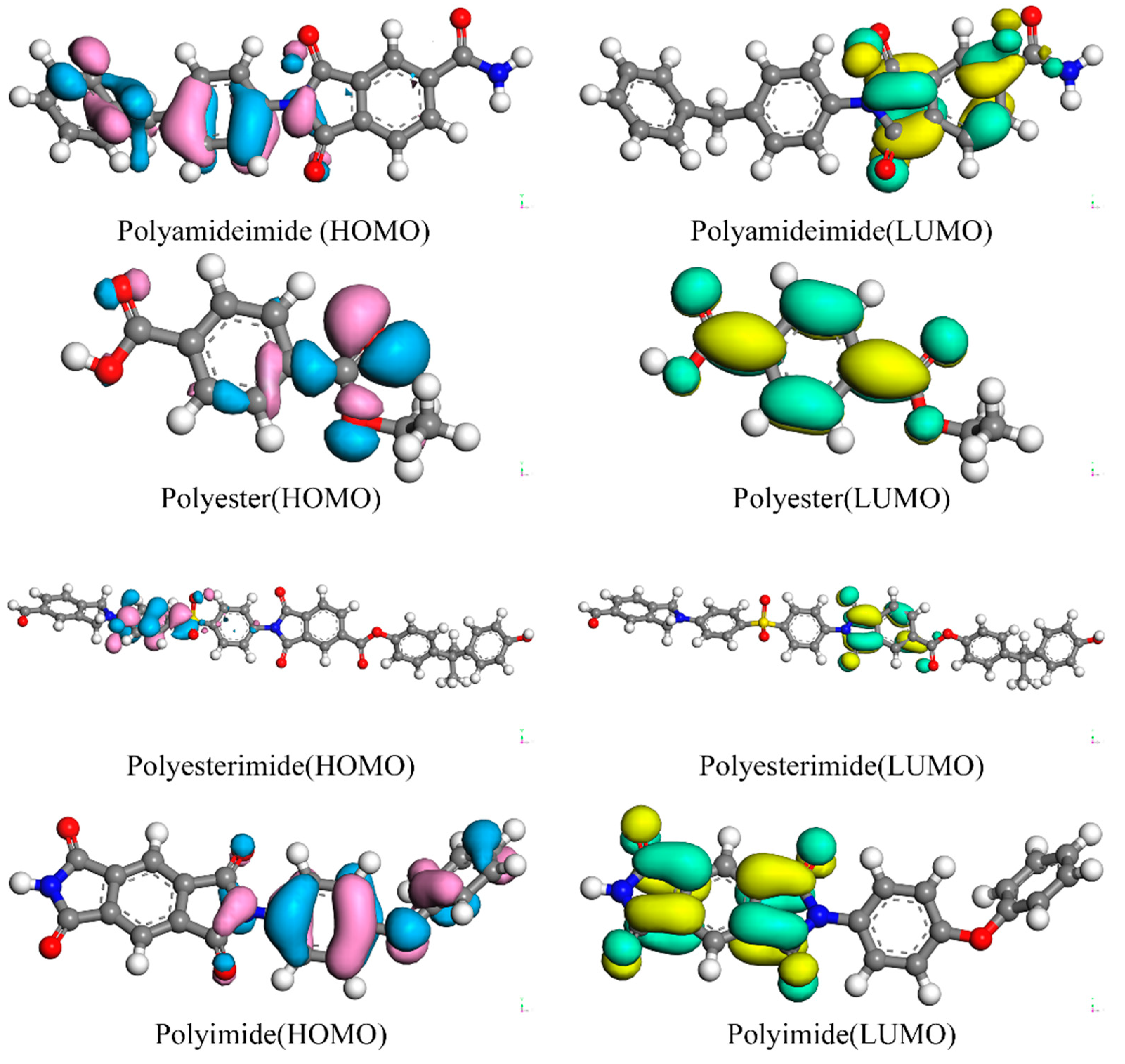
3.2. Frontier Orbitals (HOMO and LUMO)
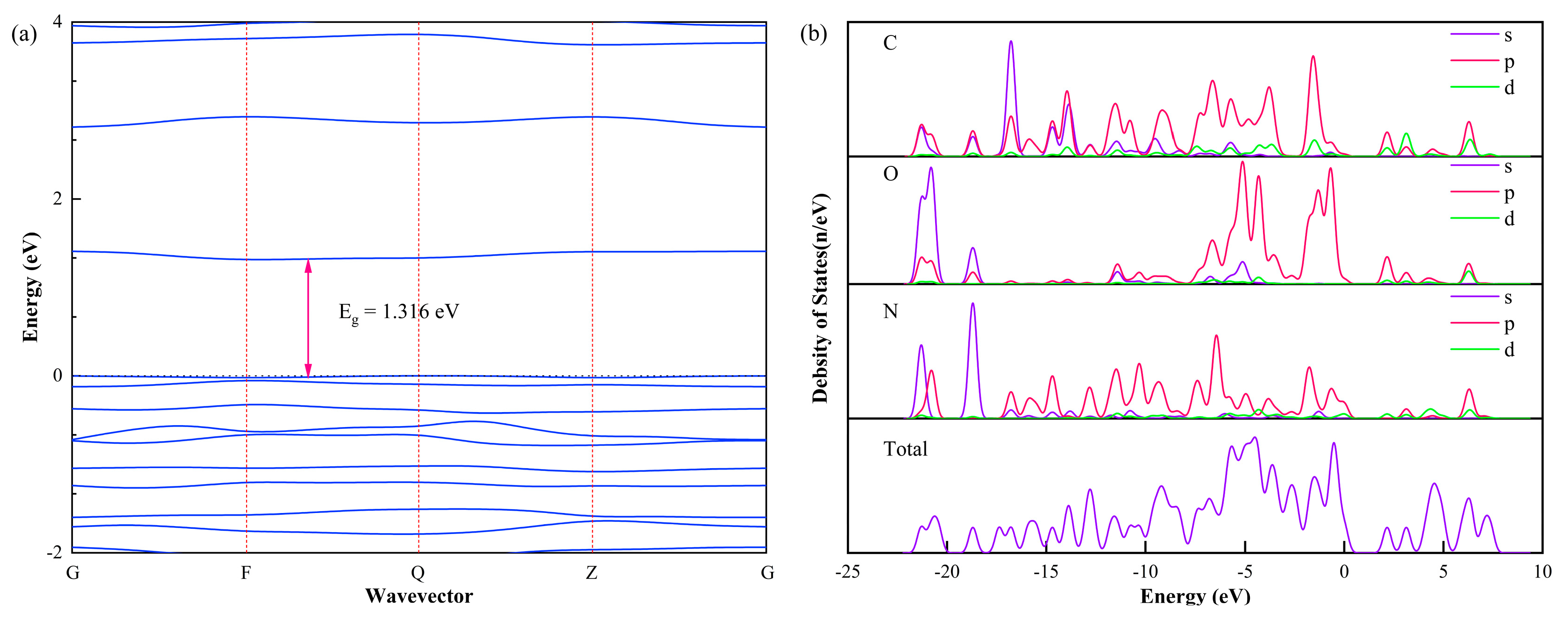
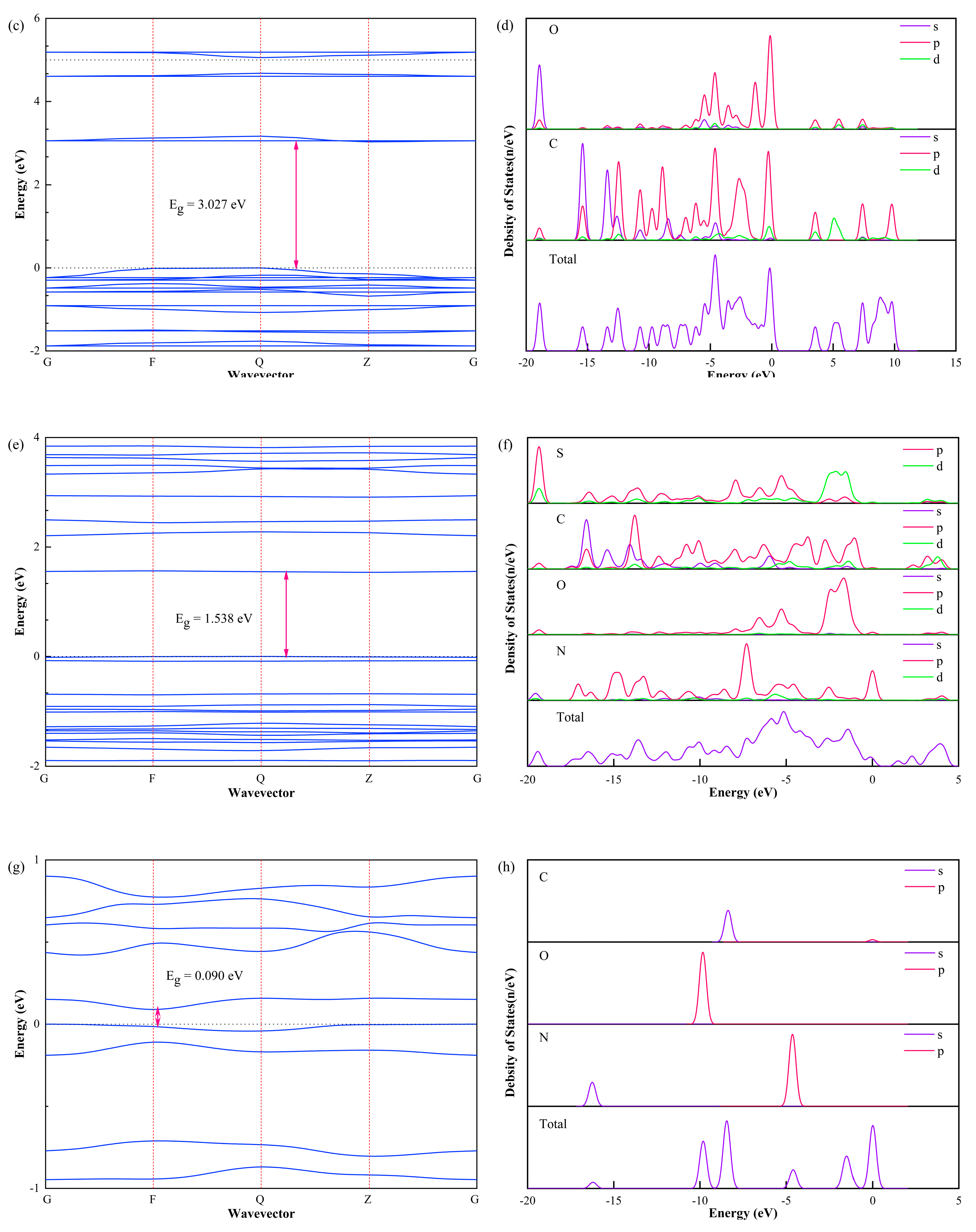
3.3. Band Structure and Density of States
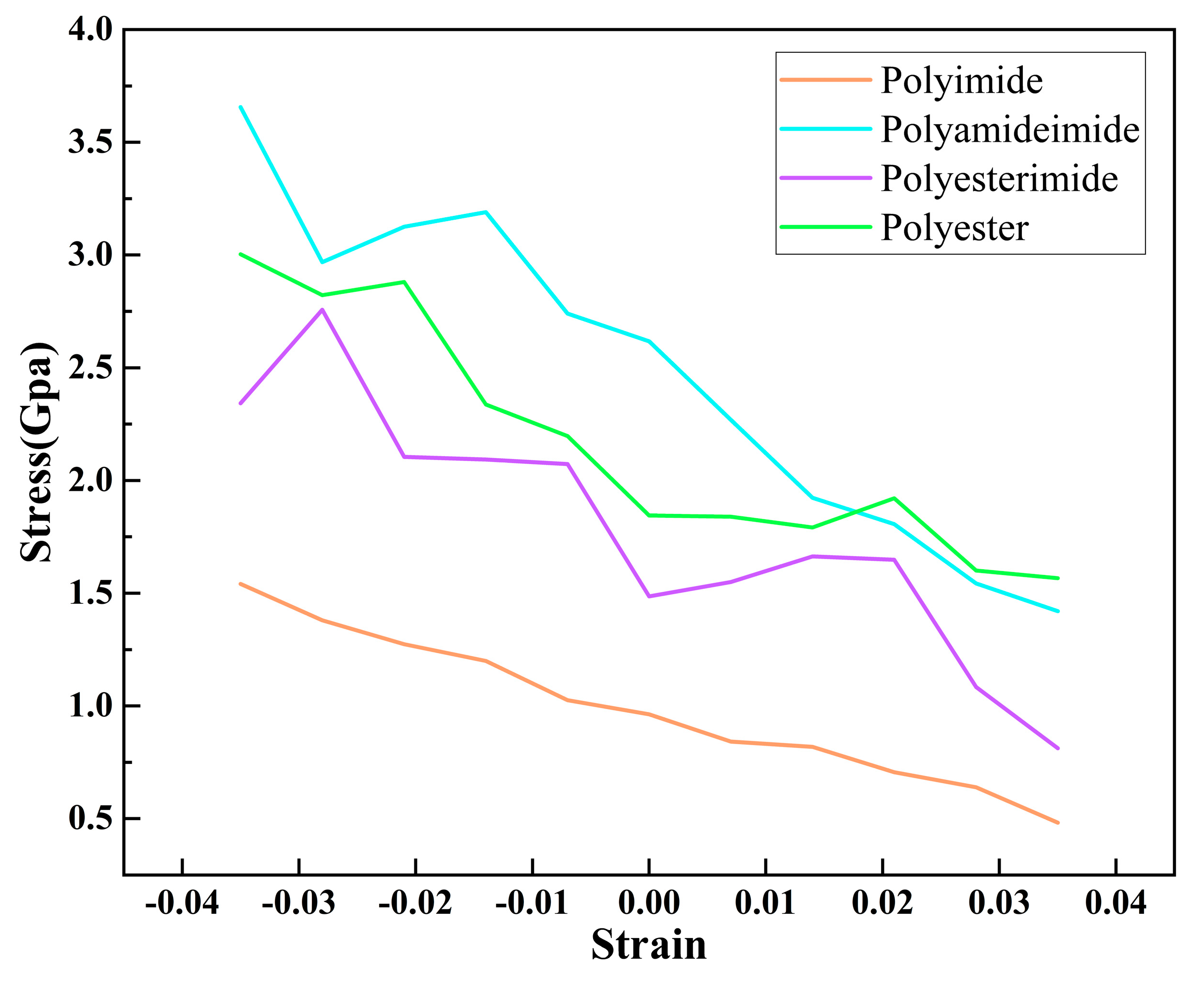
3.4. Dynamics Simulation
4. Conclusions
- The absolute value of the adsorption energy of PI and PAI molecules is relatively large, and the adsorption effect is better, so the enameled wire made of PI and PAI has better adhesion performance.
- The HOMO–LUMO gap between PET and PAI is larger, and the possibility of chemical change is lower than that of PEI and PI, indicating that PET and PAI are more stable.
- The band gap of PET is the largest among the four materials, followed by PEI, and the band gap of PI molecule is the smallest. Therefore, we can give preference to PET insulating paint when the insulating paint of the enameled wire has high requirements for insulation performance.
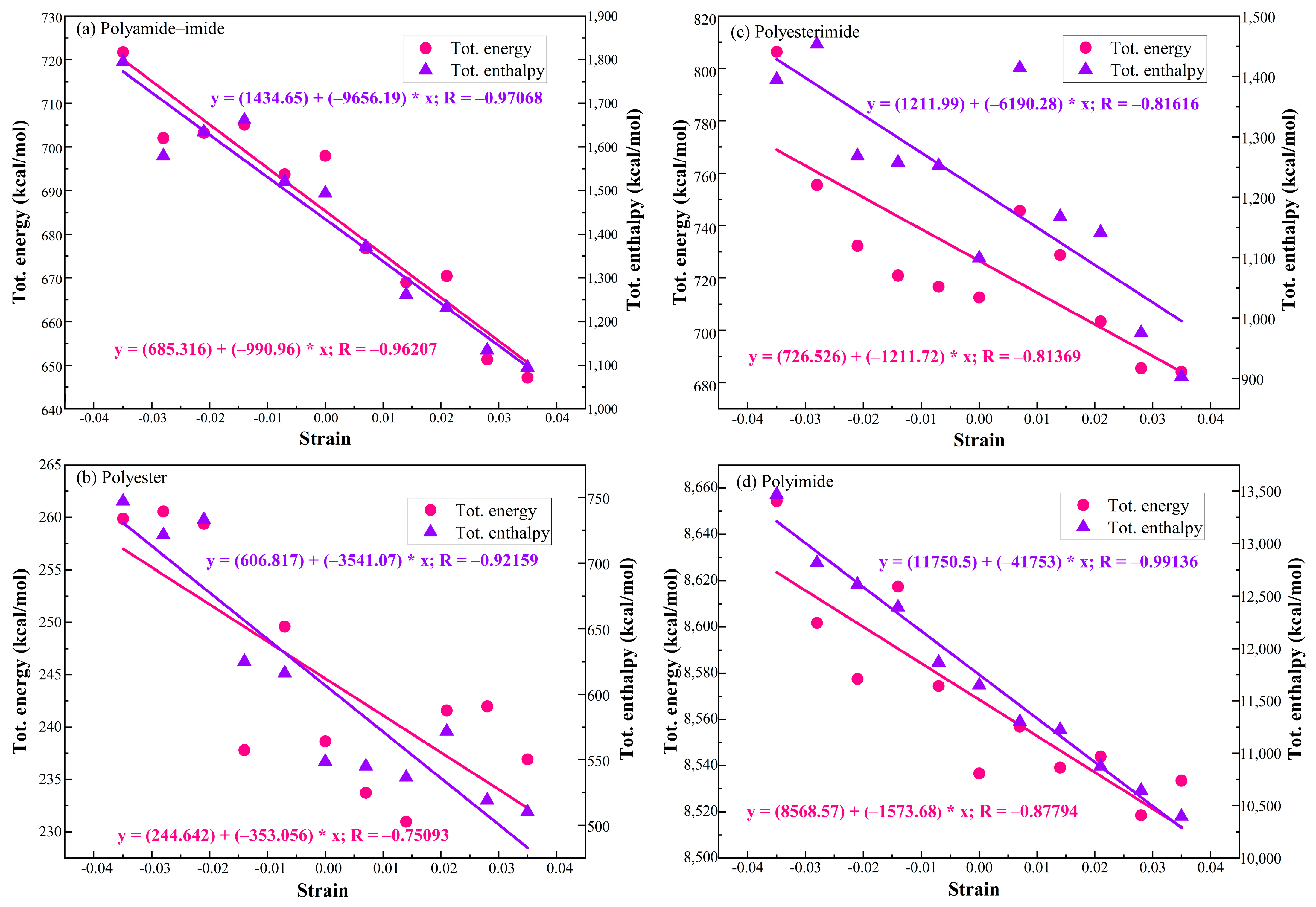
- The total energy and enthalpy of PAI, PET, PEI, and PI increases with the increase of compressive strain and decrease with the increase of tensile strain. The correlation between energy and strain of PAI and PI is relatively high, and the flexibility of the enameled wire made of PAI and PI resin paint is better than that of PET and PEI.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, H.; Liao, H.; Zhang, X.; Ni, J. Preparation and properties of water-soluble thermosetting polyester enamelled wire enamels. Insul. Mater. 2021, 54, 34–38. [Google Scholar]
- Yang, Y.; Xu, Y.; Zhang, Q. Research progress of polyimide insulating plastics for enamelled wires. Mod. Plast. Process. Appl. 2017, 29, 60–63. [Google Scholar]
- Tan, X.; Fang, M.; Wang, X. Sorption speciation of lanthanides/actinides on minerals by TRLFS, EXAFS and DFT studies: A Review. Molecules 2010, 15, 8431–8468. [Google Scholar] [CrossRef] [PubMed]
- Gavini, V.; Bhattacharya, K.; Ortiz, M. Quasi-Continuum Orbital-Free Density-Functional Theory: A route to multi-million atom non-periodic DFT calculation. J. Mech. Phys. Solids 2007, 55, 697–718. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, Y.; Shu, X.; Wang, Y.; Ran, Q. Adsorption of organic molecules on mineral surfaces studied by first-principle calculations: A review. Adv. Colloid Interface Sci. 2018, 256, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Gu, Y. Application of molecular simulation methods in the study of the relationship between the structure and properties of polyimide. Polym. Mater. Sci. Eng. 2005, 21, 15–19. [Google Scholar]
- Yin, J. Molecular Simulation and Characterization of Nano-Hybrid Film Microstructure and Electrical Properties. Ph.D. Thesis, Harbin University of Science and Technology, Harbin, China, 2006. [Google Scholar]
- Li, K.; Li, N.; Yan, N.; Wang, T.; Zhang, Y.; Song, Q.; Li, H. Adsorption of Small Hydrocarbons on Pristine, N-Doped and vacancy graphene by DFT study. Appl. Surf. Sci. 2020, 515, 146028. [Google Scholar] [CrossRef]
- Rath, S.S.; Sinha, N.; Sahoo, H.; Das, B.; Mishra, B.K. Molecular modeling studies of oleate adsorption on iron oxides. Appl. Surf. Sci. 2014, 295, 115–122. [Google Scholar] [CrossRef]
- Liu, J. Molecular Dynamics Simulation of Mechanical Properties and Deformation Mechanisms of Metal Nanocrystalline Materials. Ph.D. Thesis, Jilin University, Changchun, China, 2021. [Google Scholar]
- Cai, Y. Research on the Compression and Resilience Performance of Carbon Nanotubes Based on Molecular Dynamics Simulation. Master’s Thesis, Inner Mongolia University of Technology, Hohhot, China, 2021. [Google Scholar]
- Ji, D.; Li, T.; Hu, W.; Fuchs, H. Recent progress in aromatic polyimide dielectrics for organic electronic devices and circuits. Adv. Mater. 2019, 31, 1806070. [Google Scholar] [CrossRef] [PubMed]
- Diaham, S.; Locatelli, M.L.; Lebey, T.; Dinculescu, S. Dielectric and thermal properties of Polyamide-Imide (PAI) films. In Proceedings of the 2009 IEEE Conference on Electrical Insulation and Dielectric Phenomena, Virginia Beach, VA, USA, 18–21 October 2009; IEEE: Virginia Beach, VA, USA, 2009; pp. 482–485. [Google Scholar]
- Diaham, S.; Locatelli, M.-L. Dielectric properties of polyamide-imide. J. Phys. D Appl. Phys. 2013, 46, 185302. [Google Scholar] [CrossRef]
- Petitgas, B.; Seytre, G.; Gain, O.; Boiteux, G.; Royaud, I.; Serghei, A.; Gimenez, A.; Anton, A. High temperature aging of enamelled copper wire—Relationships between chemical structure and electrical behavior. In Proceedings of the 2011 Annual Report Conference on Electrical Insulation and Dielectric Phenomena, Cancun, Mexico, 16–19 October 2011; IEEE: Cancun, Mexico, 2011; pp. 84–88. [Google Scholar]
- Murray, T. New Technology of Polyamide-Imide Wire Enamels. The P.D. George company: St. Louis, MO, USA. Available online: elantas.com (accessed on 15 July 2022).
- Gibbs, J.W. Elementary Principles in Statistical Mechanics Developed with Especial Reference to the Rational Foundation of Thermodynamics. Available online: https://www.ams.org/journals/bull/1906-12-04/S0002-9904-1906-01319-2/ (accessed on 17 July 2022).
- Ataman, E.; Andersson, M.P.; Ceccato, M.; Bovet, N.; Stipp, S.L.S. Functional group adsorption on calcite: I. Oxygen containing and nonpolar organic molecules. J. Phys. Chem. C 2016, 120, 16586–16596. [Google Scholar] [CrossRef]
- Ataman, E.; Andersson, M.P.; Ceccato, M.; Bovet, N.; Stipp, S.L.S. Functional group adsorption on calcite: II. Nitrogen and sulfur containing organic molecules. J. Phys. Chem. C 2016, 120, 16597–16607. [Google Scholar] [CrossRef]
- First-Principles Investigation of the Lattice Vibrations in the Alkali Feldspar Solid Solution. Available online: https://link.springer.com/article/10.1007/s00269-014-0715-8 (accessed on 16 July 2022).
- Braga, L.S.; Leal, D.H.S.; Kuca, K.; Ramalho, T.C. Perspectives on the role of the Frontier Effective-for-Reaction Molecular Orbital (FERMO) in the study of chemical reactivity: An updated review. Curr. Org. Chem. 2020, 24, 314–331. [Google Scholar] [CrossRef]
- Chidiebere, C.W.; Duru, C.E.; Mbagwu, J.C. Application of computational chemistry in chemical reactivity: A review. J. Niger. Soc. Phys. Sci. 2021, 3, 292–297. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, A.; Eslami, M. A Density functional theory study on detection of amphetamine drug by silicon carbide nanotubes. Phys. E Low-Dimens. Syst. Nanostruct. 2021, 125, 114411. [Google Scholar] [CrossRef]
- Hussain, R.; Saeed, M.; Yasir Mehboob, M.; Ullah Khan, S.; Khan, M.U.; Adnan, M.; Ahmed, M.; Iqbal, J.; Ayub, K. Density functional theory study of palladium cluster adsorption on a graphene support. RSC Adv. 2020, 10, 20595–20607. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
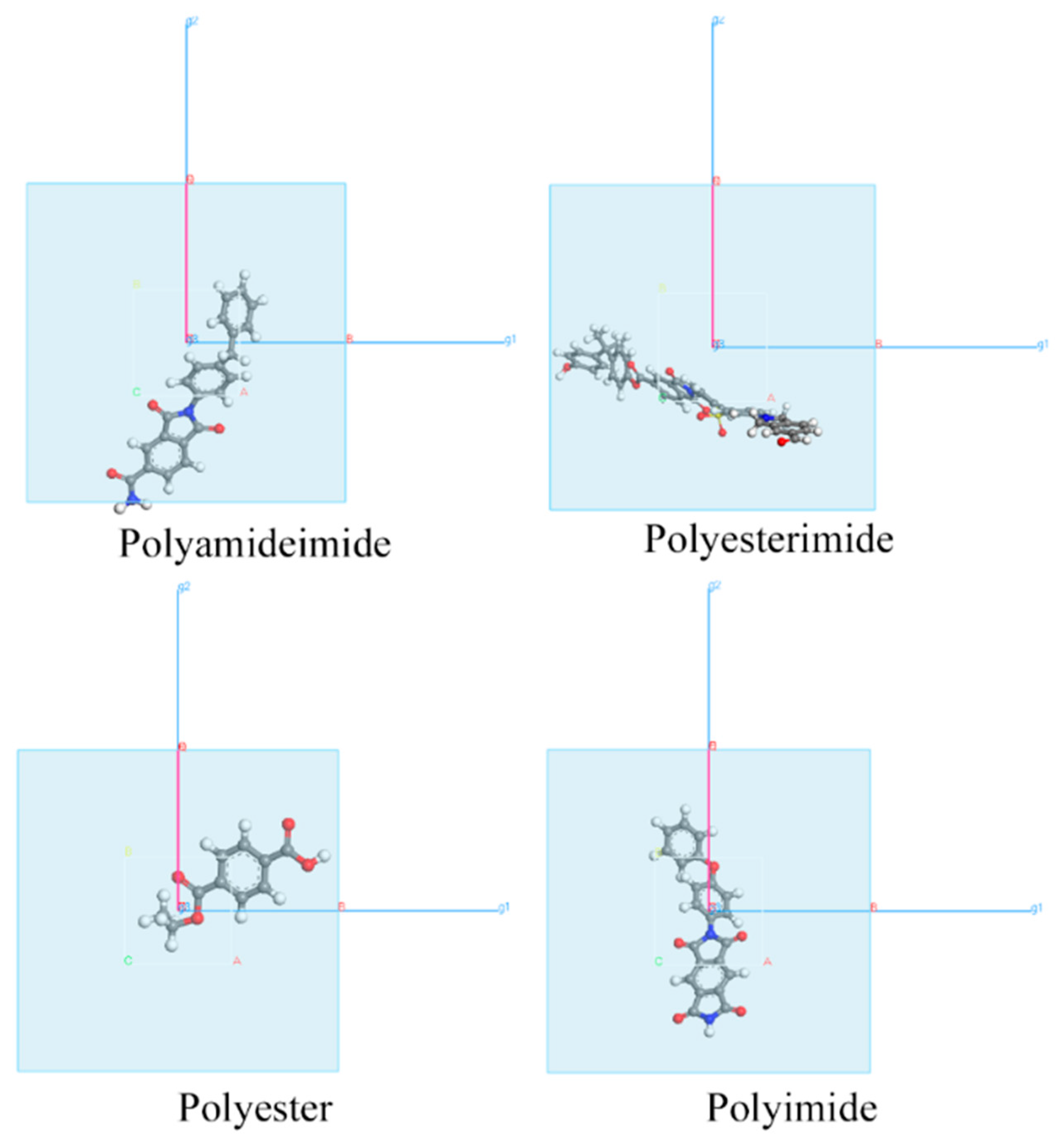{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | ||||
|---|---|---|---|---|
| Polyamide–imide | −741,827.337 | −247,235,809.960 | −247,977,765.974 | −128.677 |
| Polyester | −431,390.392 | −98,820,241.886 | −99,251,749.028 | −116.750 |
| Polyesterimide | −1,783,765.813 | −36,597,135.761 | −38,381,021.702 | −120.128 |
| Polyimide | −892,885.203 | −148,690,935.521 | −149,583,956.785 | −136.061 |
| Types | HOMO [eV] | LUMO [eV] | [eV] |
|---|---|---|---|
| Polyamide–imide | −5.5431 | −3.3525 | −2.1906 |
| Polyester | −6.4111 | −2.8654 | −3.5457 |
| Polyesterimide | −5.1866 | −3.6791 | −1.5075 |
| Polyimide | −5.3689 | −4.0600 | −1.3089 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Wu, Z.; Zhang, H.; Cheng, Y.; Ren, H. Analysis of Influence of Insulating Resin Paint Film on Enameled Wire Properties Based on Molecular Simulation. Coatings 2022, 12, 1352. https://doi.org/10.3390/coatings12091352
Zhang Z, Wu Z, Zhang H, Cheng Y, Ren H. Analysis of Influence of Insulating Resin Paint Film on Enameled Wire Properties Based on Molecular Simulation. Coatings. 2022; 12(9):1352. https://doi.org/10.3390/coatings12091352
Chicago/Turabian StyleZhang, Zhongli, Zhensheng Wu, Huiyuan Zhang, Yibin Cheng, and Hao Ren. 2022. "Analysis of Influence of Insulating Resin Paint Film on Enameled Wire Properties Based on Molecular Simulation" Coatings 12, no. 9: 1352. https://doi.org/10.3390/coatings12091352
APA StyleZhang, Z., Wu, Z., Zhang, H., Cheng, Y., & Ren, H. (2022). Analysis of Influence of Insulating Resin Paint Film on Enameled Wire Properties Based on Molecular Simulation. Coatings, 12(9), 1352. https://doi.org/10.3390/coatings12091352