
Antimicrobial Electrospun Fibers of Polyester Loaded with Engineered Cyclic Gramicidin Analogues

 ,
, 

Abstract
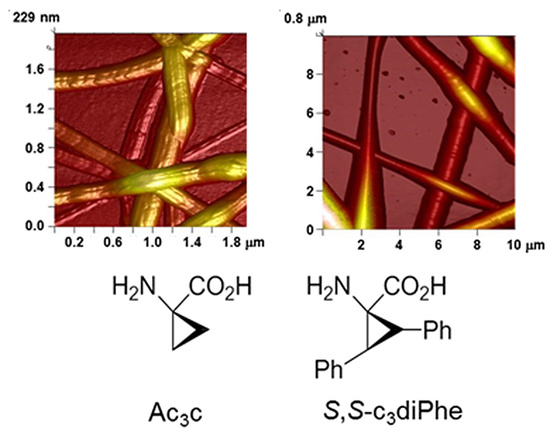
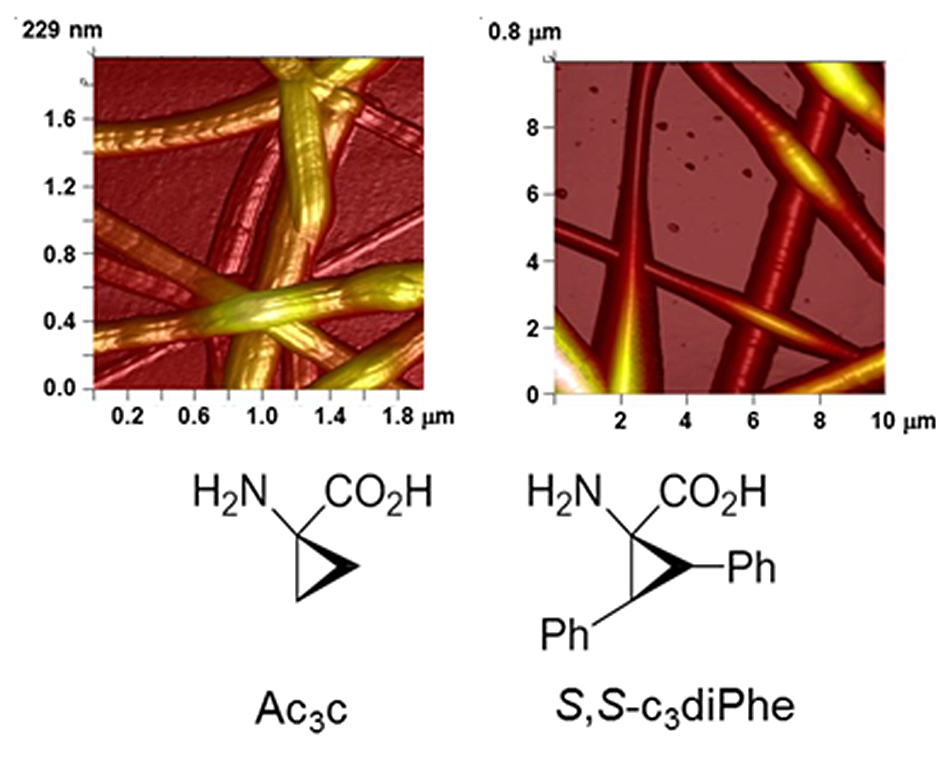
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Electrospinning
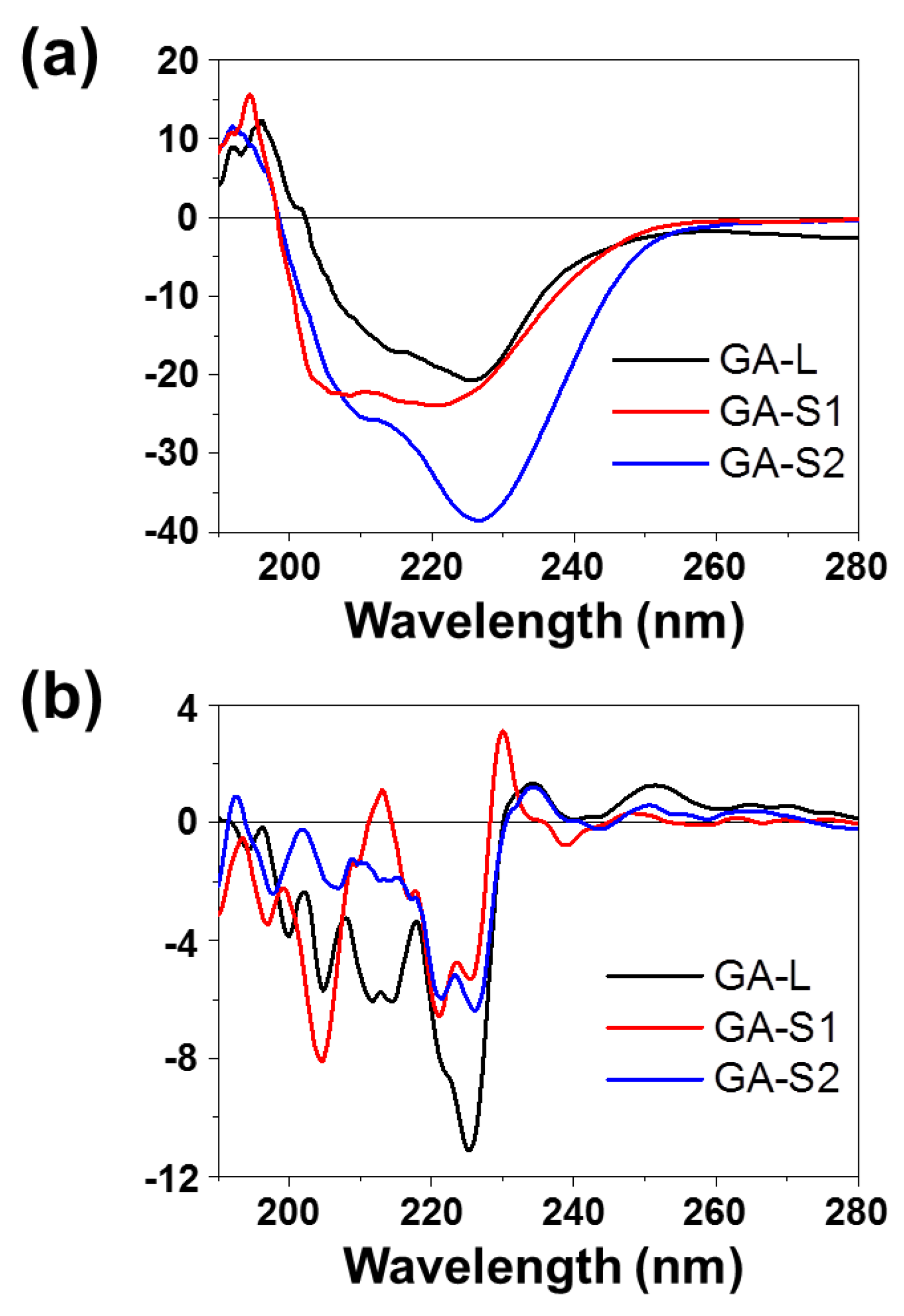
2.3. Circular Dichroism (CD)
2.4. Microscopy
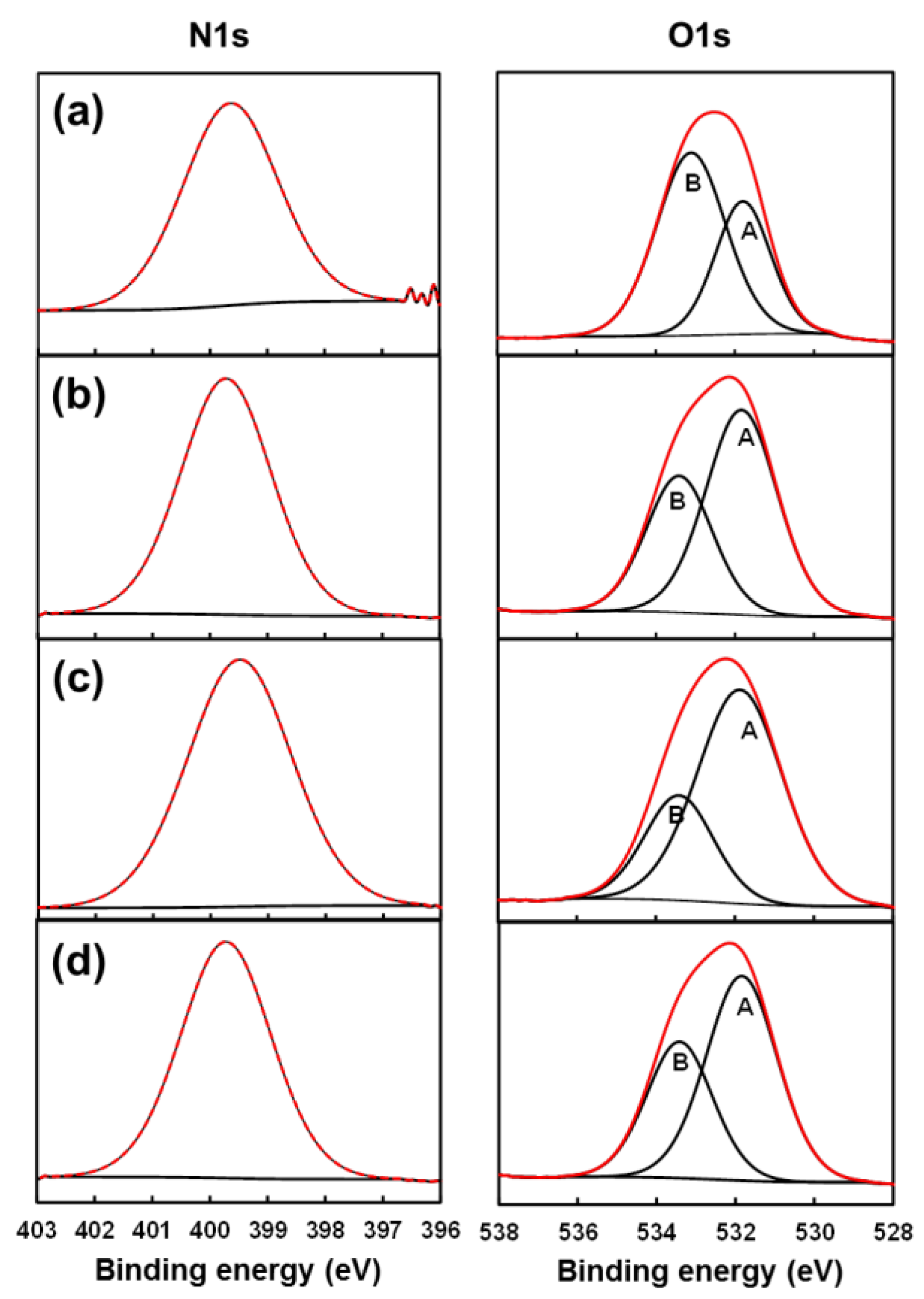
2.5. Chemical Characterization
2.6. Properties
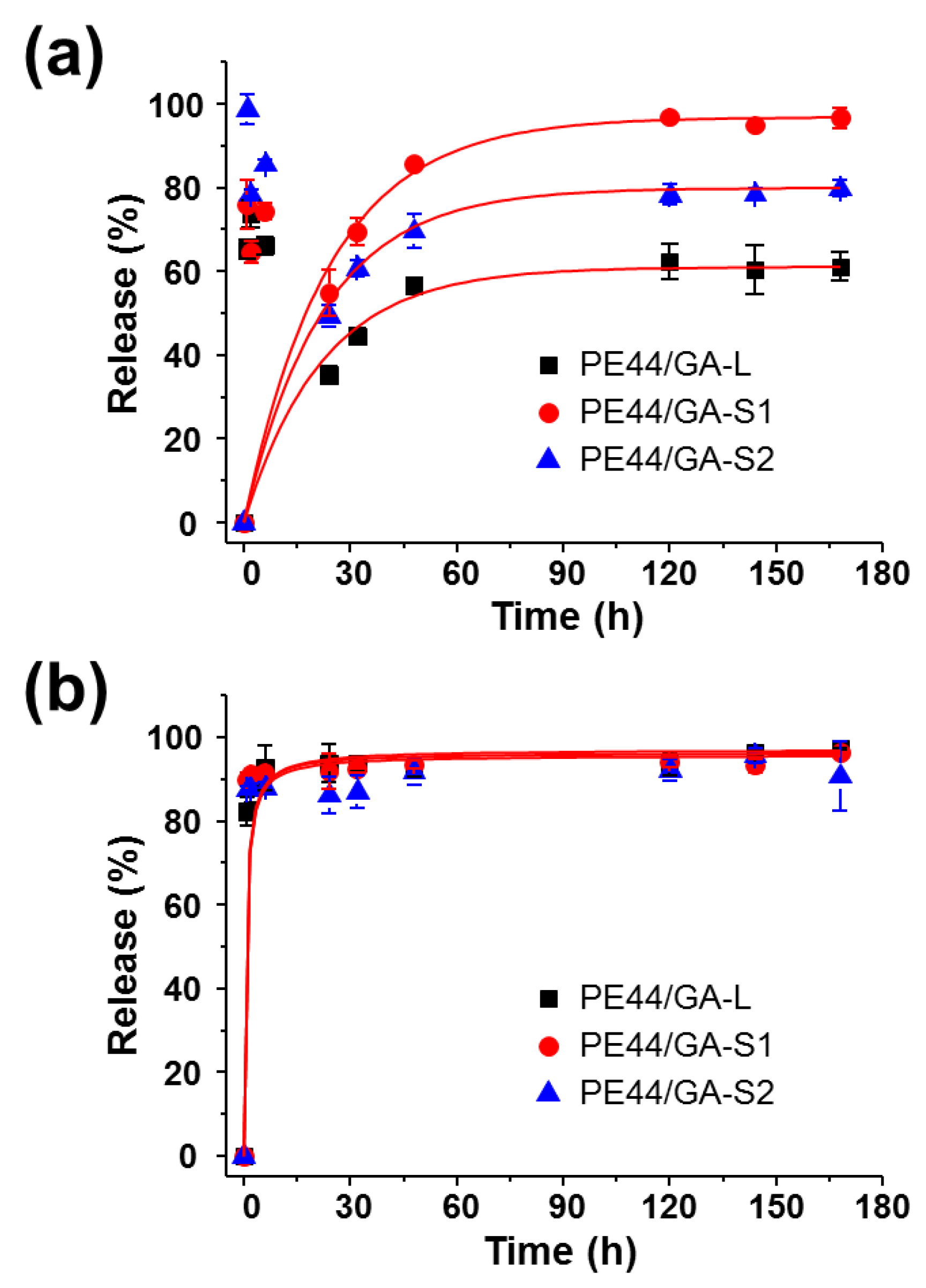
2.7. Release Experiments
2.8. Inhibition of Bacterial Growth
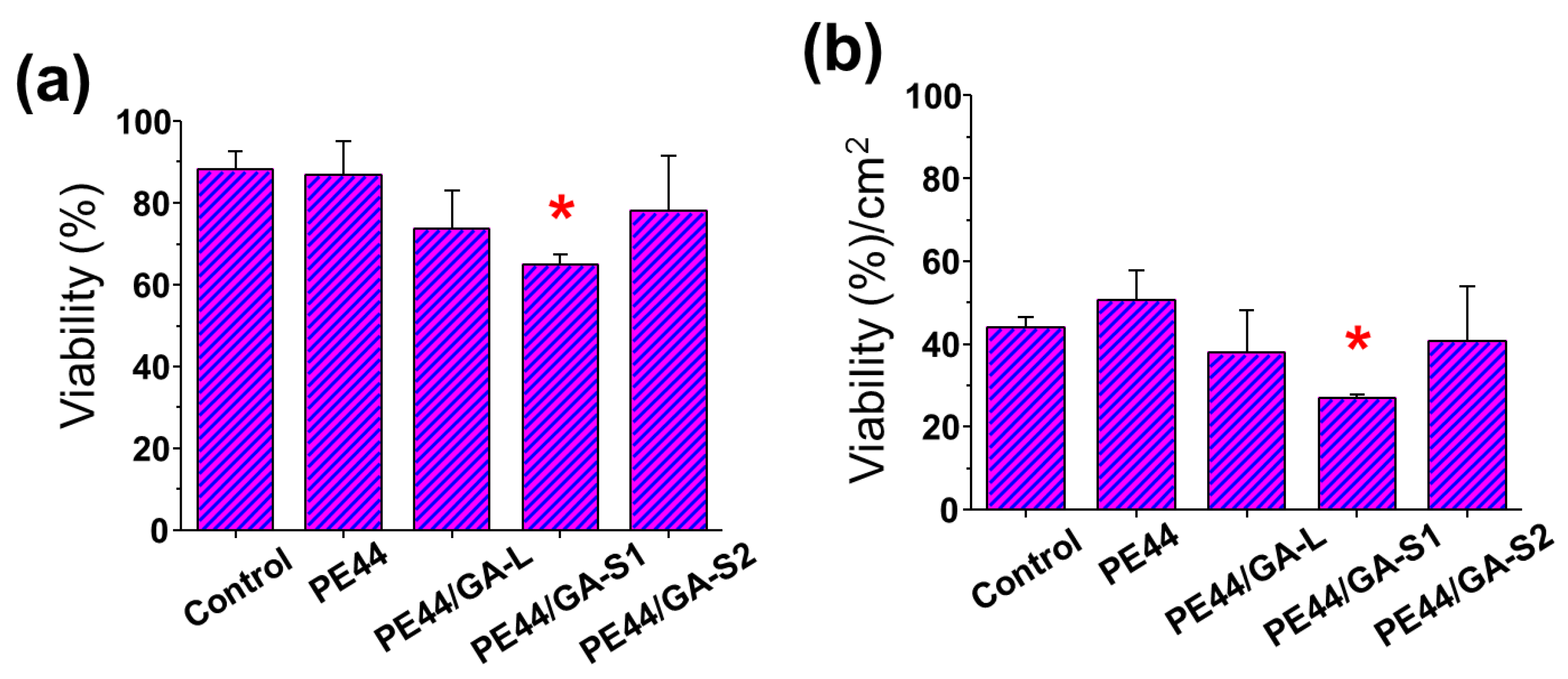
2.9. Cytotoxicity and Cell Adhesion
3. Results
3.1. Electrospinning of PE44 and Peptide Mixtures
3.2. Chemical Characterization
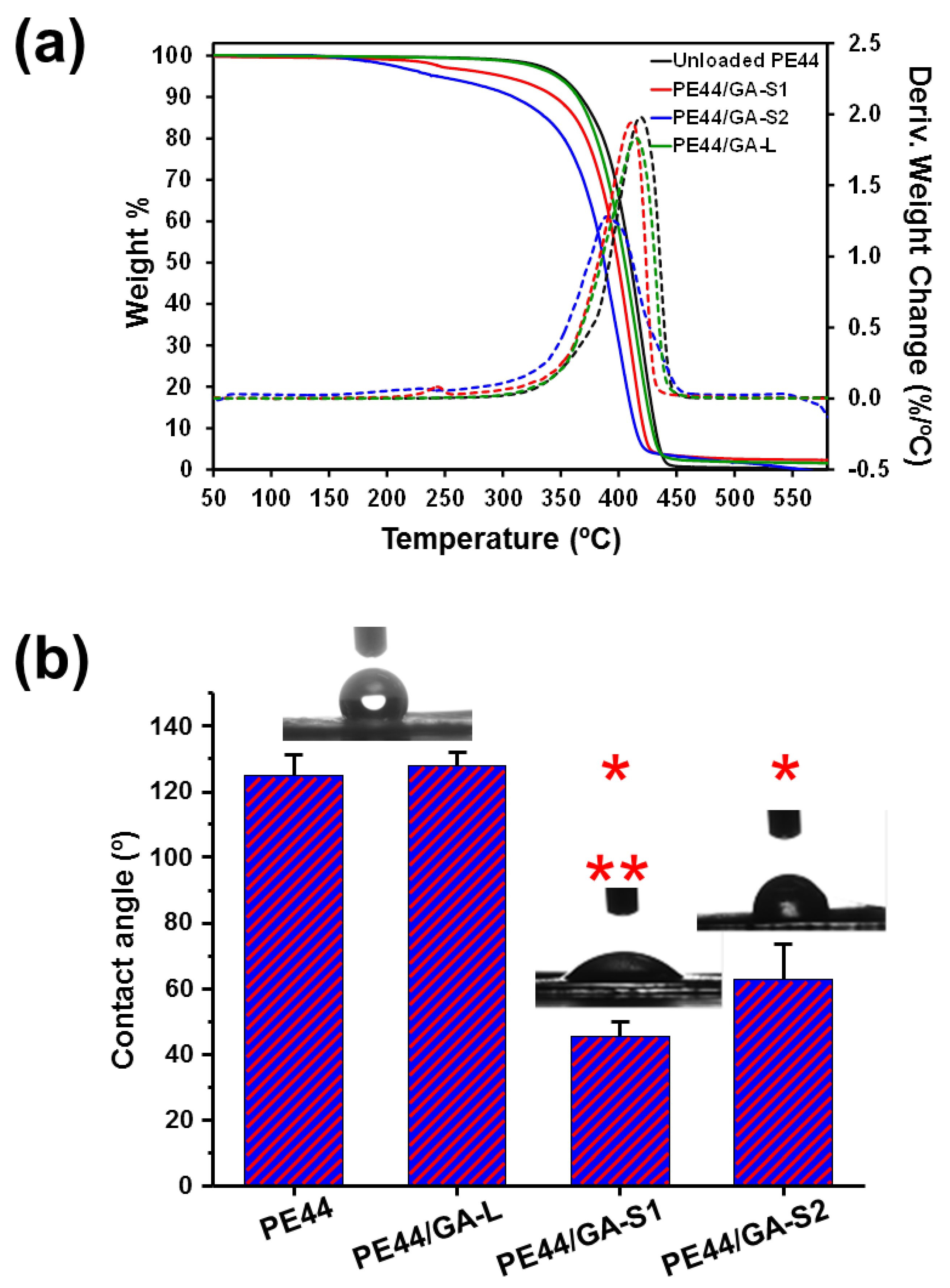
3.3. Properties
3.4. Peptide Release
3.5. Antimicrobial Activity
3.6. Biocompatibility
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wan, W.K.; Yang, L.; Padavan, D.T. Use of degradable and nondegradable nanomaterials for controlled release. Nanomedicine 2007, 2, 483–509. [Google Scholar] [CrossRef] [PubMed]
- Timko, B.P.; Kohane, S. Materials to clinical devices: Technologies for remotely triggered drug delivery. Clin. Ther. 2012, 34, S25–S35. [Google Scholar] [CrossRef] [PubMed]
- LaVan, D.A.; McGuire, T.; Langer, R. Small-scale systems for in vivo drug delivery. Nat. Biotechnol. 2003, 21, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Timko, B.P.; Dvir, T.; Kohane, D.S. Remotely triggerable drug delivery systems. Adv. Mater. 2010, 22, 4925–4943. [Google Scholar] [CrossRef] [PubMed]
- Balmert, S.C.; Little, S.R. Biomimetic delivery with micro and nanoparticles. Adv. Mater. 2012, 24, 3757–3778. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Li, W.; Guo, Y.J.; Feng, S.S. Nanomedicine strategies for sustained, controlled and targeted treatment of cancer stem cells. Nanomedicine 2016, 11, 3261–3282. [Google Scholar] [CrossRef] [PubMed]
- Orawan, S. Biomedical application of electrospun polycaprolactone fiber mats. Polym. Adv. Technol. 2016, 27, 1264–1273. [Google Scholar]
- Natarjan, J.; Dasgupta, Q.; Shetty, S.N.; Sarkar, K.; Madras, G.; Chatterjee, K. Poly(ester amide)s from Soybean Oil for Modulated Release and Bone Regeneration. ACS Appl. Mater. Interfaces 2016, 8, 25170–25184. [Google Scholar] [CrossRef] [PubMed]
- Pushpamalar, J.; Veeramachineni, A.K.; Owh, C.; Loh, X.J. Biodegradable polysaccharides for controlled drug delivery. ChemPlusChem 2016, 81, 504–514. [Google Scholar] [CrossRef]
- Cardoso, M.J.; Caridade, S.G.; Costa, R.R.; Mano, J.F. Enzymatic degradation of polysaccharide-based layer-by-layer structures. Biomacromolecules 2016, 17, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Winnacker, M.; Rieger, B. Poly(ester amide)s: Recent insights into synthesis, stability and biomedical applications. Polym. Chem. 2016, 7, 7039–7046. [Google Scholar] [CrossRef]
- Manavitehrani, I.; Fathi, A.; Badr, H.; Daly, S.; Shirazi, A.N.; Deghani, F. Biomedical applications of biodegradable polyesters. Polymers 2016, 8, 20. [Google Scholar] [CrossRef]
- Stebbins, N.D.; Faig, J.J.; Yu, W.; Guliyev, R.; Uhrich, K.E. Polyactives: Controlled and sustained bioactive release via hydrolytic degradation. Biomater. Sci. 2015, 3, 1171–1187. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Zhao, J.; Fei, J.; Cui, Y.; Li, J. Assembled microcapsules by doxorubicin and polysaccharide as high effective anticancer drug carriers. Adv. Healthc. Mater. 2013, 2, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cui, Y.; He, Q.; Yang, Y.; Fei, J.; Li, J. Selective recognition of co-assembled thrombin aptamer and docetaxel on mesoporous silica nanoparticles against tumor cell proliferation. Chem. Eur. J. 2011, 17, 13170–13174. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Hou, Y.-F.; Niu, P.-F.; Zhang, K.; Shoji, T.; Tsuboi, Y.; Yao, F.-Y.; Zhao, L.-M.; Chang, J.-B. Biodegradable PLGA nanoparticles loaded with hydrophobic drugs: Confocal Raman microspectroscopic characterization. J. Mater. Chem. B 2015, 3, 3677–3680. [Google Scholar] [CrossRef]
- Ashwanikumar, N.; Kumar, N.A.; Nair, S.A.; Kumar, G.S.V. 5-Fluorouracil-lipid conjugate: Potential candidate for drug delivery through encapsulation in hydrophobic polyester-based nanoparticles. Acta Biomater. 2014, 10, 4685–4694. [Google Scholar] [CrossRef] [PubMed]
- Maione, S.; del Valle, L.J.; Pérez-Madrigal, M.M.; Cativiela, CC.; Puiggalí, J.; Alemán, C. Electrospray loading and release of hydrophobic gramicidin in polyester microparticles. RSC Adv. 2016, 6, 73045–73055. [Google Scholar] [CrossRef]
- Pérez-Madrigal, M.M.; Llorens, E.; del Valle, L.J.; Puiggalí, J.; Armelin, E.; Alemán, C. Semiconducting, biodegradable and bioactive fibers for drug delivery. Express Polym. Lett. 2016, 10, 628–646. [Google Scholar] [CrossRef] [Green Version]
- Planellas, M.; Pérez-Madrigal, M.M.; del Valle, L.J.; Kobauri, S.; Katsarava, R.; Alemán, C.; Puiggalí, J. Microfibres of conducting polythiophene and biodegradable poly(ester urea) for scaffolds. Polym. Chem. 2015, 6, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.W.; Hung, Y.J.; Yang, C.H.; Chen, Y.C. The antimicrobial activity of Gramicidin A is associated with hydroxyl radical formation. PLoS ONE 2015, 10, e0117065. [Google Scholar] [CrossRef] [PubMed]
- Yala, J.F.; Thebault, P.; Hequet, A.; Humblot, V.; Pradier, C.M.; Berjeaud, J.M. Elaboration of antibiofilm materials by chemical grafting of an antimicrobial peptide. Appl. Microbiol. Biotechnol. 2011, 89, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Qin, L.H.; Pace, C.J.; Wong, P.; Malonis, R.; Gao, J.M. Solubilized Gramicidin A as potential systemic antibiotics. ChemBioChem 2012, 13, 51–55. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Owens, T.A.; Barwe, S.P.; Rajasekaran, A.K. Gramicidin A induces metabolic dysfunction and energy depletion leading to cell death in renal cell carcinoma cells. Mol. Cancer Ther. 2013, 12, 2296–2307. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Owens, T.A.; Inge, L.J.; Bremmer, R.M.; Rajasekaran, A.K. Gramicidin A blocks tumor growth and angiogenesis through inhibition of hypoxia-inducible factor in renal cell carcinoma. Mol. Cancer Ther. 2014, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Wijesinghe, D.; Arachchige, M.C.; Lu, A.; Reshetnyak, Y.K.; Andreev, O.A. pH dependent transfer of nano-pores into membrane of cancer cells to induce apoptosis. Sci. Rep. 2013, 3, 3650. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.K.; Liu, H.Y.; Ambudkar, S.V.; Mayer, M. A combination of curcumin with either gramicidin or ouabain selectively kills cells that express the multidrug resistance-linked ABCG2 transporter. J. Biol. Chem. 2014, 289, 31397–31410. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, H.N.; Khan, M.S.; Wu, H.-F. Graphene oxide as a nanocarrier for gramicidin (GOGD) for high antibacterial performance. RSC Adv. 2014, 4, 50035–50046. [Google Scholar] [CrossRef]
- Dittrich, C.; Meier, W. Solid peptide nanoparticles—Structural characterization and quantification of cargo encapsulation. Macromol. Biosci. 2010, 10, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Schuster, T.B.; de Bruyn Outboter, D.; Bordignon, E.; Jeschke, G.; Meier, W. Reversible peptide particle formation using a mini amino acid sequence. Soft Matter 2010, 6, 5596–5604. [Google Scholar] [CrossRef]
- Wang, K.; Ruan, J.; Song, H.; Zhang, J.; Wo, Y.; Guo, S.; Cui, D. Biocompatibility of graphene oxide. Nanoscale Res. Lett. 2011, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Biron, E.; Chatterjee, J.; Ovadia, O.; Langenegger, D.; Brueggen, J.; Hoyer, D.; Schmid, H.A.; Jelinek, R.; Gilon, C.; Hoffman, A.; et al. Improving oral bioavailability of peptides by multiple N-methylation: somatostatin analogues. Angew. Chem. Int. Ed. 2008, 47, 2595–2599. [Google Scholar] [CrossRef] [PubMed]
- Horne, W.S.; Gellman, S.H. Foldamers with heterogeneous backbones. Acc. Chem. Res. 2008, 41, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Sadowsky, J.D.; Murray, J.K.; Tomita, Y.; Gellman, S.H. Exploration of backbone space in foldamers containing α- and β-amino acid residues: Developing Protease-resistant oligomers that bind tightly to the BH3-recognition cleft of Bcl-xL. ChemBioChem 2007, 8, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Solanas, C.; de la Torre, B.G.; Fernández-Reyes, M.; Santiverri, C.; Jiménez, M.A.; Rivas, L.; Jiménez, A.I.; Andreu, D.; Cativiela, C. Sequence inversion and phenylalanine surrogates at the β-turn enhance the antibiotic activity of Gramicidin S. J. Med. Chem. 2010, 53, 4119–4129. [Google Scholar] [CrossRef] [PubMed]
- Gause, G.F. Gramicidin S and its use in the treatment of infected wounds. Nature 1944, 154, 703. [Google Scholar] [CrossRef]
- Kondejewski, L.H.; Farmer, S.W.; Wishart, D.S.; Hancock, R.E.; Hodges, R.S. Gramicidin S is active against both Gram-positive and Gram-negative bacteria. Int. J. Pept. Protein. Res. 1996, 47, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Prenner, E.J.; Lewis, R.N.; McElhaney, R.N. The interaction of the antimicrobial peptide gramicidin S with lipid bilayer model and biological membranes. Biochim. Biophys. Acta 1999, 1462, 201–221. [Google Scholar] [CrossRef]
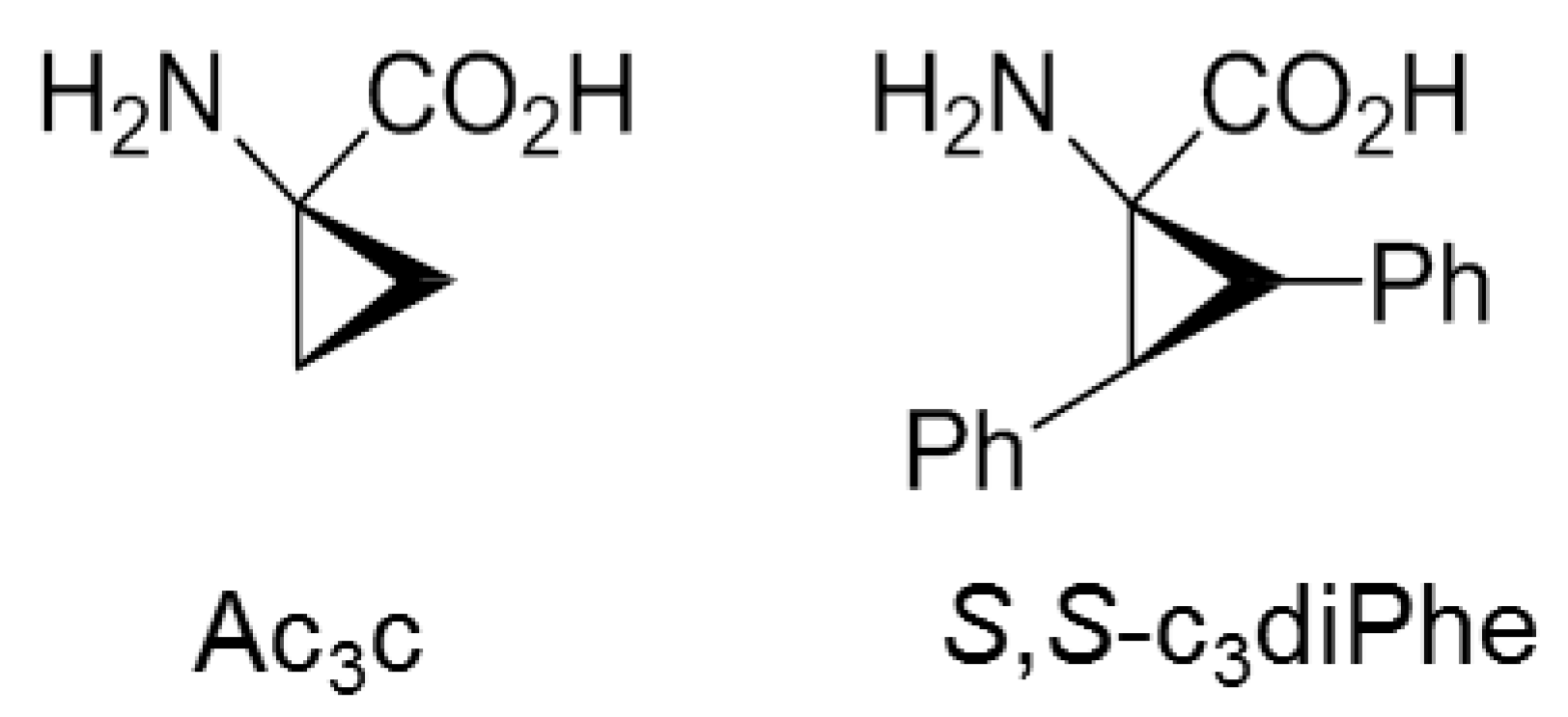
- Alemán, C. Conformational properties of α-amino acids disubstituted at the α-carbon. J. Phys. Chem. B 1997, 101, 5046–5050. [Google Scholar] [CrossRef]
- Alemán, C.; Jiménez, A.I.; Cativiela, C.; Pérez, J.J.; Casanovas, J. Influence of the phenyl side chain on the conformation of cyclopropane analogues of phenylalanine. J. Phys. Chem. B 2002, 106, 11849–11858. [Google Scholar] [CrossRef]
- Casanovas, J.; Jiménez, A.I.; Cativiela, C.; Pérez, J.J.; Alemán, C. N-Acetyl-N’-methylamide derivative of (2S,3S)-1-amino-2,3-diphenylcyclopropane carboxylic acid: Theoretical analysis of the conformational impact produced by the incorpororation of the second phenyl group to the cyclopropane analogue of phenylalanine. J. Org. Chem. 2003, 68, 7088–7091. [Google Scholar] [CrossRef] [PubMed]
- Frenot, A.; Chronakis, I.S. Polymer nanofibers assembled by electrospinning. Curr. Opin. Colloid Interface Sci. 2003, 8, 64–75. [Google Scholar] [CrossRef]
- Li, D.; Xia, Y. Electrospinning of nanofibers: Reinventing the wheel? Adv. Mater. 2004, 16, 1151–1170. [Google Scholar] [CrossRef]
- Jayaraman, K.; Kotaki, M.; Zhang, Y.; Mo, X.; Ramakrishna, S. Recent advances in polymer nanofibers. J. Nanosci. Nanotechnol. 2004, 4, 52–65. [Google Scholar] [PubMed]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, 668–673. [Google Scholar] [CrossRef] [PubMed]
- On-line analysis for protein Circular Dichroism spectra. Available online: http://dichroweb.cryst.bbk.ac.uk (accessed on 11 May 2017).
- Chen, Y.; Wallace, B.A. Solvent effects on the conformation and far UV CD spectra of gramicidin. Biopolymers 1997, 42, 771–781. [Google Scholar] [CrossRef]
- Llorens, E.; Ibañez, H.; del Valle, L.J.; Puiggalí, J. Biocompatibility and drug release behavior of scaffolds prepared by coaxial electrospinning of poly(butylene succinate) and polyethylene glycol. Mater. Sci. Eng. C 2015, 49, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Ji, J.H.; Zhang, W.; Zhang, Y.H.; Jiang, J.; Wu, Z.W.; Pu, S.H.; Chu, P.K. Biocompatibility and bioactivity of plasma-treated biodegradable poly(butylene succinate). Acta Biomater. 2009, 5, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-Ray Photoelectron Spectroscopy; Perkin-Elmer Corp: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Yang, Z.; Yuan, S.; Liang, B.; Liu, Y.; Choong, C.; Pehkonen, S.O. Chitosan microsphere scaffold tethered with RGD-conjugated poly(methacrylic acid) brushes as effective carriers for the endothelial cells. Macromol. Biosci. 2014, 14, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.C. Evaluation of a simple correction for the hydrocarbon contamination layer in quantitative surface analysis by XPS. J. Electron. Spectrosc. Relat. Phenom. 2005, 148, 21–28. [Google Scholar] [CrossRef]
- Marmur, A. Soft contact: Measurement and interpretation of contact angles. Soft Matter 2006, 2, 12–17. [Google Scholar] [CrossRef]
- Jeong, E.H.; Im, S.S.; Youk, J.H. Electrospinning and structural characterization of ultrafine poly (butylene succinate) fibers. Polymer 2005, 46, 9538–9543. [Google Scholar] [CrossRef]
- Del Valle, L.J.; Camps, R.; Díaz, A.; Franco, L.; Rodríguez-Galán, A.; Puiggalí, J. Electrospinning of polylactide and polycaprolactone mixtures for preparation of materials with tunable drug release properties. J. Polym. Res. 2011, 18, 1903–1917. [Google Scholar] [CrossRef]
- Murase, S.; Aymat, M.; Calvet, A.; del Valle, L.J.; Puiggalí, J. Electrosprayed poly (butylene succinate) microspheres loaded with indole derivatives: A system with anticancer activity. Eur. Polym. J. 2015, 71, 196–209. [Google Scholar] [CrossRef]
- Nishio, M.; Kohno, J.; Sakurai, M.; Suzuki, S.; Okada, N.; Kawano, K.; Komatsubara, S. TMC-135A and B, new triene-ansamycins, produced by Streptomyces sp. J. Antibiot. 2000, 53, 724–727. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D (nm) | RMS Rq (nm) | T50% (°C) | T70% (°C) | Tmax (°C) | θ (°) | |
|---|---|---|---|---|---|---|
| Unloaded PE44 | 246 ± 68 | 36.2 ± 8.7 | 410 | 420 | 419 | 125 ± 6 |
| PE44/GA-S1 | 106 ± 25 | 6.1 ± 2.4 | 400 | 410 | 412 | 128 ± 4 |
| PE44/GA-S2 | 146 ± 34 | 5.0 ± 1.7 | 387 | 400 | 401 | 47 ± 1 |
| PE44/GA-L | 590 ± 156 | 36.8 ± 3.6 | 405 | 416 | 415 | 68 ± 2 |
| C 1s (%) | N 1s (%) | O 1s (%) | |
|---|---|---|---|
| Unloaded PE44 | 69.23 | 0.26 | 30.51 |
| PE44/GA-S1 | 67.77 | 5.58 | 26.65 |
| PE44/GA-S2 | 66.63 | 3.11 | 30.26 |
| PE44/GA-L | 70.13 | 6.52 | 23.25 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maione, S.; Del Valle, L.J.; Pérez-Madrigal, M.M.; Cativiela, C.; Puiggalí, J.; Alemán, C. Antimicrobial Electrospun Fibers of Polyester Loaded with Engineered Cyclic Gramicidin Analogues. Fibers 2017, 5, 34. https://doi.org/10.3390/fib5030034
Maione S, Del Valle LJ, Pérez-Madrigal MM, Cativiela C, Puiggalí J, Alemán C. Antimicrobial Electrospun Fibers of Polyester Loaded with Engineered Cyclic Gramicidin Analogues. Fibers. 2017; 5(3):34. https://doi.org/10.3390/fib5030034
Chicago/Turabian StyleMaione, Silvana, Luis Javier Del Valle, Maria M. Pérez-Madrigal, Carlos Cativiela, Jordi Puiggalí, and Carlos Alemán. 2017. "Antimicrobial Electrospun Fibers of Polyester Loaded with Engineered Cyclic Gramicidin Analogues" Fibers 5, no. 3: 34. https://doi.org/10.3390/fib5030034
APA StyleMaione, S., Del Valle, L. J., Pérez-Madrigal, M. M., Cativiela, C., Puiggalí, J., & Alemán, C. (2017). Antimicrobial Electrospun Fibers of Polyester Loaded with Engineered Cyclic Gramicidin Analogues. Fibers, 5(3), 34. https://doi.org/10.3390/fib5030034