The Role of DNA Methylation in Common Skeletal Disorders

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
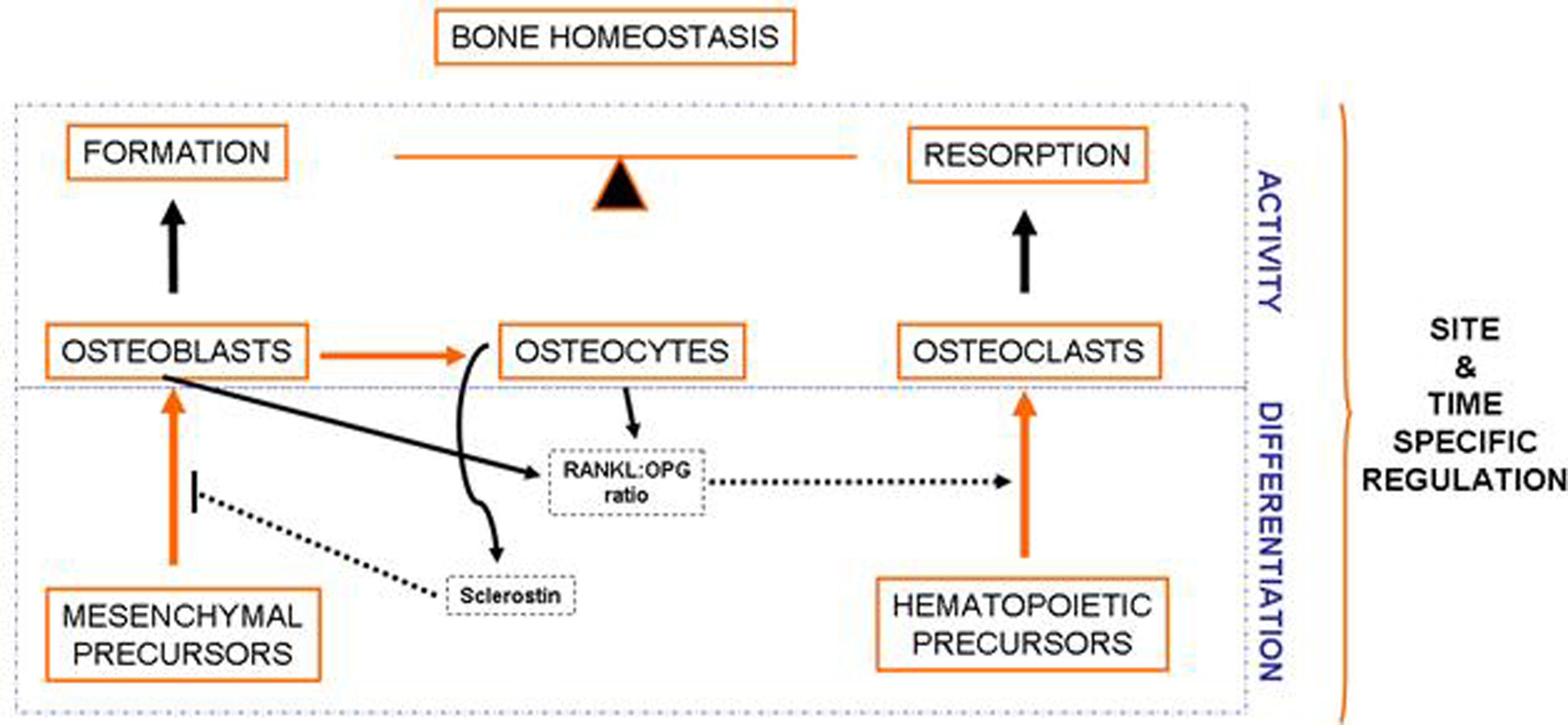
1. Bone Cells and Bone Remodeling in Health and Disease

2. DNA Methylation Influences Gene Expression
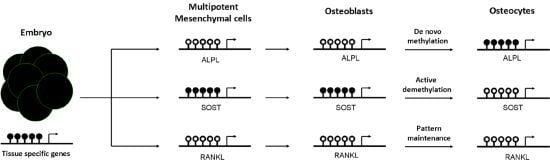
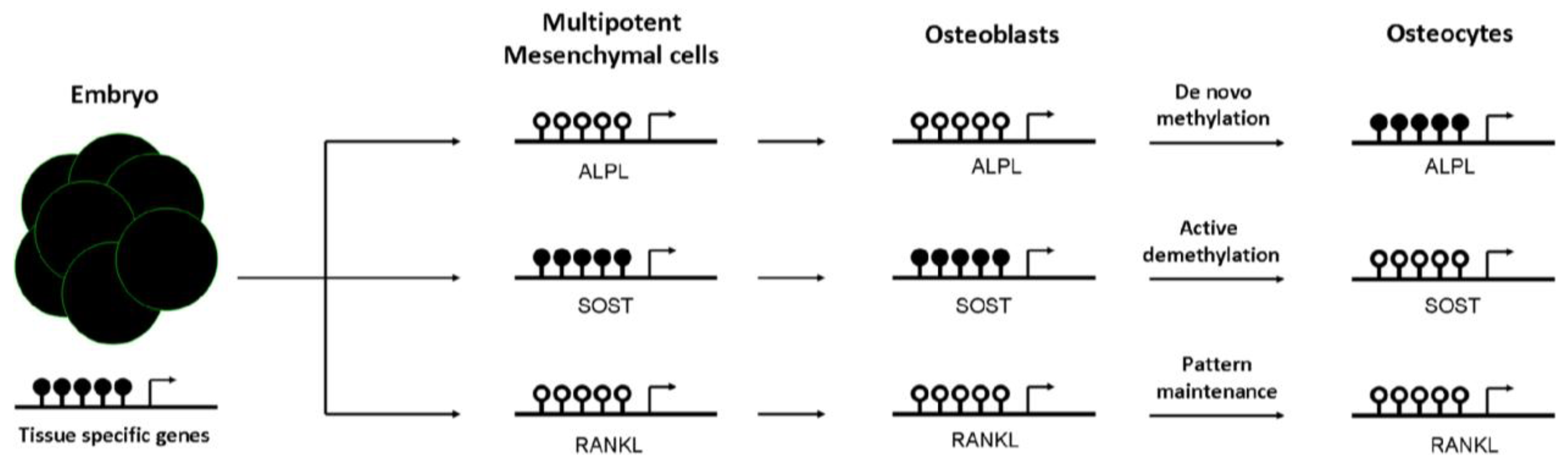
3. Role of DNA Methylation in Establishing a Bone Cell Phenotype

4. Methylation Marks and Common Skeletal Diseases
4.1. Osteoporosis
4.2. Osteoarthritis
4.3. Tumors and Bone
5. Concluding Remarks

Acknowledgments
References
- Matsuo, K.; Irie, N. Osteoclast-osteoblast communication. Arch. Biochem. Biophys. 2008, 473, 201–209. [Google Scholar] [CrossRef]
- Jackson, L.; Jones, D.R.; Scotting, P.; Sottile, V. Adult mesenchymal stem cells: Differentiation potential and therapeutic applications. J. Postgrad. Med. 2007, 53, 121–127. [Google Scholar]
- Vaananen, H.K.; Zhao, H.; Mulari, M.; Halleen, J.M. The cell biology of osteoclast function. J. Cell. Sci. 2000, 113, 377–381. [Google Scholar]
- Dallas, S.L.; Bonewald, L.F. Dynamics of the transition from osteoblast to osteocyte. Ann. NY Acad. Sci. 2010, 1192, 437–443. [Google Scholar]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef]
- Zarrinkalam, M.R.; Mulaibrahimovic, A.; Atkins, G.J.; Moore, R.J. Changes in osteocyte density correspond with changes in osteoblast and osteoclast activity in an osteoporotic sheep model. Osteoporos. Int. 2012, 23, 1329–1336. [Google Scholar]
- O’Brien, C.A.; Plotkin, L.I.; Galli, C.; Goellner, J.J.; Gortazar, A.R.; Allen, M.R.; Robling, A.G.; Bouxsein, M.; Schipani, E.; Turner, C.H.; et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One 2008, 3, e2942. [Google Scholar]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003, 22, 6267–6276. [Google Scholar]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. The RANKL/RANK/OPG pathway. Curr. Osteoporos. Rep. 2007, 5, 98–104. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar]
- Hadjidakis, D.J.; Androulakis, I.I. Bone remodeling. Ann. NY Acad. Sci. 2006, 1092, 385–396. [Google Scholar] [CrossRef]
- Sims, N.A.; Gooi, J.H. Bone remodeling: Multiple cellular interactions required for coupling of bone formation and resorption. Semin. Cell Dev. Biol. 2008, 19, 444–451. [Google Scholar]
- Raggatt, L.J.; Partridge, N.C. Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef]
- Dequeker, J.; Aerssens, J.; Luyten, F.P. Osteoarthritis and osteoporosis: Clinical and research evidence of inverse relationship. Aging Clin. Exp. Res. 2003, 15, 426–439. [Google Scholar]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: how osteoblasts become osteocytes. Dev. Dyn. 2006, 235, 176–190. [Google Scholar] [CrossRef]
- Probst, A.V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell. Biol. 2009, 10, 192–206. [Google Scholar]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Buiting, K.; Barnicoat, A.; Lich, C.; Pembrey, M.; Malcolm, S.; Horsthemke, B. Disruption of the bipartite imprinting center in a family with Angelman syndrome. Am. J. Hum. Genet. 2001, 68, 1290–1294. [Google Scholar]
- Buiting, K.; Gross, S.; Lich, C.; Gillessen-Kaesbach, G.; El Maarri, O.; Horsthemke, B. Epimutations in Prader-Willi and Angelman syndromes: A molecular study of 136 patients with an imprinting defect. Am. J. Hum. Genet. 2003, 72, 571–577. [Google Scholar] [CrossRef]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef]
- Bird, A.P. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980, 8, 1499–1504. [Google Scholar]
- Illingworth, R.S.; Gruenewald-Schneider, U.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Smith, C.; Harrison, D.J.; Andrews, R.; Bird, A.P. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010, 6, e1001134. [Google Scholar]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef]
- Hermann, A.; Gowher, H.; Jeltsch, A. Biochemistry and biology of mammalian DNA methyltransferases. Cell. Mol. Life. Sci. 2004, 61, 2571–2587. [Google Scholar]
- Leonhardt, H.; Page, A.W.; Weier, H.U.; Bestor, T.H. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 1992, 71, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar]
- Hashimshony, T.; Zhang, J.; Keshet, I.; Bustin, M.; Cedar, H. The role of DNA methylation in setting up chromatin structure during development. Nat. Genet. 2003, 34, 187–192. [Google Scholar]
- Razin, A.; Cedar, H. Distribution of 5-methylcytosine in chromatin. Proc. Natl. Acad. Sci. USA 1977, 74, 2725–2728. [Google Scholar] [CrossRef]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Frank, D.; Keshet, I.; Shani, M.; Levine, A.; Razin, A.; Cedar, H. Demethylation of CpG islands in embryonic cells. Nature 1991, 351, 239–241. [Google Scholar]
- Straussman, R.; Nejman, D.; Roberts, D.; Steinfeld, I.; Blum, B.; Benvenisty, N.; Simon, I.; Yakhini, Z.; Cedar, H. Developmental programming of CpG island methylation profiles in the human genome. Nat. Struct. Mol. Biol. 2009, 16, 564–571. [Google Scholar] [CrossRef]
- Lienert, F.; Wirbelauer, C.; Som, I.; Dean, A.; Mohn, F.; Schubeler, D. Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet. 2011, 43, 1091–1097. [Google Scholar]
- Epsztejn-Litman, S.; Feldman, N.; Abu-Remaileh, M.; Shufaro, Y.; Gerson, A.; Ueda, J.; Deplus, R.; Fuks, F.; Shinkai, Y.; Cedar, H.; et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 2008, 15, 1176–1183. [Google Scholar]
- Feldman, N.; Gerson, A.; Fang, J.; Li, E.; Zhang, Y.; Shinkai, Y.; Cedar, H.; Bergman, Y. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat. Cell. Biol. 2006, 8, 188–194. [Google Scholar] [CrossRef]
- Niehrs, C.; Schafer, A. Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol. 2012, 22, 220–227. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Kerkel, K.; Spadola, A.; Yuan, E.; Kosek, J.; Jiang, L.; Hod, E.; Li, K.; Murty, V.V.; Schupf, N.; Vilain, E.; et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat. Genet. 2008, 40, 904–908. [Google Scholar] [CrossRef]
- Tycko, B. Allele-specific DNA methylation: Beyond imprinting. Hum. Mol. Genet. 2010, 19, R210–R220. [Google Scholar] [CrossRef]
- Bell, J.T.; Pai, A.A.; Pickrell, J.K.; Gaffney, D.J.; Pique-Regi, R.; Degner, J.F.; Gilad, Y.; Pritchard, J.K. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011, 12, R10. [Google Scholar]
- Hellman, A.; Chess, A. Extensive sequence-influenced DNA methylation polymorphism in the human genome. Epigenetics Chromatin 2010, 3, 11. [Google Scholar] [CrossRef]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Jiang, Y.; Mishima, H.; Sakai, S.; Liu, Y.K.; Ohyabu, Y.; Uemura, T. Gene expression analysis of major lineage-defining factors in human bone marrow cells: Effect of aging, gender, and age-related disorder. J. Orthop. Res. 2008, 26, 910–917. [Google Scholar] [CrossRef]
- Kang, M.I.; Kim, H.S.; Jung, Y.C.; Kim, Y.H.; Hong, S.J.; Kim, M.K.; Baek, K.H.; Kim, C.C.; Rhyu, M.G. Transitional CpG methylation between promoters and retroelements of tissue-specific genes during human mesenchymal cell differentiation. J. Cell. Biochem. 2007, 102, 224–239. [Google Scholar] [CrossRef]
- Zhang, R.P.; Shao, J.Z.; Xiang, L.X. GADD45A protein plays an essential role in active DNA demethylation during terminal osteogenic differentiation of adipose-derived mesenchymal stem cells. J. Biol. Chem. 2011, 286, 41083–41094. [Google Scholar]
- Locklin, R.M.; Oreffo, R.O.; Triffitt, J.T. Modulation of osteogenic differentiation in human skeletal cells in vitro by 5-azacytidine. Cell Biol. Int. 1998, 22, 207–215. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Sanudo, C.; Bolado, A.; Fernandez, A.F.; Arozamena, J.; Pascual-Carra, M.A.; Rodriguez-Rey, J.C.; Fraga, M.F.; Bonewald, L.F.; Riancho, J.A. DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J. Bone Miner. Res. 2012, 27, 926–937. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Sanudo, C.; Sanchez-Verde, L.; Garcia-Renedo, R.J.; Arozamena, J.; Riancho, J.A. Epigenetic regulation of alkaline phosphatase in human cells of the osteoblastic lineage. Bone 2011, 49, 830–838. [Google Scholar] [CrossRef]
- Arnsdorf, E.J.; Tummala, P.; Castillo, A.B.; Zhang, F.; Jacobs, C.R. The epigenetic mechanism of mechanically induced osteogenic differentiation. J. Biomech. 2010, 43, 2881–2886. [Google Scholar] [CrossRef]
- Villagra, A.; Gutierrez, J.; Paredes, R.; Sierra, J.; Puchi, M.; Imschenetzky, M.; Wijnen, A.A.; Lian, J.; Stein, G.; Stein, J.; et al. Reduced CpG methylation is associated with transcriptional activation of the bone-specific rat osteocalcin gene in osteoblasts. J. Cell Biochem. 2002, 85, 112–122. [Google Scholar] [CrossRef]
- Dansranjavin, T.; Krehl, S.; Mueller, T.; Mueller, L.P.; Schmoll, H.J.; Dammann, R.H. The role of promoter CpG methylation in the epigenetic control of stem cell related genes during differentiation. Cell Cycle 2009, 8, 916–924. [Google Scholar] [CrossRef]
- Loeser, R.F.; Im, H.J.; Richardson, B.; Lu, Q.; Chubinskaya, S. Methylation of the OP-1 promoter: Potential role in the age-related decline in OP-1 expression in cartilage. Osteoarthr. Cartil. 2009, 17, 513–517. [Google Scholar]
- Lee, J.Y.; Lee, Y.M.; Kim, M.J.; Choi, J.Y.; Park, E.K.; Kim, S.Y.; Lee, S.P.; Yang, J.S.; Kim, D.S. Methylation of the mouse DIx5 and Osx gene promoters regulates cell type-specific gene expression. Mol. Cells 2006, 22, 182–188. [Google Scholar]
- Penolazzi, L.; Lambertini, E.; Giordano, S.; Sollazzo, V.; Traina, G.; del Senno, L.; Piva, R. Methylation analysis of the promoter F of estrogen receptor alpha gene: Effects on the level of transcription on human osteoblastic cells. J. Steroid Biochem.Mol. Biol. 2004, 91, 1–9. [Google Scholar] [CrossRef]
- Demura, M.; Bulun, S.E. CpG dinucleotide methylation of the CYP19 I.3/II promoter modulates cAMP-stimulated aromatase activity. Mol. Cell Endocrinol. 2008, 283, 127–132. [Google Scholar] [CrossRef]
- Thaler, R.; Agsten, M.; Spitzer, S.; Paschalis, E.P.; Karlic, H.; Klaushofer, K.; Varga, F. Homocysteine suppresses the expression of the collagen cross-linker lysyl oxidase involving IL-6, Fli1, and epigenetic DNA methylation. J. Biol. Chem. 2011, 286, 5578–5588. [Google Scholar]
- Teitell, M.A.; Mikkola, H.K. Transcriptional activators, repressors, and epigenetic modifiers controlling hematopoietic stem cell development. Pediatr. Res. 2006, 59, 33–39. [Google Scholar] [CrossRef]
- Riancho, J.A.; Delgado-Calle, J. Osteoblast-osteoclast interaction mechanisms. Reumatol. Clin. 2011, 7, S1–S4. [Google Scholar]
- Delgado-Calle, J.; Sanudo, C.; Fernandez, A.F.; Garcia-Renedo, R.; Fraga, M.F.; Riancho, J.A. Role of DNA methylation in the regulation of the RANKL-OPG system in human bone. Epigenetics 2012, 7, 83–91. [Google Scholar] [CrossRef]
- Kitazawa, R.; Kitazawa, S. Methylation status of a single CpG locus 3 bases upstream of TATA-box of receptor activator of nuclear factor-kappaB ligand RANKL. Gene promoter modulates cell- and tissue-specific RANKL expression and osteoclastogenesis. Mol. Endocrinol. 2007, 21, 148–158. [Google Scholar]
- Yasui, T.; Hirose, J.; Aburatani, H.; Tanaka, S. Epigenetic regulation of osteoclast differentiation. Ann. NY Acad. Sci. 2011, 1240, 7–13. [Google Scholar]
- Delgado-Calle, J.; Garmilla, P.; Riancho, J.A. Do epigenetic marks govern bone homeostasis? Curr. Genomics 2012, 13, 252–263. [Google Scholar]
- Kato, S.; Inoue, K.; Youn, M.I. Emergence of the osteo-epigenome in bone biology. IBMS BoneKEy 2010, 7, 314–324. [Google Scholar] [CrossRef]
- Earl, S.C.; Harvey, N.; Cooper, C. The epigenetic regulation of bone mass. IBMS BoneKEy 2010, 7, 54–62. [Google Scholar] [CrossRef]
- Riggs, B.L.; Khosla, S.; Melton, L.J. Sex steroids and the construction and conservation of the adult skeleton. Endocr. Rev. 2002, 23, 279–302. [Google Scholar] [CrossRef]
- Manolagas, S.C. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010, 31, 266–300. [Google Scholar] [CrossRef]
- Liu, L.; van Groen, T.; Kadish, I.; Li, Y.; Wang, D.; James, S.R.; Karpf, A.R.; Tollefsbol, T.O. Insufficient DNA methylation affects healthy aging and promotes age-related health problems. Clin. Epigenetics 2011, 2, 349–360. [Google Scholar] [CrossRef]
- Rodriguez-Rodero, S.; Fernandez-Morera, J.L.; Fernandez, A.F.; Menendez-Torre, E.; Fraga, M.F. Epigenetic regulation of aging. Discov. Med. 2010, 10, 225–233. [Google Scholar]
- Fraga, M.F.; Esteller, M. Epigenetics and aging: The targets and the marks. Trends Genet. 2007, 23, 413–418. [Google Scholar] [CrossRef]
- Fraga, M.F. Genetic and epigenetic regulation of aging. Curr. Opin. Immunol. 2009, 21, 446–453. [Google Scholar] [CrossRef]
- Calvanese, V.; Lara, E.; Kahn, A.; Fraga, M.F. The role of epigenetics in aging and age-related diseases. Ageing Res. Rev. 2009, 8, 268–276. [Google Scholar]
- Huidobro, C.; Fernandez, A.F.; Fraga, M.F. Aging epigenetics: Causes and consequences. Mol. Asp. Med. 2012, in press. [Google Scholar]
- Mahon, P.; Harvey, N.; Crozier, S.; Inskip, H.; Robinson, S.; Arden, N.; Swaminathan, R.; Cooper, C.; Godfrey, K. Low maternal vitamin D status and fetal bone development: Cohort study. J. Bone Miner. Res. 2010, 25, 14–19. [Google Scholar] [CrossRef]
- Oreffo, R.O.; Lashbrooke, B.; Roach, H.I.; Clarke, N.M.; Cooper, C. Maternal protein deficiency affects mesenchymal stem cell activity in the developing offspring. Bone 2003, 33, 100–107. [Google Scholar] [CrossRef]
- Lillycrop, K.A.; Phillips, E.S.; Torrens, C.; Hanson, M.A.; Jackson, A.A.; Burdge, G.C. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR alpha promoter of the offspring. Br. J. Nutr. 2008, 100, 278–282. [Google Scholar]
- Lillycrop, K.A.; Slater-Jefferies, J.L.; Hanson, M.A.; Godfrey, K.M.; Jackson, A.A.; Burdge, G.C. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br. J. Nutr. 2007, 97, 1064–1073. [Google Scholar] [CrossRef]
- Lillycrop, K.A.; Phillips, E.S.; Jackson, A.A.; Hanson, M.A.; Burdge, G.C. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J. Nutr. 2005, 135, 1382–1386. [Google Scholar]
- Brandt, K.D.; Dieppe, P.; Radin, E.L. Etiopathogenesis of osteoarthritis. Rheum. Dis. Clin. North Am. 2008, 34, 531–559. [Google Scholar]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar]
- Goldring, S.R. The role of bone in osteoarthritis pathogenesis. Rheum.Dis. Clin. North Am. 2008, 34, 561–571. [Google Scholar] [CrossRef]
- Bellido, M.; Lugo, L.; Roman-Blas, J.A.; Castaneda, S.; Calvo, E.; Largo, R.; Herrero-Beaumont, G. Improving subchondral bone integrity reduces progression of cartilage damage in experimental osteoarthritis preceded by osteoporosis. Osteoarthr. Cartil. 2011, 19, 1228–1236. [Google Scholar] [CrossRef]
- Goldring, M.B.; Goldring, S.R. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann. NY Acad. Sci. 2010, 1192, 230–237. [Google Scholar] [CrossRef]
- Herrero-Beaumont, G.; Roman-Blas, J.A.; Largo, R.; Berenbaum, F.; Castaneda, S. Bone mineral density and joint cartilage: Four clinical settings of a complex relationship in osteoarthritis. Ann.Rheum. Dis. 2011, 70, 1523–1525. [Google Scholar] [CrossRef]
- Suri, S.; Walsh, D.A. Osteochondral alterations in osteoarthritis. Bone 2012, 51, 204–211. [Google Scholar] [CrossRef]
- Roach, H.I.; Yamada, N.; Cheung, K.S.; Tilley, S.; Clarke, N.M.; Oreffo, R.O.; Kokubun, S.; Bronner, F. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005, 52, 3110–3124. [Google Scholar] [CrossRef]
- Roach, H.I.; Aigner, T. DNA methylation in osteoarthritic chondrocytes: A new molecular target. Osteoarthr. Cartil. 2007, 15, 128–137. [Google Scholar] [CrossRef]
- Zimmermann, P.; Boeuf, S.; Dickhut, A.; Boehmer, S.; Olek, S.; Richter, W. Correlation of COL10A1 induction during chondrogenesis of mesenchymal stem cells with demethylation of two CpG sites in the COL10A1 promoter. Arthritis Rheum. 2008, 589, 2743–2753. [Google Scholar]
- Barter, M.J.; Bui, C.; Young, D.A. Epigenetic mechanisms in cartilage and osteoarthritis: DNA methylation, histone modifications and microRNAs. Osteoarthr. Cartil. 2012, 20, 339–349. [Google Scholar]
- Goldring, M.B.; Marcu, K.B. Epigenomic and microRNA-mediated regulation in cartilage development, homeostasis, and osteoarthritis. Trends Mol. Med. 2012, 18, 109–118. [Google Scholar] [CrossRef]
- Bui, C.; Barter, M.J.; Scott, J.L.; Xu, Y.; Galler, M.; Reynard, L.N.; Rowan, A.D.; Young, D.A. cAMP response element-binding CREB recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. FASEB J. 2012, 26, 3000–3011. [Google Scholar]
- Poschl, E.; Fidler, A.; Schmidt, B.; Kallipolitou, A.; Schmid, E.; Aigner, T. DNA methylation is not likely to be responsible for aggrecan down regulation in aged or osteoarthritic cartilage. Ann. Rheum. Dis. 2005, 64, 477–480. [Google Scholar]
- Zuscik, M.J.; Baden, J.F.; Wu, Q.; Sheu, T.J.; Schwarz, E.M.; Drissi, H.; O’Keefe, R.J.; Puzas, J.E.; Rosier, R.N. 5-azacytidine alters TGF-beta and BMP signaling and induces maturation in articular chondrocytes. J. Cell Biochem. 2004, 922, 316–331. [Google Scholar]
- Javaid, M.K.; Lane, N.E.; Mackey, D.C.; Lui, L.Y.; Arden, N.K.; Beck, T.J.; Hochberg, M.C.; Nevitt, M.C. Changes in proximal femoral mineral geometry precede the onset of radiographic hip osteoarthritis: The study of osteoporotic fractures. Arthritis Rheum. 2009, 60, 2028–2036. [Google Scholar] [CrossRef]
- Baker-Lepain, J.C.; Lynch, J.A.; Parimi, N.; McCulloch, C.E.; Nevitt, M.C.; Corr, M.; Lane, N.E. Variant alleles of the WNT antagonist FRZB are determinants of hip shape and modify the relationship between hip shape and osteoarthritis. Arthritis Rheum. 2012, 64, 1457–1465. [Google Scholar] [CrossRef]
- Schiffern, A.N.; Stevenson, D.A.; Carroll, K.L.; Pimentel, R.; Mineau, G.; Viskochil, D.H.; Roach, J.W. Total hip arthroplasty; hip osteoarthritis; total knee arthroplasty; and knee osteoarthritis in patients with developmental dysplasia of the hip and their family members: A kinship analysis report. J. Pediatr. Orthop. 2012, 32, 609–612. [Google Scholar]
- Sandell, L.J. Etiology of osteoarthritis: genetics and synovial joint development. Nat. Rev. Rheumatol. 2012, 8, 77–89. [Google Scholar]
- Bos, S.D.; Slagboom, P.E.; Meulenbelt, I. New insights into osteoarthritis: Early developmental features of an ageing-related disease. Curr. Opin. Rheumatol. 2008, 20, 553–559. [Google Scholar] [CrossRef]
- Aspden, R.M. Osteoarthritis: A problem of growth not decay? Rheumatology 2008, 47, 1452–1460. [Google Scholar] [CrossRef]
- Panoutsopoulou, K.; Southam, L.; Elliott, K.S.; Wrayner, N.; Zhai, G.; Beazley, C.; Thorleifsson, G.; Arden, N.K.; Carr, A.; Chapman, K.; et al. Insights into the genetic architecture of osteoarthritis from stage 1 of the arcOGEN study. Ann. Rheum. Dis. 2011, 70, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Arcogen Consortium. Identification of new susceptibility loci for osteoarthritis arcOGEN: A genome-wide association study. Lancet 2012, 380, 815–823. [CrossRef] [Green Version]
- Valdes, A.M.; Spector, T.D. Genetic epidemiology of hip and knee osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 23–32. [Google Scholar] [CrossRef]
- Reynard, L.N.; Bui, C.; Canty-Laird, E.G.; Young, D.A.; Loughlin, J. Expression of the osteoarthritis-associated gene GDF5 is modulated epigenetically by DNA methylation. Hum. Mol. Genet. 2011, 20, 3450–3460. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Fernandez, A.F.; Sainz, J.; Zarrabeitia, M.T.; Garcia-Renedo, R.J.; Perez-Nunez, M.I.; Garcia-Ibarbia, C.; Fraga, M.F.; Riancho, J.A. Genome-wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis Rheum. 2012. [Google Scholar] [CrossRef]
- Saha, B.; Kaur, P.; Tsao-Wei, D.; Naritoku, W.Y.; Groshen, S.; Datar, R.H.; Jones, L.W.; Imam, S.A. Unmethylated E-cadherin gene expression is significantly associated with metastatic human prostate cancer cells in bone. Prostate 2008, 68, 1681–1688. [Google Scholar]
- Tost, J.; Hamzaoui, H.; Busato, F.; Neyret, A.; Mourah, S.; Dupont, JM.; Bouizar, Z. Methylation of specific CpG sites in the P2 promoter of parathyroid hormone-related protein determines the invasive potential of breast cancer cell lines. Epigenetics 2011, 6, 1035–1046. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Delgado-Calle, J.; Riancho, J.A. The Role of DNA Methylation in Common Skeletal Disorders. Biology 2012, 1, 698-713. https://doi.org/10.3390/biology1030698
Delgado-Calle J, Riancho JA. The Role of DNA Methylation in Common Skeletal Disorders. Biology. 2012; 1(3):698-713. https://doi.org/10.3390/biology1030698
Chicago/Turabian StyleDelgado-Calle, Jesús, and José A. Riancho. 2012. "The Role of DNA Methylation in Common Skeletal Disorders" Biology 1, no. 3: 698-713. https://doi.org/10.3390/biology1030698
APA StyleDelgado-Calle, J., & Riancho, J. A. (2012). The Role of DNA Methylation in Common Skeletal Disorders. Biology, 1(3), 698-713. https://doi.org/10.3390/biology1030698