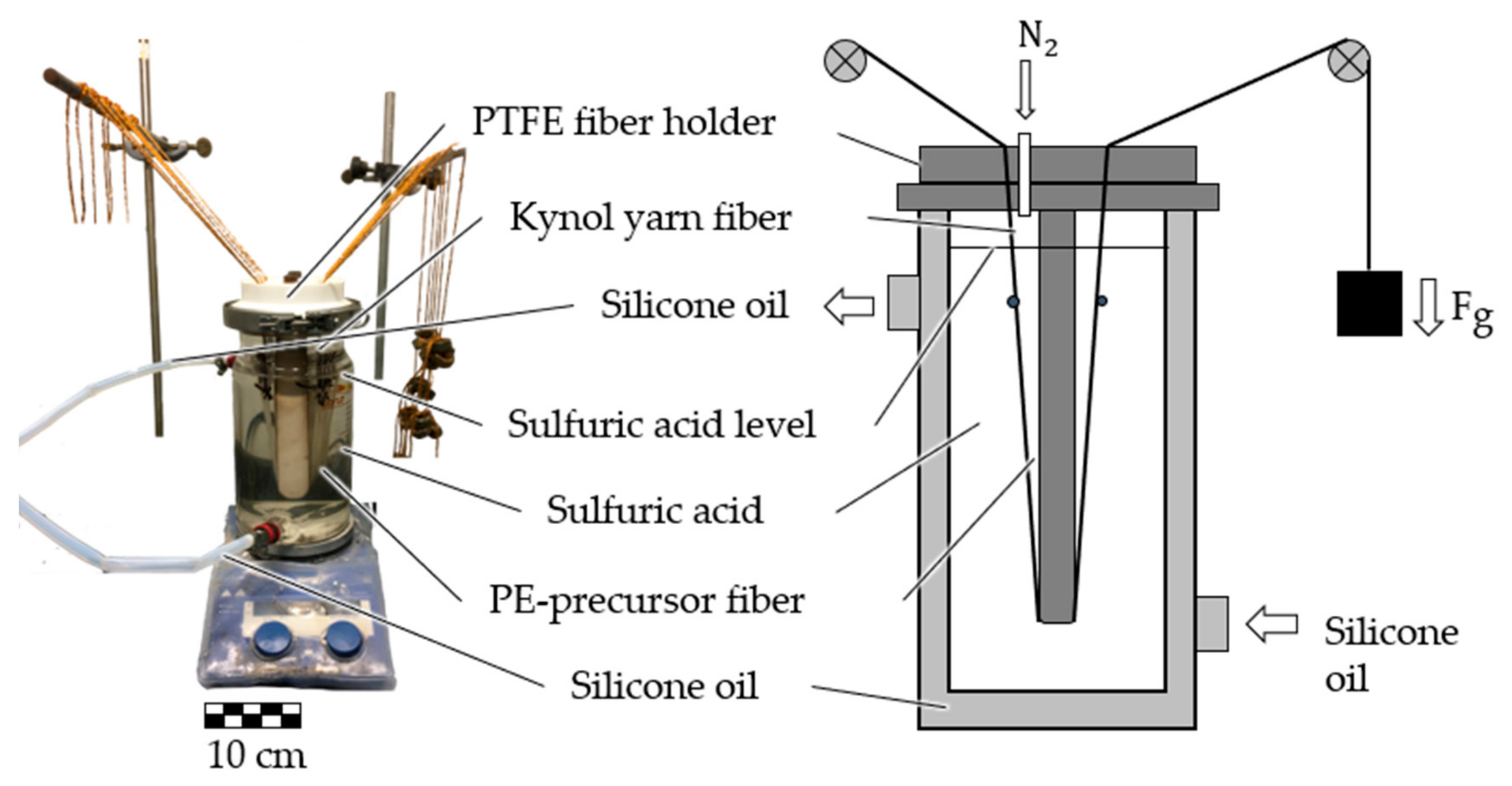
3.2.1. Batch Sulfonation with Higher Fiber Tension
In the batch sulfonation trials with increased fiber tension, the dissolution of PA6 and the sulfonation steps are analyzed. The stabilization parameters, TGA and gas pycnometry results are listed in
Table 11.
The HDPE-INS precursor has a density of 1.073 ± 0.001 g/cm3. The TGA measurements show a mass loss value of 84% for ΔmPE and 99% for Δmtotal. For the 40/2 samples, the density increases with increasing sulfonation time. The density values are 1.006 ± 0.001 g/cm3 (1 h), 1.044 ± 0.002 g/cm3 (2 h), 1.212 ± 0.002 g/cm3 (3 h) and 1.397 ± 0.002 g/cm3 (4 h). Initially, the density drops below the precursor density due to the dissolution of PA6, which has a higher density (1.12–1.15 g/cm3). After 3 h of sulfonation, the fiber density considerably increases due to cross-linking and integration of sulfuric acid groups. The cross-linking of the polymer chains contributes to the thermal stability of the PE. As the cross-linking increases, the mass loss during TGA decreases, specifically in the temperature range of 400–600 °C. The TGA results of the 40/2 samples reflect this trend, showing a decrease in ΔmPE and Δmtotal values with longer sulfonation time. The values decrease from 83%/98% (1 h) to 76%/92% (2 h), 38%/69% (3 h), and 17%/60% (4 h).
Similarly, the 60/2 samples exhibit an increasing trend in density with increasing sulfonation time. The density values are 1.020 ± 0.001 g/cm3 (1 h), 1.062 ± 0.002 g/cm3 (2 h), 1.235 ± 0.002 g/cm3 (3 h), and 1.416 ± 0.003 g/cm3 (4 h). The dissolution of PA6 at 60 °C appears to result in a higher degree of cross-linking during sulfonation, possibly due to faster dissolution of PA6 or enhanced swelling of the PE filaments due to uptake of sulfur-containing groups. The faster removal of PA6 allows for easier and faster dissolution of sulfuric acid into the PE filaments, leading to higher cross-linking and density values. The TGA analysis of the 60/2 samples shows increasing ΔmPE/Δmtotal values, which are lower than those of the 40/2 sample. The values for the 60/2 sample are: 78%/95% (1 h), 74%/91% (2 h), 42%/70% (3 h) and 19%/58% (4 h).
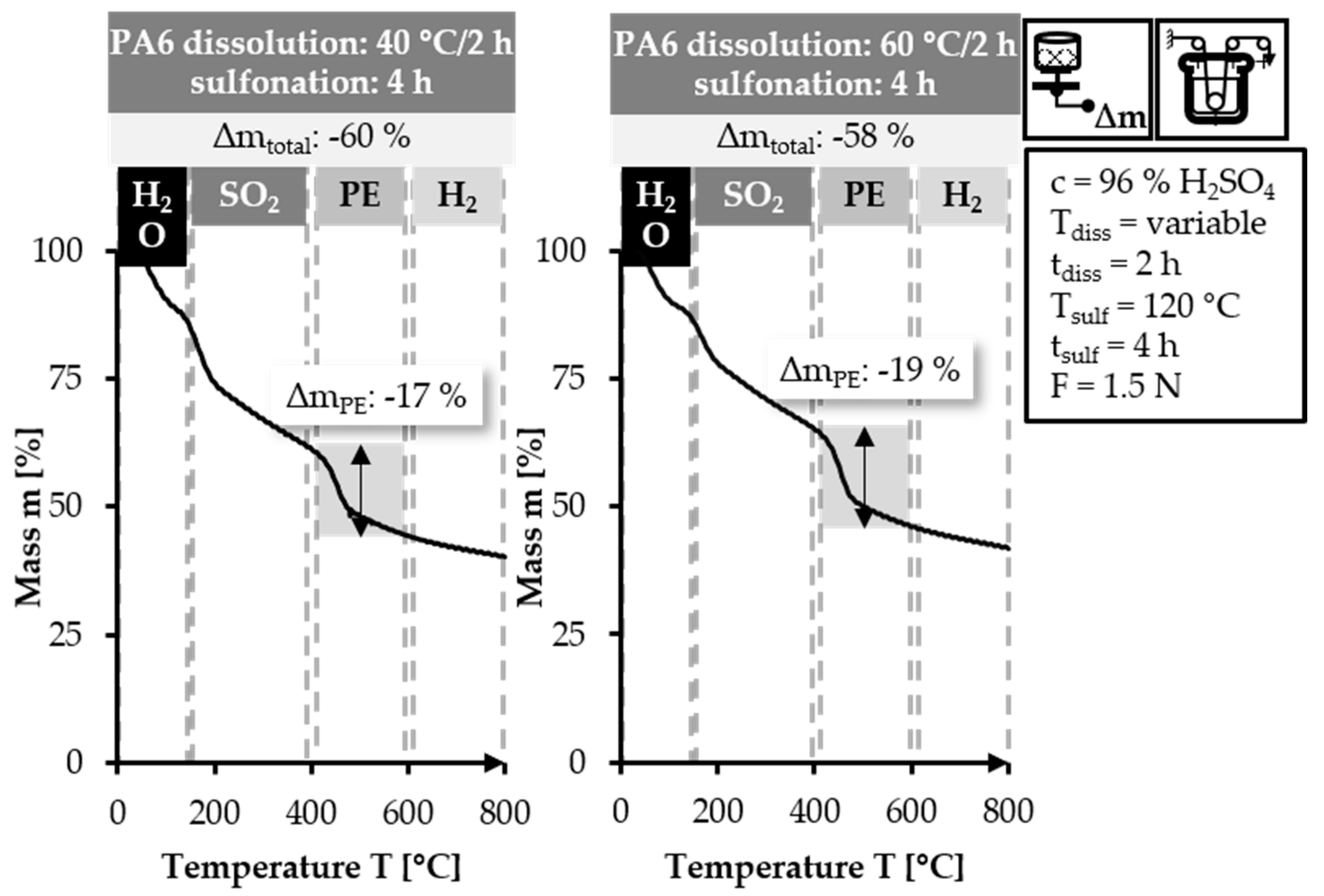
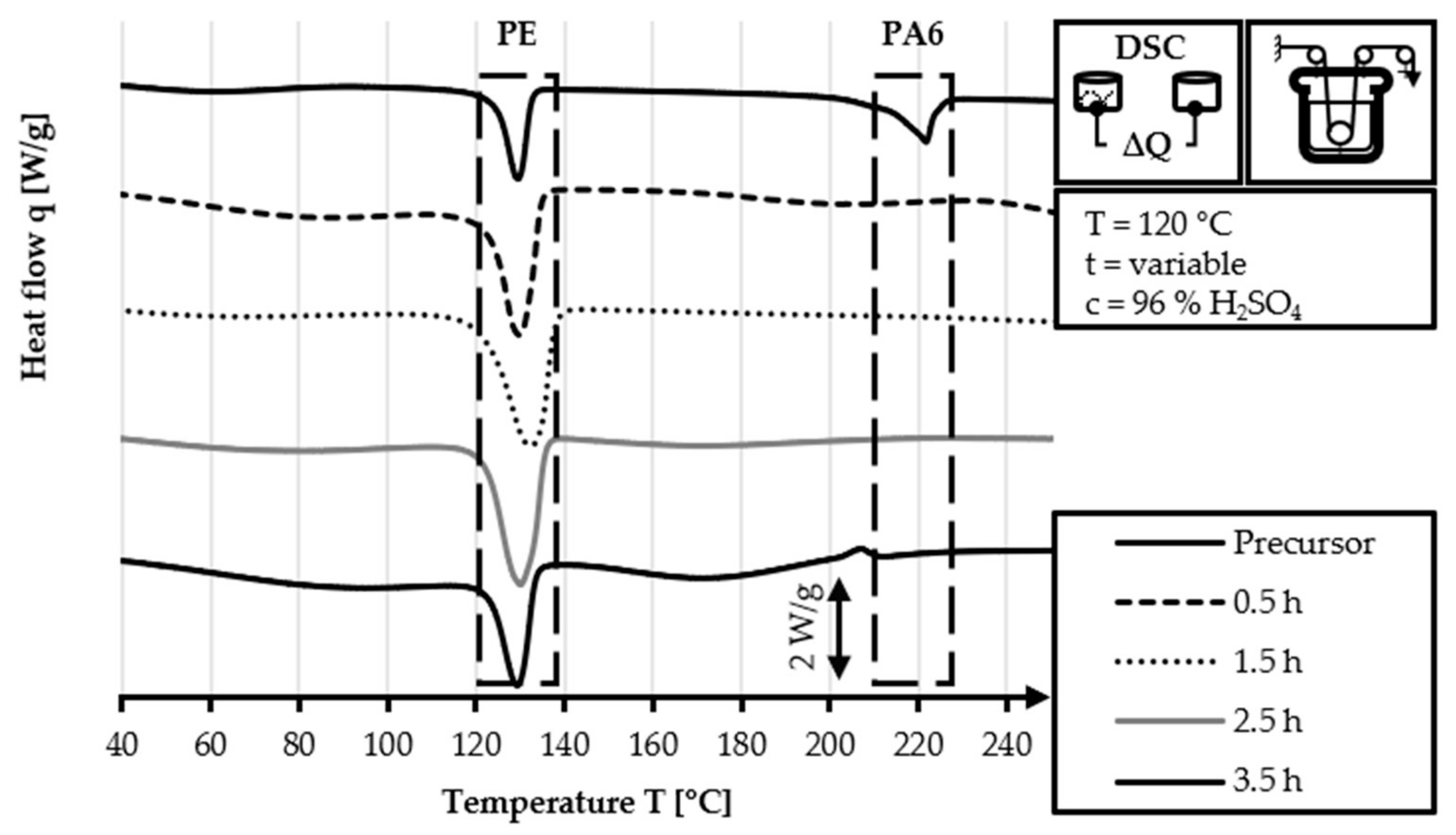
The TGA curves in
Figure 10 are similar to each other. The 40/2 sample exhibits a slightly steeper weight loss in the 120–400 °C range. In the temperature range of 400–600 °C, the 40/2 sample has a lower mass loss value (17%) compared to the
60/2 sample (19%). The mass loss in the final temperature range of 600–800 °C is similar in both samples. The two samples shown in
Figure 10 have higher values for Δm
PE and Δm
total than the equivalent samples that are stabilized with lower fiber tension. This is caused by the higher applied fiber tension, which impedes the diffusion of the sulfuric acid into and through the PE filaments [
34,
38]. This leads to a lower degree of cross-linking levels for the same reaction time and is indicated by higher values for Δm
PE and Δm
total. Nevertheless, the higher fiber tension is preferred, as it leads to enhanced parallel alignment of the polymer chains and consequently improved mechanical properties.
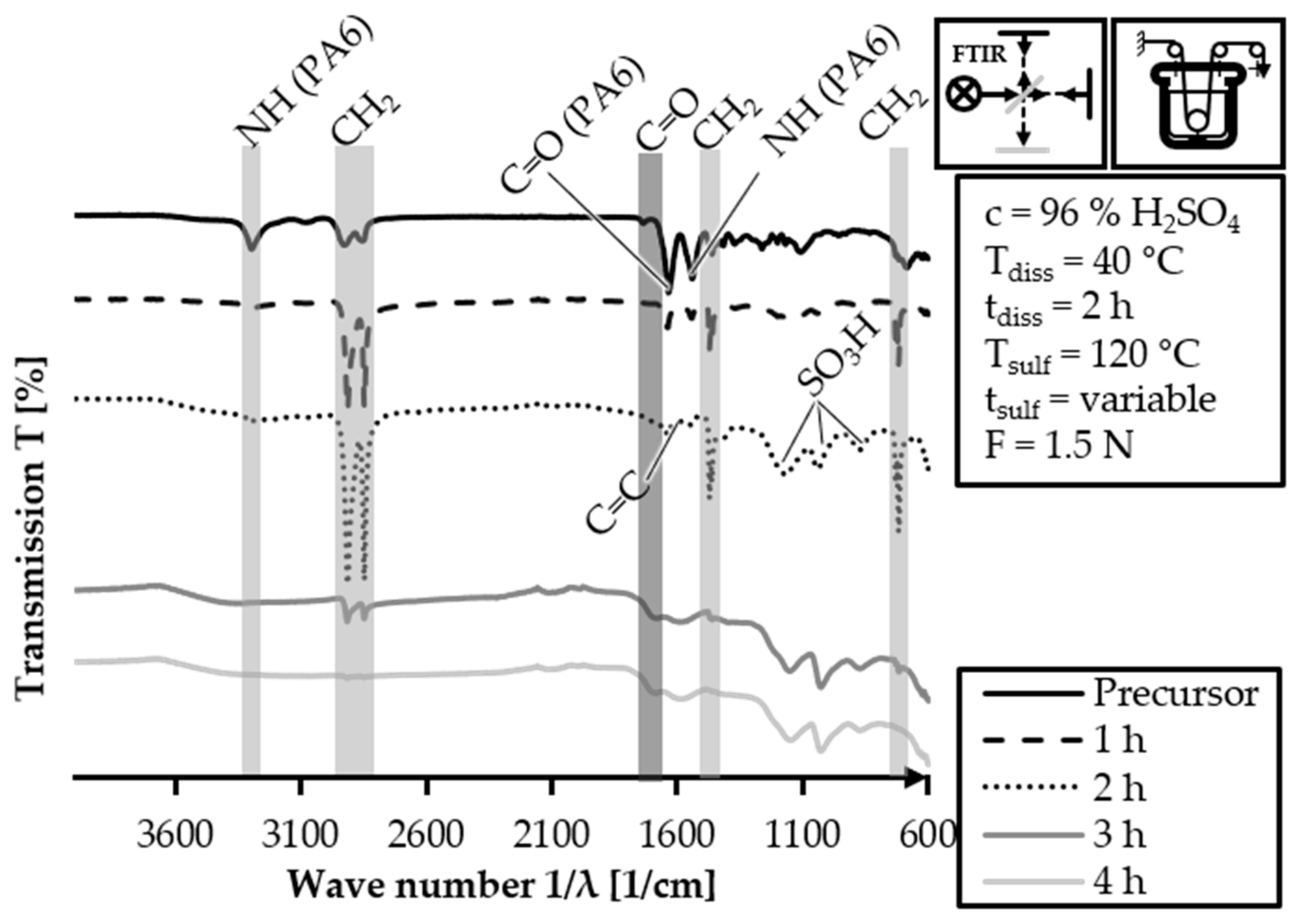
The batch sulfonation trial samples with HDPE-INS are analyzed via FTIR to investigate the evolution of functional groups in the fiber. The FTIR spectra of the HDPE-INS precursor fiber and of the 40/2 samples for different sulfonation times are shown in a stacked manner in
Figure 11. The FTIR spectrum of the HDPE-INS precursor exhibits signals related to PE and PA6. The evolution of the structure of sulfonated PE throughout the sulfonation reaction [
21] and the resulting FTIR signals [
34] have been reported previously in detail in the literature. The PE-related signals appear in the regions of 3000–2840 cm
−1 (C-H stretching), 1470–1460 cm
−1(CH2 deformation), and 770–720 cm
−1 (CH2 deformation). Additionally, PA6-related peaks are observed at 3300 cm
−1 (N-H stretching), 1650 cm
−1 (C=O stretching) and 1550 cm
−1 (N-H stretching). In the spectrum of the 1 h sulfonation sample, the PA6-related peaks are reduced compared to the precursor, but they are still present. This is unexpected since the pretreatment dissolution step should have eliminated any PA6 peaks. However, there is an increase in the intensity of PE-related peaks (3000–2840 cm
−1; 1470–1460 cm
−1; and 770–720 cm
−1) and the appearance of subtle peaks related to sulfones (1150–1050 cm
−1: S=O symmetric and asymmetric stretching), indicating the occurrence of sulfonation reactions. In the 2 h sulfonation sample, the PA6-related peaks further diminish in intensity (3300 cm
−1; 1650 cm
−1; 1550 cm
−1), with only very low intensity observed. The alkane-related peaks of PE at 3000–2840 cm
−1 (C-H stretching) and 1470–1460 cm
−1 (CH
2 deformation) become more prominent in this sample. The sulfone-related peak intensities increase (1150–1050 cm
−1: S=O symmetric and asymmetric stretching) compared to the 1 h sulfonation sample. In the FTIR spectrum of the 3 h sulfonation sample, the PA6 related peaks are no longer observable. The intensity of the alkane related peaks decreases (3000–2840 cm
−1 and 1470–1460 cm
−1) compared to the 2 h sulfonation sample, indicating increased cross-linking and sulfonation degree within the fiber. This is demonstrated by the increased intensity of sulfone peaks (1150–1050 cm
−1), the appearance of peaks created by the C=C stretching of double bonds in the polymer chains (1620–1570 cm
−1), and peaks related to C=O stretching of carboxylic groups (1710–1690 cm
−1). The spectrum of the 4 h sulfonation sample shows no peaks related to PA6 and significantly reduced alkane-related peaks (3000–2840 cm
−1 and 1470–1460 cm
−1). The sulfone related peaks (1150–1050 cm
−1), the double bond peaks (1620–1570 cm
−1) and the carboxylic group related peak (1710–1690 cm
−1) are clearly observable. These results indicate an advanced sulfonation and cross-linking degree within the fiber. Overall, the FTIR analysis confirms the progression of sulfonation and cross-linking reactions during the sulfonation process, leading to the formation of sulfone groups and increased cross-linking within the fiber samples.
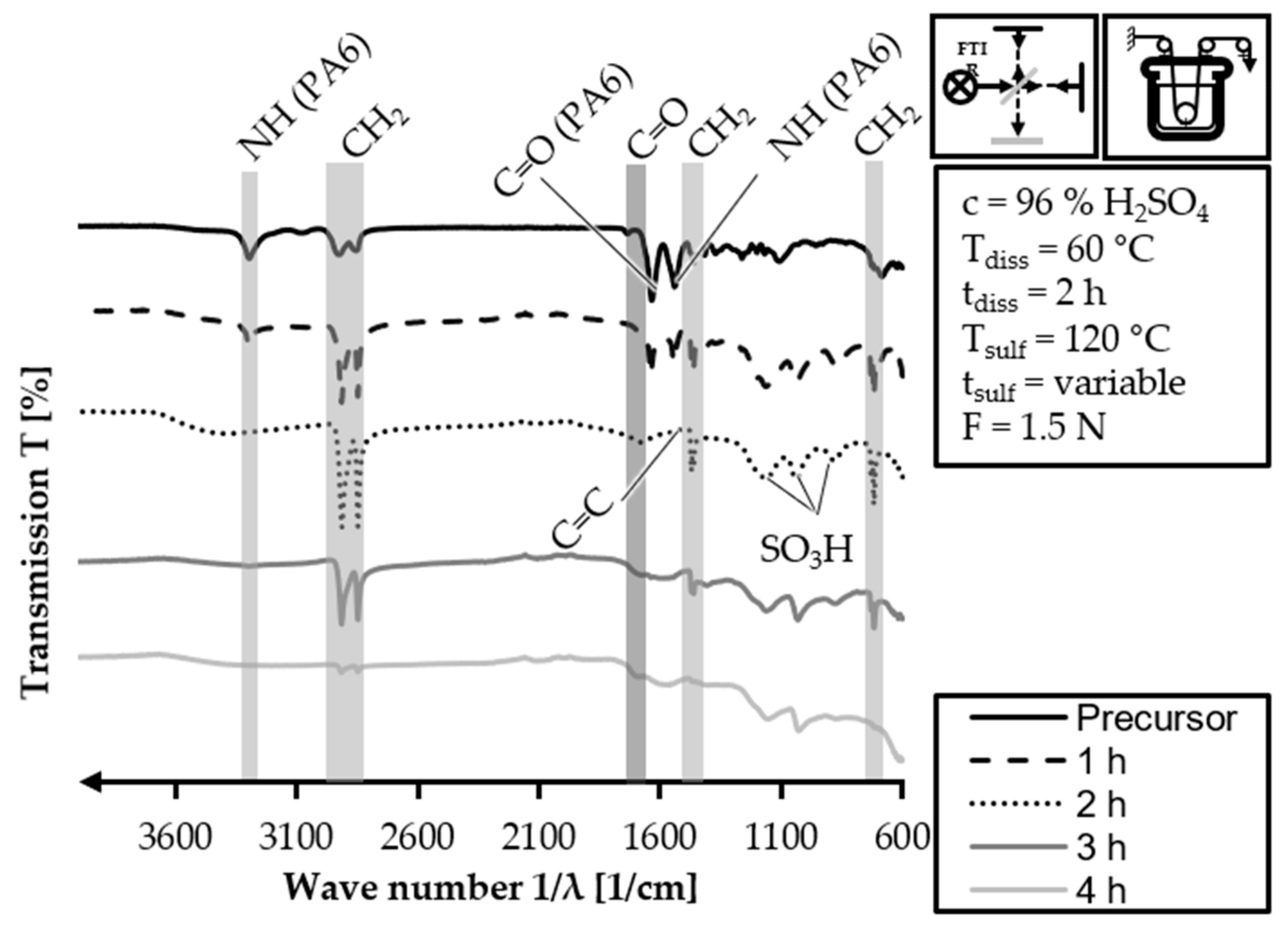
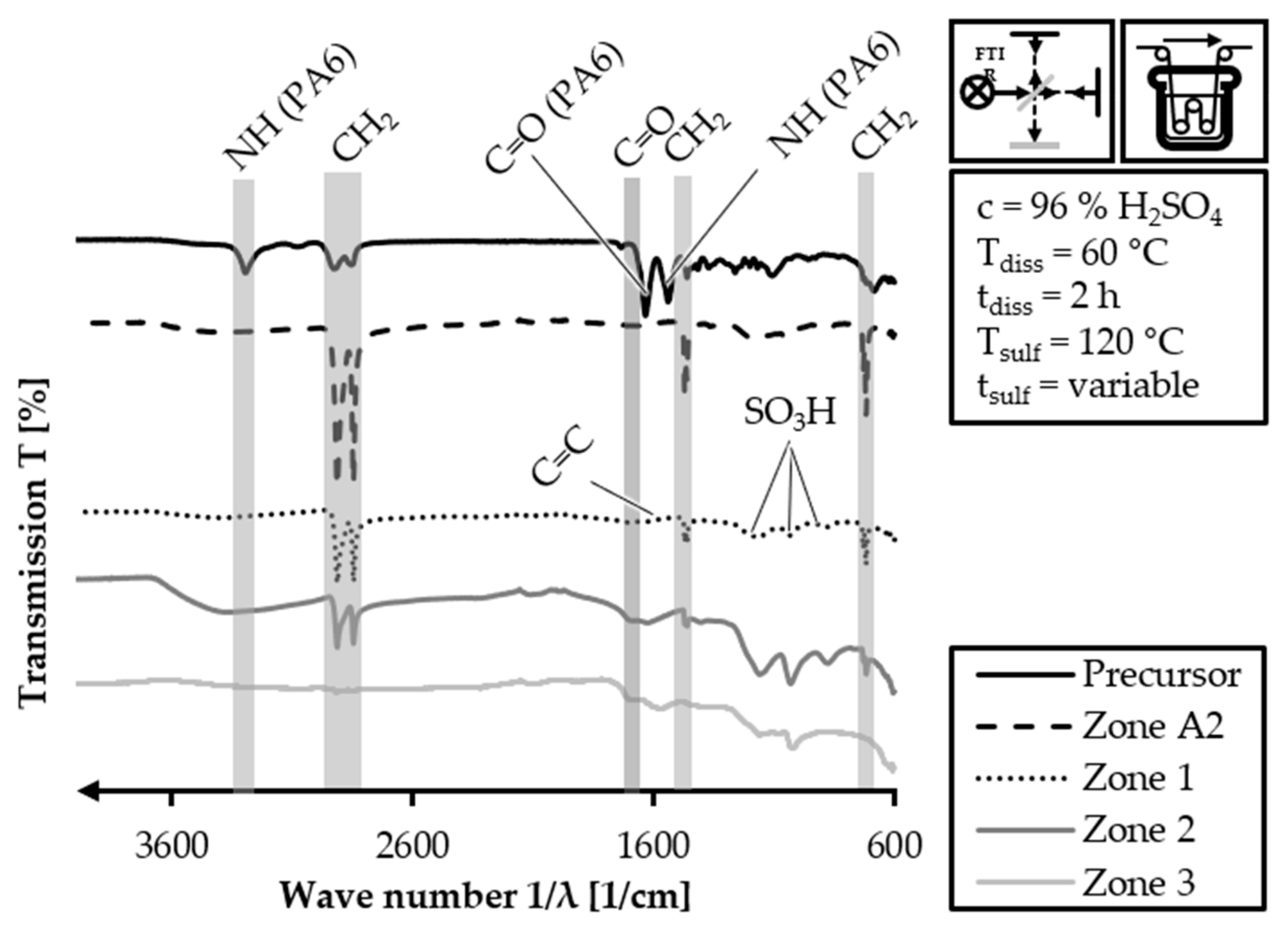
The FTIR analysis is also carried out for the
60/2 samples of the batch sulfonation trials with the HDPE-INS precursor. The stacked FTIR spectra of the
60/2 samples and the HDPE-INS precursor are shown in
Figure 12. The FTIR spectrum of the HDPE-INS precursor is consistent with what was expected and described in
Figure 11. However, the presence of PA6 related peaks in the 1 h sulfonation sample (3300 cm
−1; 1650 cm
−1; 1550 cm
−1) suggests that the pretreatment did not completely dissolve PA6. The alkane related peaks (3000–2840 cm
−1; 1470–1460 cm
−1; and 770–720 cm
−1) exhibit increased intensity, and the spectrum presents the sulfone related peaks (1150–1050 cm
−1) induced by the sulfonation process. In the 2 h sulfonation sample spectrum, there are no observable PA6 related peaks. The alkane- (3000–2840 cm
−1; 1470–1460 cm
−1; and 770–720 cm
−1) and sulfone- (1150–1050 cm
−1) related peaks show increased intensity compared to the 1 h sulfonation sample. Additionally, a peak corresponding to the C=O stretching of the carboxylic groups (1710–1690 cm
−1) introduced into the polymer via sulfonation is present. In the 3 h sulfonation sample, the PA6 related peaks are absent. Furthermore, the intensity of the alkane-related peaks decreases (3000–2840 cm
−1; 1470–1460 cm
−1; and 770–720 cm
−1), while a peak for C=C stretching of cross-linking induced carboxylic groups (1620–1570 cm
−1) becomes observable. The peak for the carboxylic groups induced by sulfonation (1710–1690 cm
−1) is also present. The FTIR spectrum of the 4 h sulfonation sample does not exhibit any peaks related to PA6, and the intensities of the alkane-related peaks are significantly reduced (3000–2840 cm
−1; 1470–1460 cm
−1; and 770–720 cm
−1). However, the peaks for the carboxylic groups, double bonds and sulfones in the fiber polymers show a slight increase compared to the 3 h sulfonation sample. These trends indicate that the cross-linking and sulfonation degree of the fiber increase with longer sulfonation reaction time, as expected.
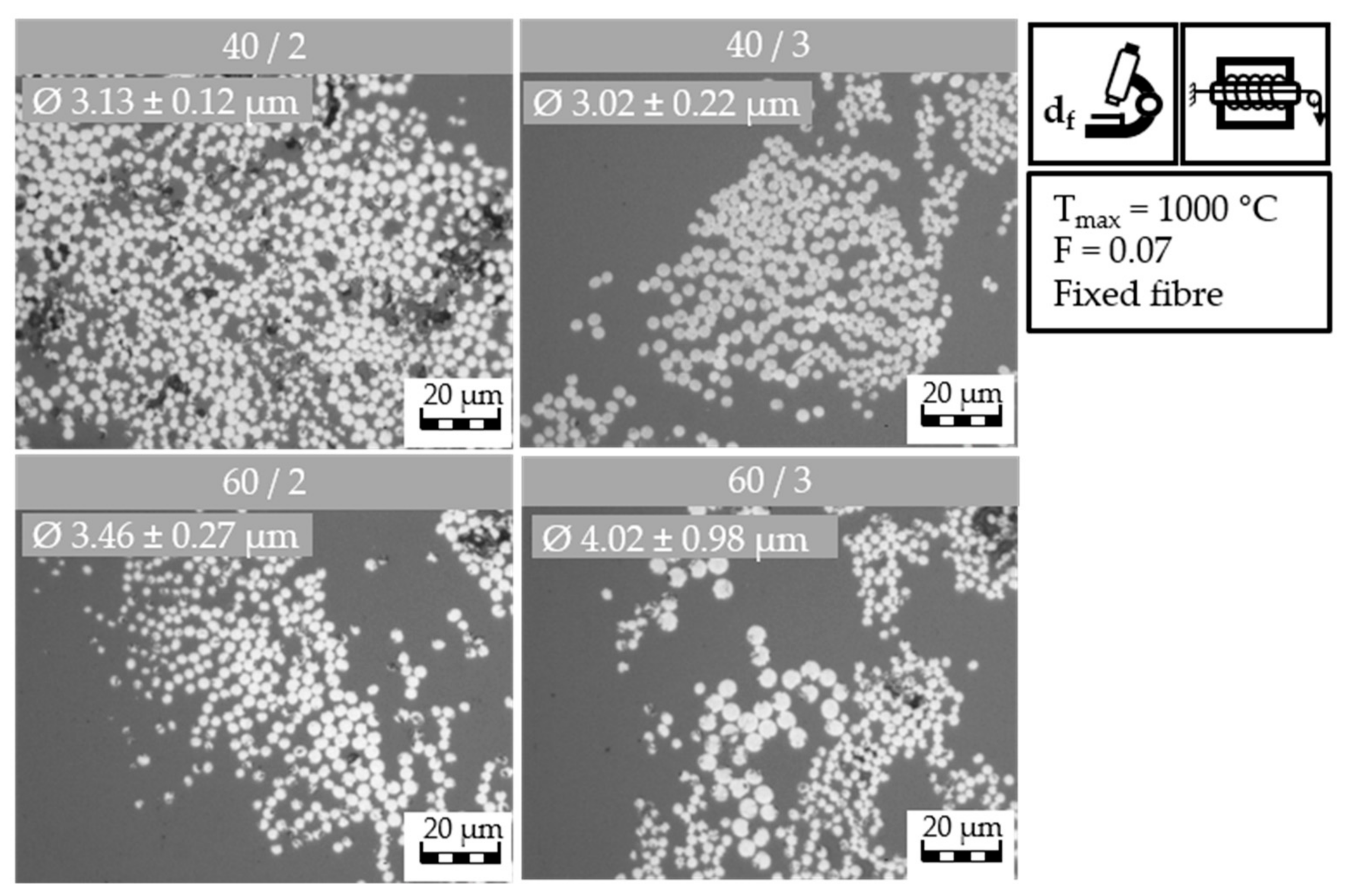
The samples from the batch sulfonation trials with the HDPE-INS precursor are investigated using light microscopy. The images can be found in the appendix (
Figure A5 and
Figure A6), while the measured filament diameters of the
40/2 and
60/2 samples are shown in
Table 12.
The 40/2 samples all present round filaments with the following filament diameters: 3.09 ± 0.37 μm (1 h), 3.48 ± 0.47 μm (2 h), 3.60 ± 0.26 μm (3 h), and 3.97 ± 0.3 μm (4 h). The filament diameter increases with longer sulfonation time due to swelling and incorporation of sulfur-containing groups. In the light microscopy image of the 1 h sulfonation sample of the 60/2 samples, filaments with a diameter of 3.61 ± 0.29 μm are observed. The 2 h sample in the light microscopy image shows measured filament diameters of 4.10 ± 0.36 μm. These two samples again exhibit some blended contrast, possibly caused by the reaction of the remaining sulfuric acid with the resin used during sample preparation. The 3 h sulfonation sample does not show this effect; instead, isolated and clearly defined mostly round filaments with a filament diameter of 4.05 ± 0.50 μm are observed. Lastly, the image of the 4 h sample presents round filaments with a cross-section diameter of 4.05 ± 0.30 μm. The filament diameter increases to a lesser degree after 2 h of sulfonation, as cross-linking reactions happen parallel to the uptake of new sulfur-containing groups, thereby mitigating the swelling effect. The increase in filament diameter is caused by swelling and the incorporation of sulfur-containing groups. The 60/2 samples exhibit a larger filament diameter compared to the 40/2 samples, suggesting that the fibers incorporate a higher number of sulfur containing groups, which result in higher water uptake through the hygroscopic character of sulfur-containing groups. This could potentially result in a higher sulfonation degree, which could lead to a higher cross-linking degree of the fibers and ultimately result in CF with improved mechanical properties after carbonization.
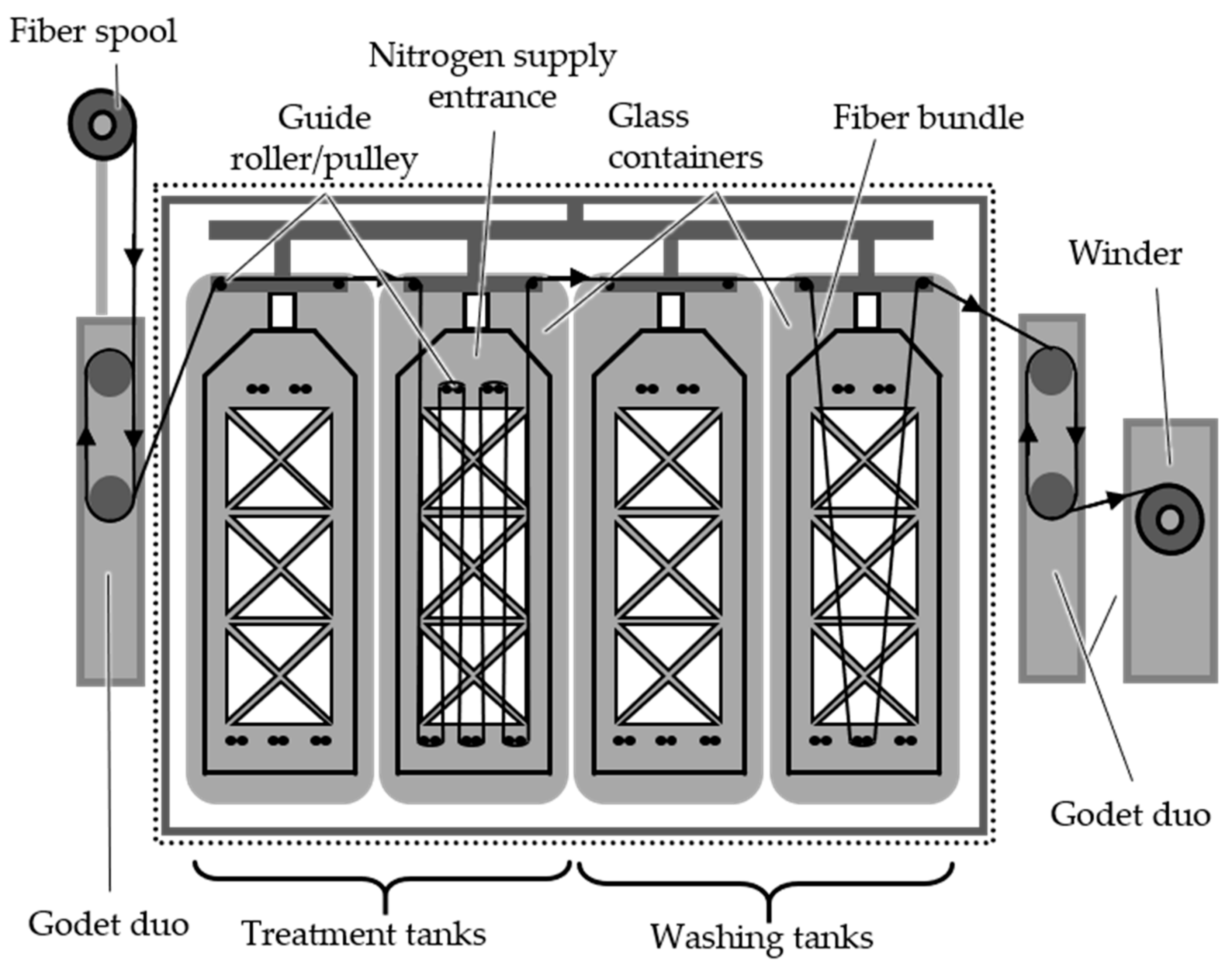
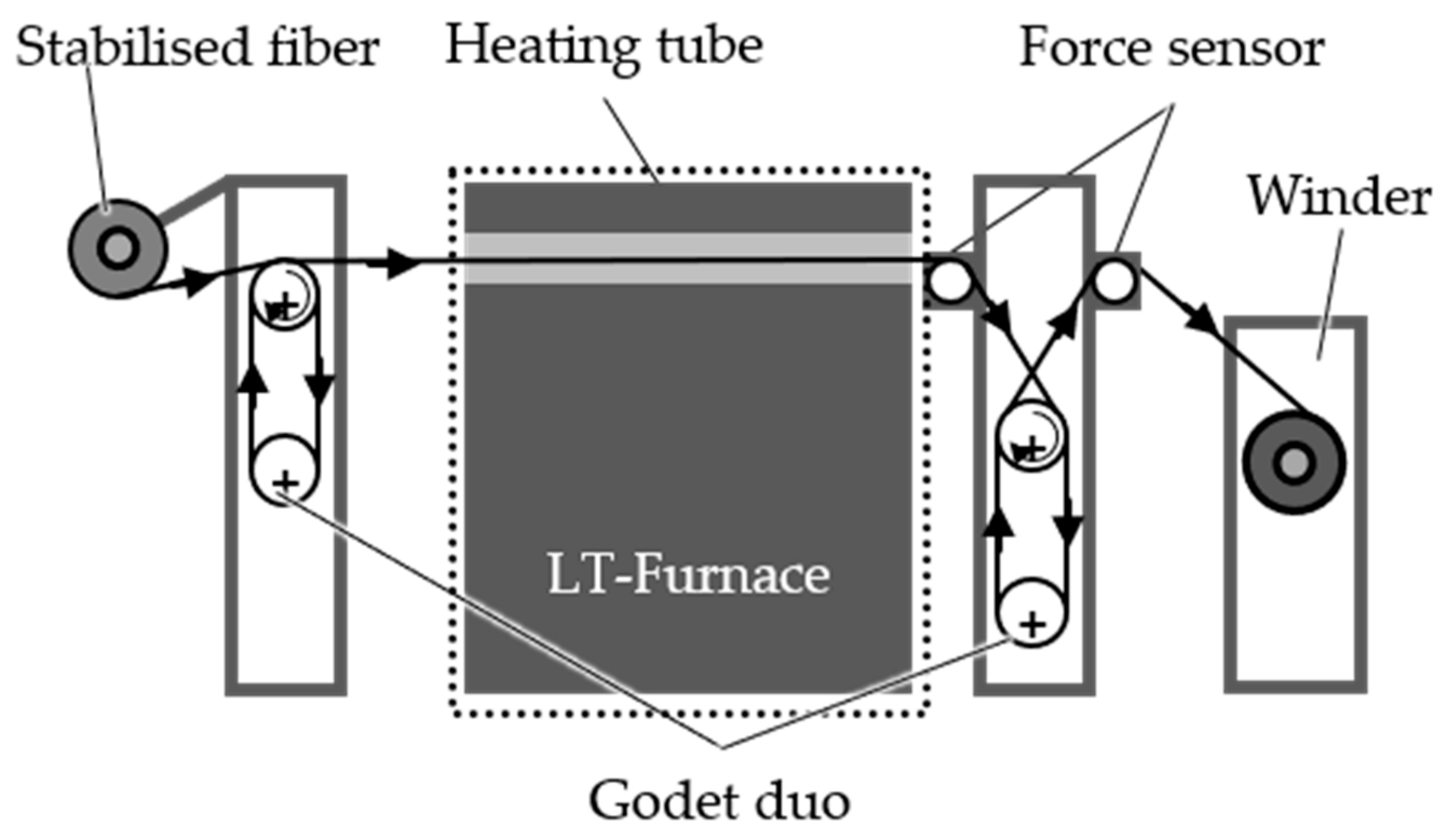
3.2.2. Continuous Stabilization
The batch stabilization trials confirm the feasibility of conducting stabilization with higher fiber tension, which facilitates the implementation of continuous stabilization and subsequent continuous carbonization processes. The first step involves investigating continuous stabilization. The continuous dissolution of PA6 is conducted at 40 °C and 60 °C for 2 h, while sulfonation takes place at 120 °C. Fiber samples are collected after the dissolution treatment (40 °C: zone LA.2; 60 °C: zone A.2) and after zones 1, 2 and 3 of the sulfonation (40 °C: zones L1, L2 and L3; 60 °C: zones 1,2 and 3). The TGA and gas pycnometry results for the continuous stabilization trials using the HDPE-INS precursor are presented in
Table 13.
The HDPE-INS precursor has a density of 1.073 ± 0.001 g/cm3. The TGA analysis provides values for ΔmPE and Δmtotal of 84% and 99%, respectively. The density of the samples obtained from the continuous dissolution of PA6 at 40 °C and the subsequent continuous sulfonation are as follows: 0.960 ± 0.001 g/cm3 (zone LA.2), 1.047 ± 0.001 g/cm3 (zone L1), 1.217 ± 0.001 g/cm3 (zone L2) and 1.494 ± 0.003 g/cm3 (zone L3). For the continuous stabilization trials with a dissolution temperature of 60 °C, the density values are as follows: 0.977 ± 0.001 g/cm3 (zone A.2), 1.046 ± 0.002 g/cm3 (zone 1), 1.225 ± 0.001 g/cm3 (zone 2) and 1.494 ± 0.003 g/cm3 (zone 3). In both cases, the density values exhibit a clear increasing trend, which is expected as heavier sulfur-containing groups replace lighter hydrogen atoms and cross-linking occurs within the fibers polymer. Comparing the equivalent density values for both cases, it can be observed that different dissolution temperatures result in similar fiber density values. This indicates that both approaches yield stabilized PE fibers with similarly high-density values after the third sulfonation zone.
The TGA results of the continuous stabilization trials with a dissolution temperature of 40 °C demonstrate the expected decreasing trend in the values of ΔmPE and Δmtotal for the four stabilization zone samples. The highest values are observed in the zone LA.2 sample (ΔmPE = 97%; Δmtotal = 100%) and decrease from zone A1 (ΔmPE = 91%; Δmtotal = 94%) to zone A2 (ΔmPE = 38%; Δmtotal = 71%), reaching the lowest values in zone A3 (ΔmPE = 12%; Δmtotal = 51%). Similarly, the TGA results for the batch stabilization trial samples with a dissolution temperature of 60 °C also exhibit the same decreasing trend in ΔmPE and Δmtotal values. The zone A.2 sample has the highest values (ΔmPE = 94%; Δmtotal = 99%), followed by zone 1 (ΔmPE = 72%; Δmtotal = 90%), zone 2 (ΔmPE = 34%; Δmtotal = 64%) and finally zone 3 (ΔmPE = 12%; Δmtotal = 53%). The most significant differences in the TGA values are observed in the samples from the first sulfonation zone. Zone L1 has a ΔmPE value of 91%, while zone 1 has a ΔmPE value of 72%. This difference can be explained by the incomplete dissolution of PA6 at a dissolution temperature of 40 °C. As a result, in the first sulfonation zone L1, the remaining PA6 needs to be fully dissolved first. The presence of the remaining PA6 prevents the direct occurrence of the sulfonation reaction, hindering the access of sulfuric acid to the PE filaments and resulting in a higher ΔmPE value.
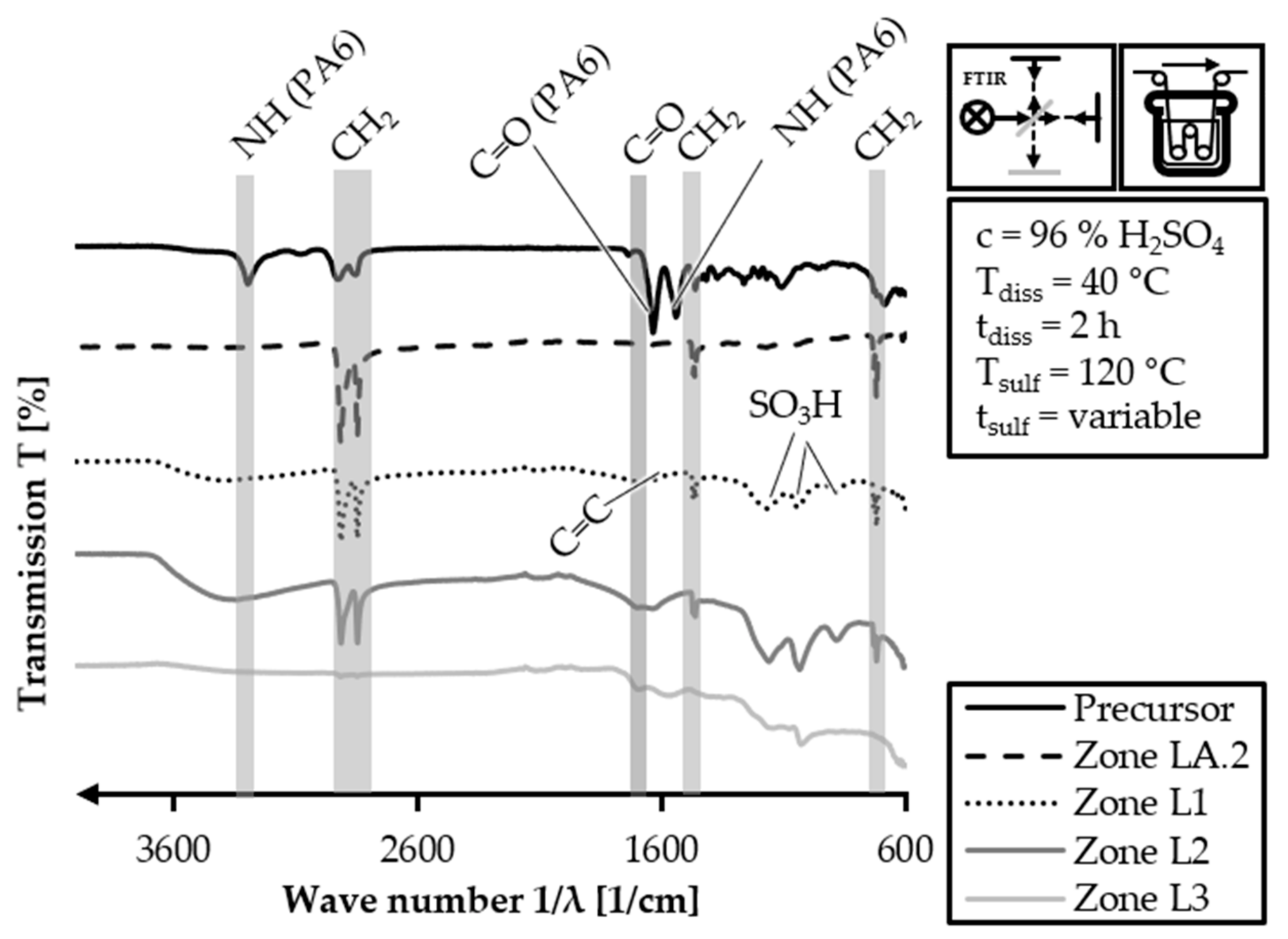
The stacked FTIR spectra of the HDPE-INS precursor and the continuous stabilization trial samples with a dissolution temperature of 40 °C are depicted in
Figure 13.
The four samples of the continuous stabilization trials do not exhibit any signals associated with PA6. In the FTIR curve of the zone LA.2 sample, only peaks related to PE are observed. These peaks correspond to the wave numbers 3000–2840 cm−1 (C-H stretching), 1470–1460 cm−1 (CH2 deformation) and 770–720 cm−1 (CH2 deformation). This indicates that the PA6 and its functional groups (amide and carboxylic groups) have dissolved to the extent that they are not detectable in the FTIR analysis. In the FTIR spectra of all three sulfonation samples, there are no peaks associated with PA6. The spectrum of zone L1 exhibits the alkane-related peaks with lower intensity (3000–2840 cm−1 and 1470–1460 cm−1) compared to the zone LA.2 spectrum. However, it shows peaks for sulfone structures (1150–1050 cm−1: S=O symmetric and asymmetric stretching) and carboxylic acid groups (1710–1690 cm−1: C=O stretching). In the FTIR spectrum of zone L2, the peak intensities for alkane structures are further decreased (3000–2840 cm−1 and 1470–1460 cm−1), while the peaks caused by sulfonation increase in intensity (1150–1050 cm−1: S=O symmetric and asymmetric stretching, 1710–1690 cm−1: C=O stretching). Finally, the FTIR spectrum for zone L3 shows almost no observable alkane-related peaks, while the intensities of the sulfone and carboxylic group peaks increase (1150–1050 cm−1: S=O symmetric and asymmetric stretching, 1710–1690 cm−1: C=O stretching). Additionally, a peak related to the C=C stretching of double bonds is identifiable at 1620–1570 cm−1, which is caused by cross-linking and desulfonation reactions.
Similarly, the samples of the continuous stabilization trials with a dissolution temperature of 60 °C are also analyzed using FTIR. The spectra of these samples are shown in a stacked manner in
Figure 14.
In the FTIR spectrum of the zone A2 sample, no identifiable peaks related to PA6 are observed, similar to the remaining sulfonation zones samples. The spectrum of zone A2 only exhibits peaks associated with PE, with high intensity. The peaks are located at 3000–2840 cm−1 (C-H stretching), 1470–1460 cm−1 (CH2 deformation) and 770–720 cm−1 (CH2 deformation). The FTIR spectrum for zone 1 shows alkane-related peaks (3000–2840 cm−1; 1470–1460 cm−1; and 770–720 cm−1) with lower intensity. Additionally, peaks caused by sulfones are present (1150–1050 cm−1: S=O symmetric and asymmetric stretching). The intensity trend of the observed peaks is continuous in the spectrum of zone 2, with a further reduction in the intensity of alkane-related peaks (3000–2840 cm−1; 1470–1460 cm−1; and 770–720 cm−1), an increase in the intensity of the sulfone-related peaks (1150–1050 cm−1) and the appearance of a peak caused by C=O stretching of carboxylic groups (1710–1690 cm−1). In the spectrum of zone 3, the alkane-related peaks are only marginally present (3000–2840 cm−1; 1470–1460 cm−1; and 770–720 cm−1). The peaks associated with sulfones (1150–1050 cm−1) and carboxylic groups (1710–1690 cm−1) are still present. Additionally, the peak representing the C=C stretching of double bonds in the polymer chains (1620–1570 cm−1) is identifiable, indicating the advanced sulfonation and cross-linking degree of the fiber after zone 3 of sulfonation.
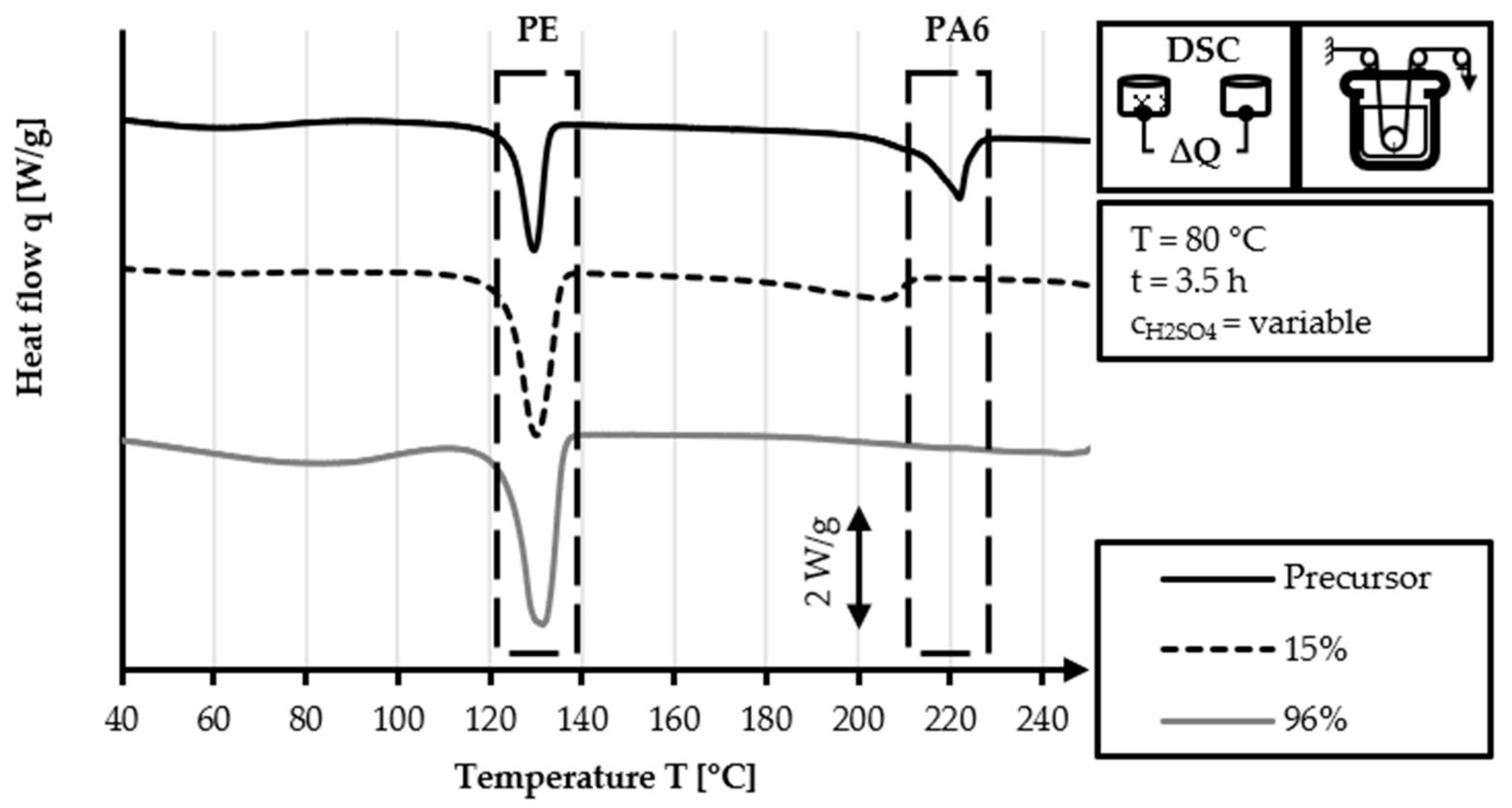
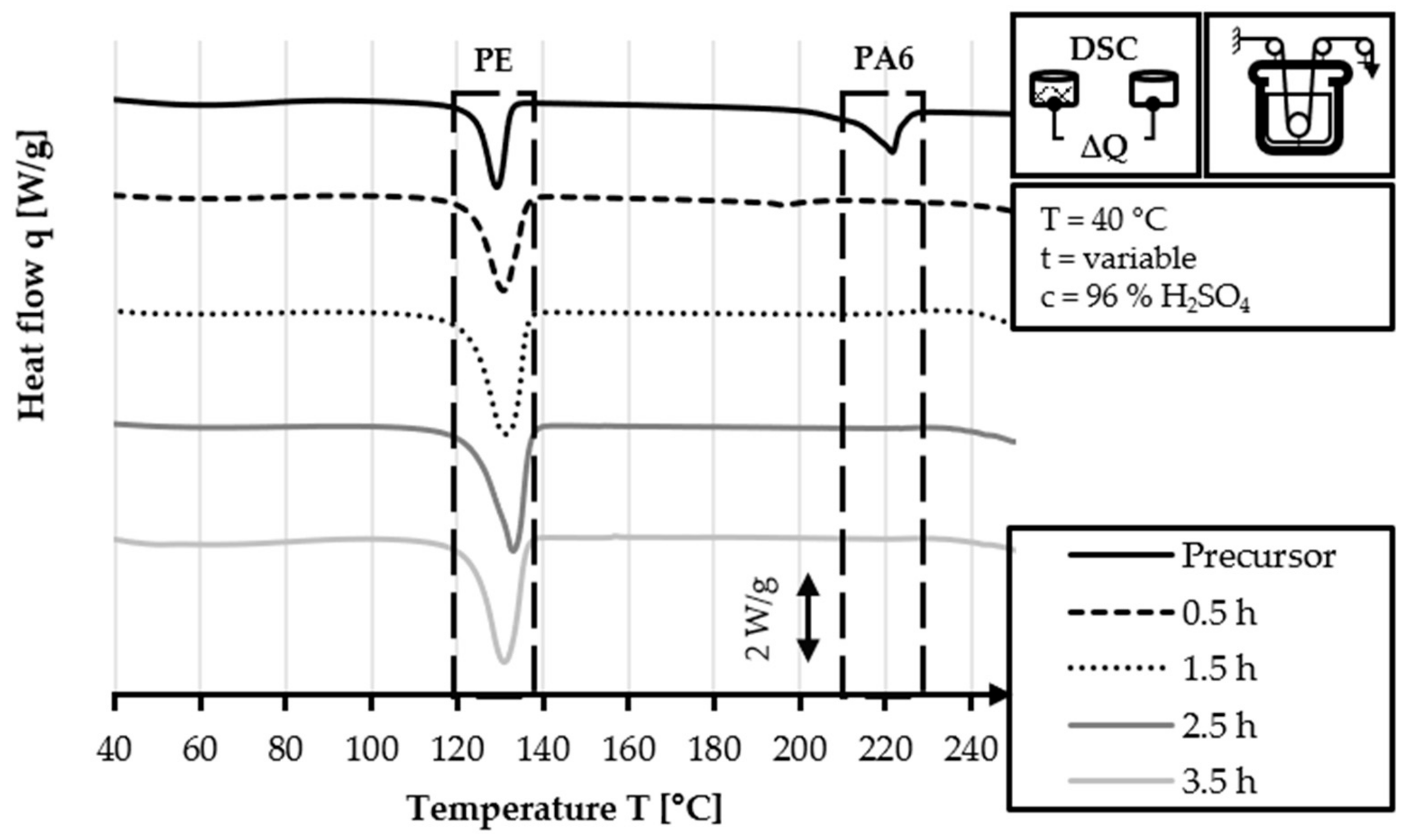
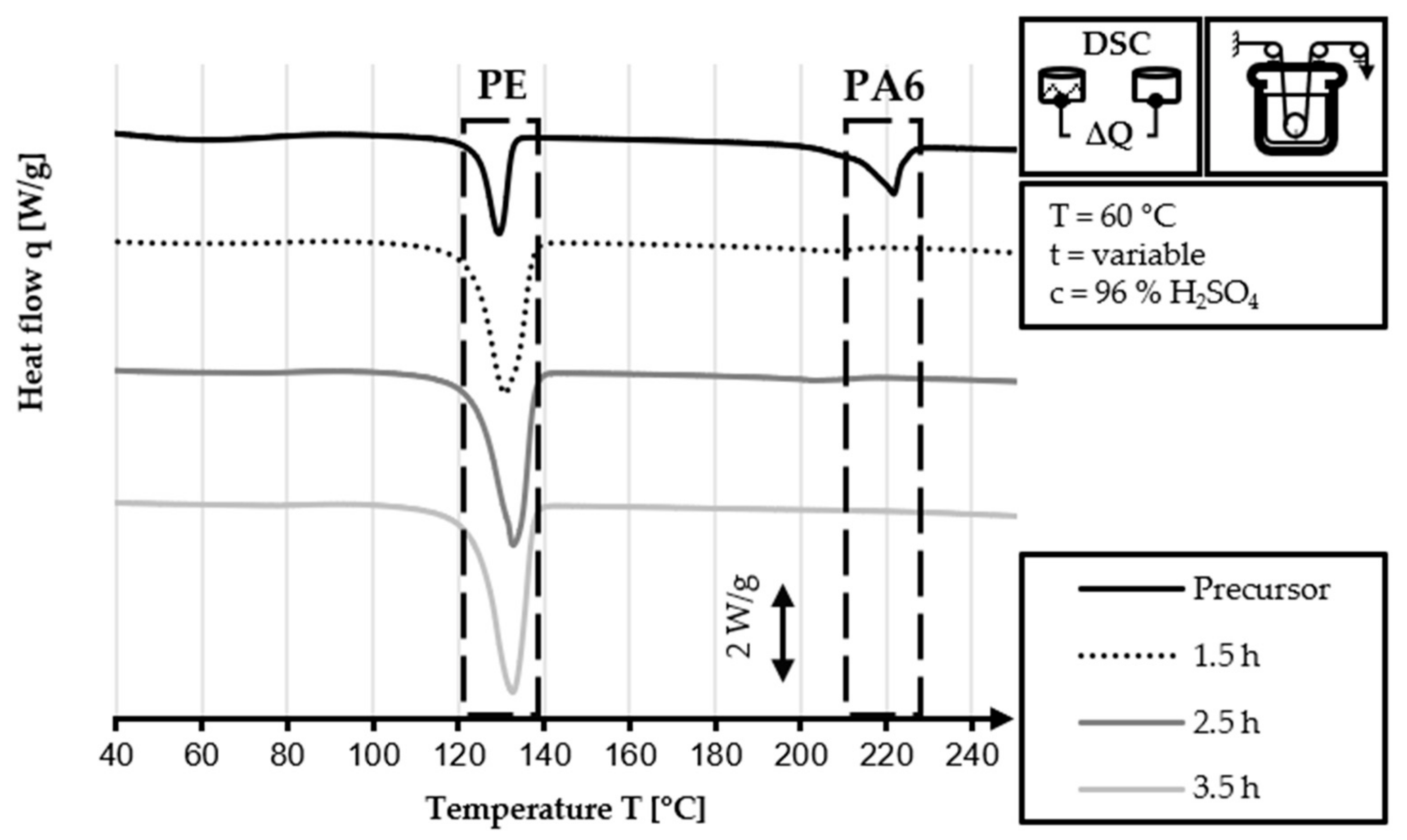
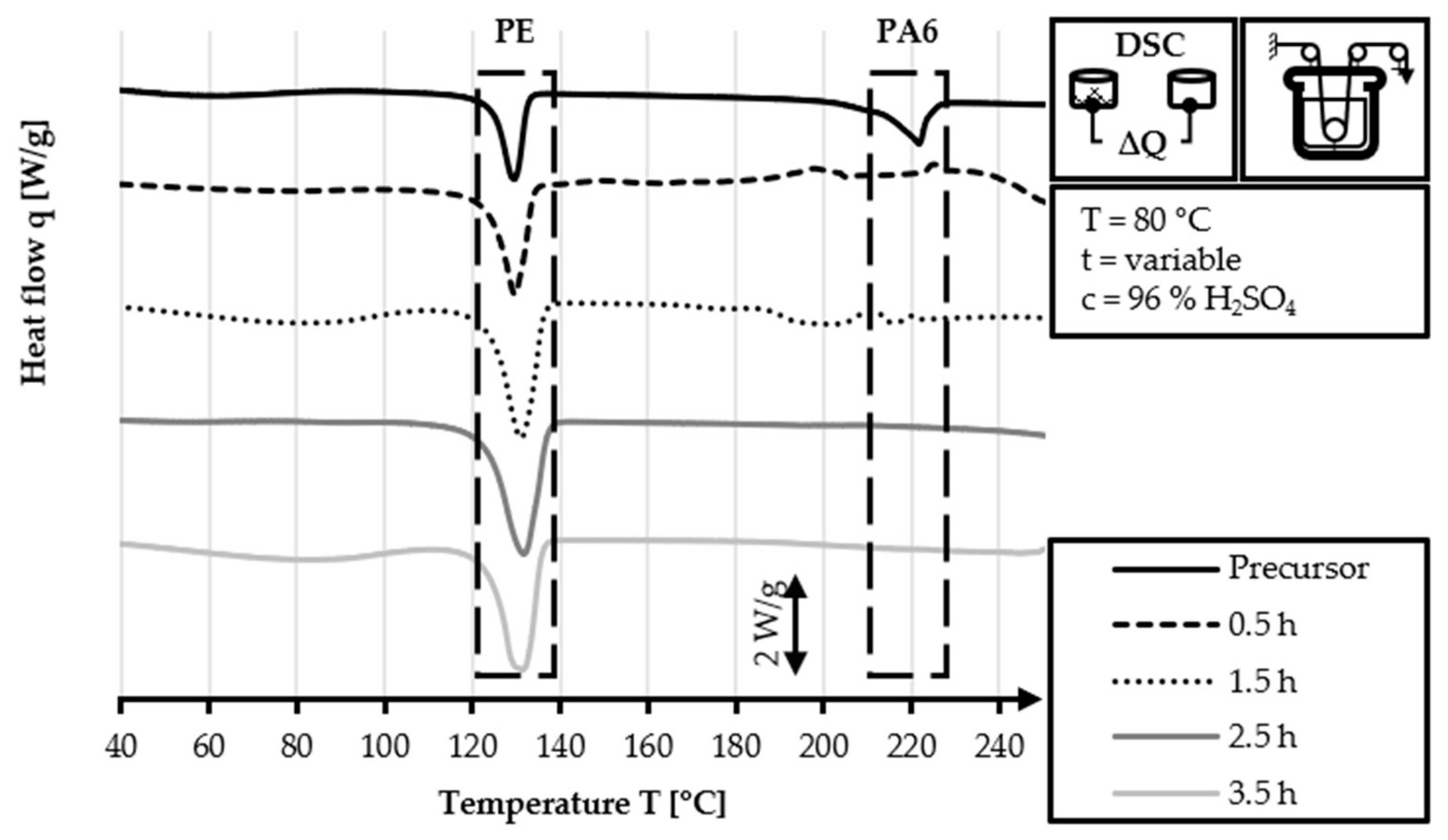
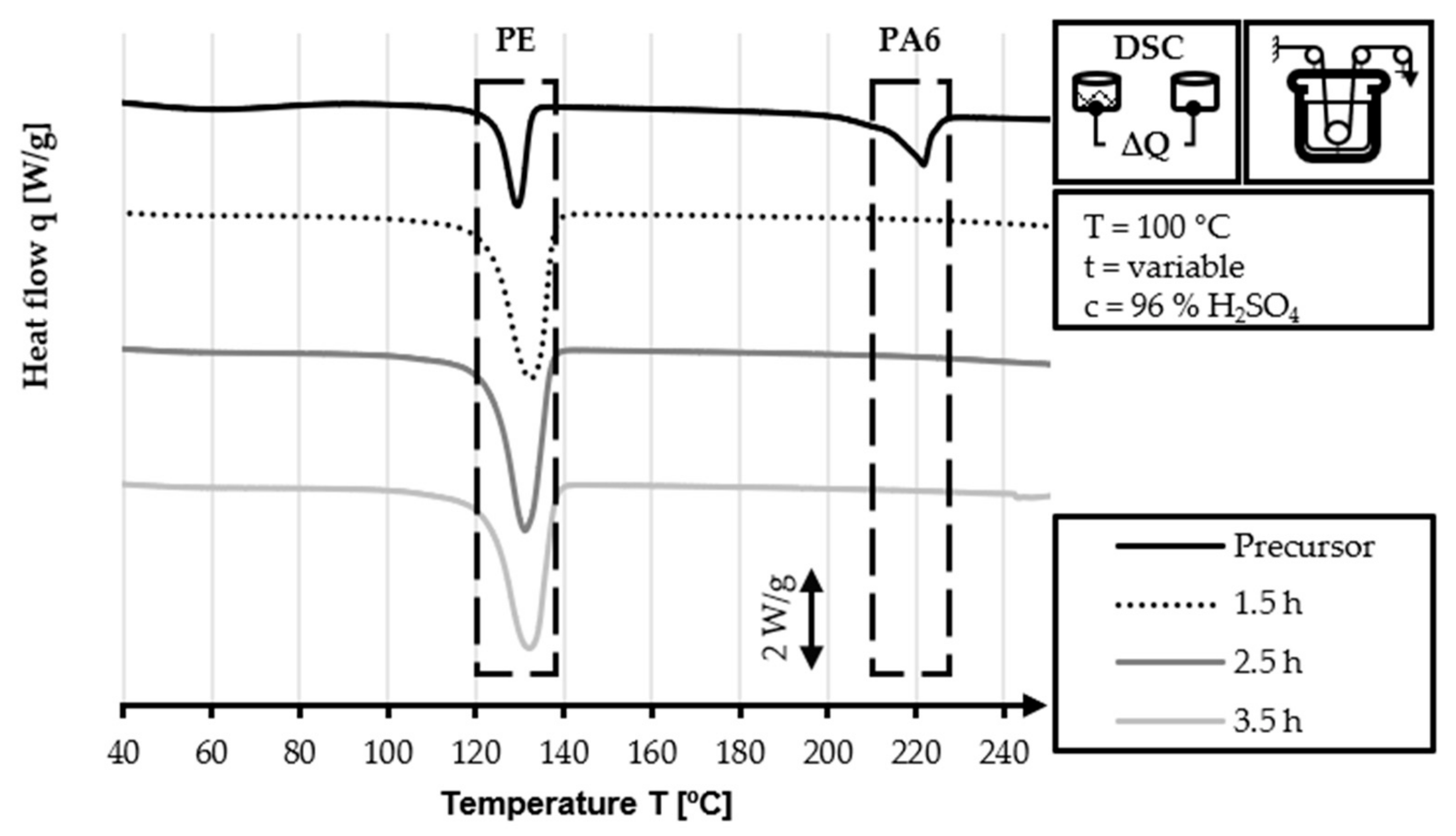
The DSC analysis of the 40/2 and 60/2 samples from the continuous stabilization trials does not exhibit any peaks related to PA6 (220 °C) in their DSC curves, unlike that of the HDPE-INS precursor. Additionally, the sharp PE precursor peak at 130 °C diminishes in size as the sulfonation process progresses and eventually disappears completely in the DSC curve of the samples of zone 3 and zone L3. In the DSC curves of zones 2 and 3 (L2 and L3, respectively), shallow endothermic depressions can be observed in the 60–130 °C and 160–225 °C temperature ranges. These depressions are probably caused by the hygroscopic behavior of sulfonic acid groups incorporated by sulfonation. The DSC measurements of sulfonated PE give only an indication of the dismantling of the crystalline structure and do not provide specific information regarding the cross-linking density and stabilization level, as is the case in stabilized PAN fibers. Comparing the DSC curves of both approaches (40 °C or 60 °C dissolution temperature), no significant identifiable differences are observed.
Light microscopy is employed to observe and investigate cross-sections of the filaments. The light microscopy images and the measured filament diameters of the continuous stabilization trials with the dissolution temperature of 40 °C are presented in
Figure 15.
The light microscopy image of the continuous stabilization sample from zone LA.2 reveals round filaments with an average filament diameter of 3.71 ± 0.40 μm. In zone L1, the observed filaments are also round, with a slightly smaller average diameter of 3.29 ± 0.47 μm. Moving to zone L2, the filament diameter increases to an average of 3.87 ± 0.30 μm. Lastly, in zone L3, the round filaments exhibit an average diameter of 3.49 ± 0.40 μm. The filament diameter of the continuous stabilization trial samples with 40 °C dissolution temperature follows an interesting trend. From zone LA.2 to zone L1, the diameter shows a slight decrease, suggesting that some residuals from PA6 may still be present in zone LA.2 and that in that zone the swelling of the filaments is more pronounced, resulting in thicker filaments. The swelling is influenced by many factors, such as the applied tension on the fiber and the uptake of water from the washing bath. The latter could contribute to the thicker filaments from zone LA.2. The diameter then increases from zone L1 to zone L2, indicating swelling and incorporation of sulfur-containing groups. In zone L3, the filament diameter becomes slightly smaller compared to zone L2, which could be attributed to desulfonation reactions leading to cross-linking and resulting in thinner filaments. Overall, the filaments obtained from the continuous stabilization trials are thinner, compared to the filament diameters of the fibers from the batch sulfonation trials. This can be attributed to the increased tensile stress applied during the continuous stabilization process.
The light microscopy images and filament diameter values for the continuous stabilization trials with 60 °C dissolution temperature are presented in
Figure 16.
The filament diameters of the samples from the continuous stabilization trials with a dissolution temperature of 60 °C follow a clear upward trend. The sample from zone A.2 exhibits the smallest filament diameter, measuring 3.04 ± 0.30 μm. The filament diameter increases for the sample from zone 1 (3.24 ± 0.58 μm). The light microscopy image of the sample from zone 2 reveals round filaments with an average filament diameter of 3.79 ± 0.32 μm. Lastly, zone 3 displays thicker round filaments with an average diameter of 3.89 ± 0.53 μm. The filament diameter increases throughout the stabilization processes as a result of swelling and the incorporation of sulfur-containing groups. Comparing the filaments from the continuous stabilization approach to those from batch stabilization, the former provides smaller diameters due to the higher tensile stress applied in the continuous process, which limits filament swelling, by making the incorporation of for example water from the washing bath more difficult. A comparison of the filament diameters for both approaches is presented in
Table 14.
The filament diameter of the 60/2 approach exhibits a consistently increasing trend, which aligns with expectations, as swelling and the incorporation of sulfur-containing groups are induced by the sulfonation process. However, the samples from the 40/2 approach deviate from this trend. Specifically, the filament diameter of the fibers after the dissolution of PA6 (zone A.2) should be the lowest, resembling the diameter of PE filaments in the virgin HDPE-INS precursor (2.93 μm). However, this is not the case, as the filament diameter measures 3.71 ± 0.40 μm. This discrepancy suggests that the dissolution of PA6 may not be complete, and a layer of PA6 still coats the individual PE filaments, requiring further dissolution in the first sulfonation zone and delaying the initiation of the sulfonation reactions.
In general, the continuous stabilization process is successful. The fibers stabilized through continuous dissolution of PA6 and continuous sulfonation exhibit round filaments with a diameter of less than 4 μm and demonstrate a high degree of sulfonation and cross-linking. Fibers obtained from zones L3 and zone 3 exhibit thermal stability, making them suitable for carbonization trials to produce CF without significant structural defects or hollow structures.
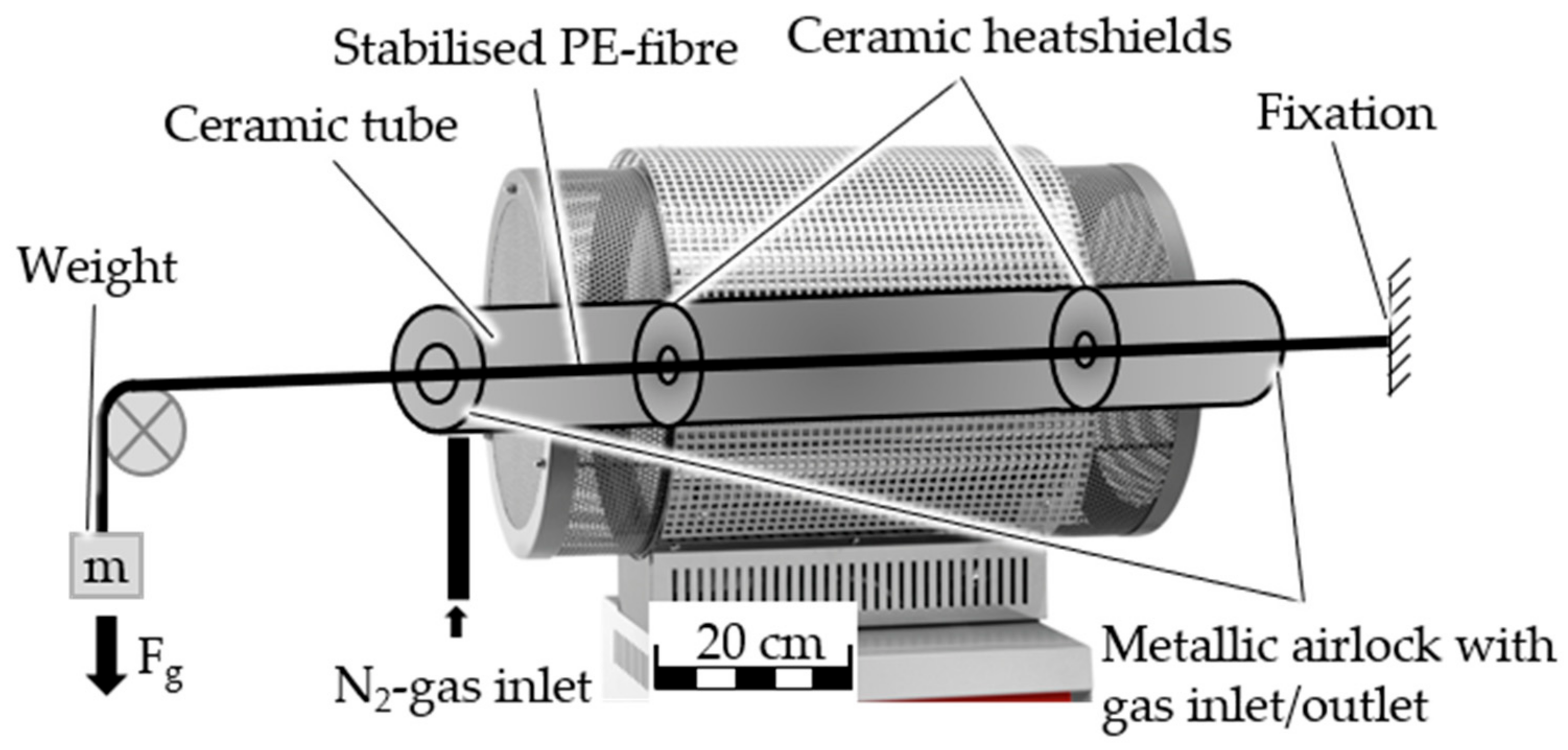
3.2.3. Carbonization
After the successful continuous stabilization of the HDPE-INS precursor, the stabilized fibers undergo carbonization trials to validate the developed concept for ultra-thin CF production. Initially, batch carbonization trials are conducted using both stabilized fibers. The results, including fiber shrinkage, CF filament diameters, and measured fiber density, are summarized in
Table 15.
The stabilized fiber from zone L3 undergoes carbonization up to 900 °C with a force of 0.5 N applied to the fiber. During the carbonization process, the 40/2-zL3-900 °C fiber experiences a 13% shrinkage in length, attributed to desulfonation reactions, cross-linking reactions, and thermal shrinkage. The resulting CF has a fiber density of 1.532 ± 0.002 g/cm
3 and filaments diameter of 2.45 ± 0.43 μm. When carbonized up to 1100 °C, the 40/2-zL3-1100 °C fiber exhibits an increased fiber shrinkage of 14%. The fiber density increases to 1.634 ± 0.009 g/cm
3, while the filament diameter remains similar at 2.51 ± 0.25 μm. These changes align with expectations as higher carbonization temperatures enhance cross-linking and remove non-carbon atoms, leading to increased fiber density, while the applied force on the fiber contributes to further density increases. The release of the sulfur-containing groups happens in the temperature range of 120–400 °C and results in a decrease in fiber density. Nevertheless, the density decrease in this temperature range is compensated by the cross-linking reactions combined with the release of hydrogen atoms from 600 °C onwards. The differences in fiber density of samples carbonized at 900 or 1100 °C are therefore mostly caused by hydrogen release and cross-linking reactions, which indeed increase the fiber density. An extensive description and analysis of the reactions happening in the different temperature ranges throughout the carbonization process were described by Barton et al. [
21,
34]. Notably, CF with filament diameters below 3 μm is successfully synthesized in both cases. Similarly, the fiber from zone 3 of the continuous stabilization with a dissolution temperature of 60 °C undergoes batch stabilization up to 900 °C (60/2-z3-900 °C) and 1100 °C (60/2-z3-1100 °C). Carbonization up to 900 °C results in a 14% fiber shrinkage. The CF exhibits a density of 1.464 ± 0.004 g/cm
3 and a filament diameter of 2.46 ± 0.31 μm. With carbonization up to 1100 °C, the fiber experiences an increased shrinkage of 15%, resulting in CF with a filament diameter of 2.72 ± 0.28 μm and a density of 1.844 ± 0.005 g/cm
3. Consistent with the previous case, the higher carbonization temperature leads to increased fiber density due to the removal of non-carbon atoms. Notably, the filament diameters of all samples fall within the desired range of 3 μm. Among the samples, the 60/2-z3-1100 °C CF exhibits the highest fiber density. Both approaches highlight that carbonization up to 900 °C is insufficient to achieve CF densities within the range of 1.8 g/cm
3.
Figure 17 displays electron microscopy images of the CF produced in the batch carbonization trials.
The electron microscopy image of the 40/2-zL3-900 °C CF reveals the presence of small pores on the CF surface, indicating surface defects. These defects become more pronounced in the sample carbonized up to 1100 °C (40/2-zL3-1100 °C), where the filaments exhibit a rough surface with a higher number of pores. These incorporated pores, created during carbonization, have a lower density compared to CF and therefore result in a density decrease. This could potentially impact the mechanical properties of the CF. In contrast, the electron microscopy image of the 60/2-z3-900 °C CF, carbonized up to 900 °C, shows a smooth surface with no identifiable surface defects. Similarly, the filaments of the CF carbonized up to 1100 °C (60/2-z3-1100 °C) also display a smooth surface with no discernible surface defects or pores. These observations explain the higher fiber density values obtained for the 60/2-z3-1100 °C CF from zone 3, compared to the equivalent CF from zone L3. These images validate that the approach with continuous dissolution of PA6 at 60 °C followed by continuous sulfonation results in CF with improved properties compared to the 40 °C dissolution temperature approach.
Due to the thin and brittle nature of the CF filaments, it is not feasible to investigate their mechanical properties through a single filament tensile strength test, as the isolation of single filaments is not possible. Instead, mechanical roving tests could be considered as an alternative method. However, for such tests, the CF would require surface activation and the application of a sizing resin.
In the continuous carbonization trial, the stabilized fiber from continuous stabilization with a dissolution temperature of 60 °C and from zone 3 of the continuous sulfonation undergoes a continuous carbonization trial. Therein, the stabilized fiber undergoes continuous carbonization up to 900 °C in the LT furnace, followed by batch carbonization up to 1100 °C. Samples are taken after both carbonization steps. The carbonization parameters and the gas pycnometry results of the CF are presented in
Table 16.
The carbonization process in the LT furnace up to 900 °C (60/2-z3-conti900 °C) results in a fiber shrinkage of 12%. The resulting CF exhibits a fiber density of 1.622 ± 0.003 g/cm3, confirming the findings from the batch carbonization trials. However, carbonization up to 900 °C is insufficient to achieve the desired CF density. Subsequent batch carbonization up to 1100 °C (60/2-z3-conti900–1100 °C) only induces a further 1% fiber shrinkage but increases the density to 1.733 ± 0.017 g/cm3. This increase in density is attributed to enhanced cross-linking of the polymer and the removal of non-carbon atoms from the fiber. Notably, the fiber density achieved through carbonization in the LT furnace is considerably higher than that obtained through batch carbonization up to 900 °C (1.464 ± 0.004 g/cm3). However, the resulting density of 1.622 ± 0.003 g/cm3 remains lower than the typical CF density of approximately 1.8 g/cm3. It is plausible that a similar higher density could be attained through carbonization up to 1100 °C if the process is conducted in a continuous high-temperature furnace (HT-furnace).
The CF filament diameter is determined via light microscopy imaging. The light microscopy image of the CF produced in the LT furnace and carbonized up to 900 °C reveals individual round filaments with a diameter of 3.67 ± 0.30 μm. Subsequent batch carbonization up to 1100 °C causes a reduction in filament diameter, with the round filaments reaching a diameter of 2.54 ± 0.28 μm. This reduction in filament diameter can be attributed to the applied tension during the process and the removal of additional non-carbon atoms from the fiber.
The samples are further analyzed via electron microscopy to assess the structural integrity of the samples in the form of surface structuration or pores in the filament side walls. The electron microscopy images of both CF samples from the continuous carbonization trials are depicted in
Figure 18.
The electron microscopy images of the CF produced in the LT furnace exhibit a smooth surface with no discernible structural defects. This observation holds for the CF samples that underwent additional carbonization up to 1100 °C in the batch carbonization furnace. The mechanical properties of the produced CFs cannot be investigated via a single filament tensile strength test, as the isolation of singe filaments from the CF is not possible. The filaments are too thin and brittle. Alternatively, the CF could be investigated with mechanical roving tests. Therefore, the CF would need to undergo surface activation, followed by the application of a sizing resin.
In summary, the continuous carbonization has been successful in producing ultra-thin PE-based carbon fibers with filament diameters below 3 μm and no significant observable defects, even at temperatures up to 1100 °C. Furthermore, CF with a density of 1.622 ± 0.003 g/cm3 has been achieved through continuous carbonization in the LT furnace up to 900 °C. By subjecting the fibers to batch carbonization up to 1100 °C, the density is increased to 1.733 ± 0.017 g/cm. Additionally, even higher CF densities could be attained through continuous carbonization in a HT-furnace up to 1100 °C or 1350 °C.
In conclusion, the feasibility of producing ultra-thin PE-based CF has been demonstrated in both batch and continuous set-ups, resulting in CF with a filament diameter of less than 3 μm. The use of the HDPE-INS precursor approach has successfully reduced the required sulfonation reaction time from 4 h to 3 h. However, the complete stabilization process does take longer due to the 2 h PA6-dissolution step. The PE-INS precursor provides ultra-thin PE precursor filaments after the dissolution of the PA6 matrix, which can be effectively stabilized through sulfonation and transformed into CF via carbonization. As a result, the two main objectives of this study have been successfully achieved.
This work serves as a crucial foundation for the innovative approach of producing ultra-thin PE-based CF. However, there are still some unanswered questions and opportunities for improvement. Firstly, the optimization of the PA6 matrix dissolution is necessary, which could involve exploring different parameter conditions, particularly lower fiber tension during this step. Reducing tension would enhance the diffusion of sulfuric acid through the INS precursor, potentially shortening the required reaction time for a complete dissolution of the PA6 component. Alternatively, using a different sea component material that allows for the physical separation of the two components could be considered. Regarding the continuous carbonization of the stabilized fibers, further investigation into higher maximum temperatures is needed to improve the mechanical properties of the resulting CF. Additionally, the use of combinatory analytical methods, such as MS-TGA, which combines TGA with Mass Spectrometry (MS), could be used in future studies, to gain further insight into the structure evolution by directly identifying which molecules are separated during carbonization. This leads to most significant area requiring improvement, namely the development of a new sample preparation method for single filament tensile strength tests. Alternatively, the CF could be analyzed using mechanical roving tests. Addressing these open questions and improvement opportunities would enhance the understanding and performance of the ultra-thin PE-based CF production process.




























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}