Genetic and Epigenetic Regulation of CCR5 Transcription

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introductions
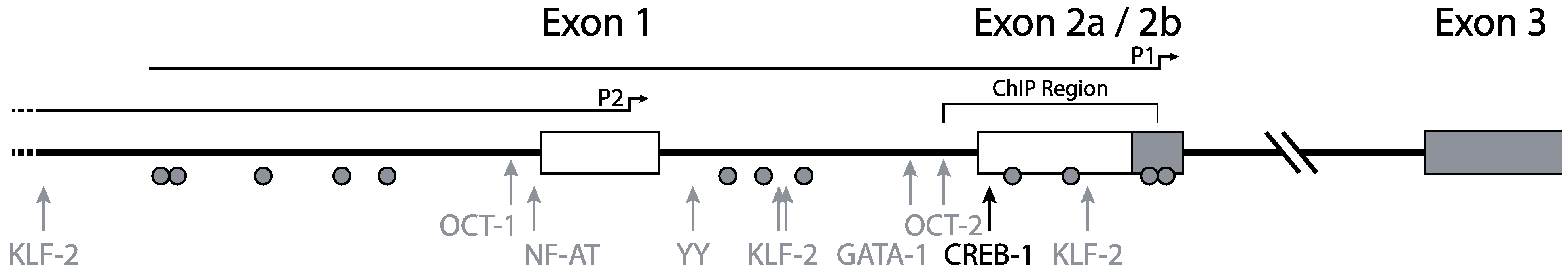
2. Genomic Organization

3. CCR5 Regulation by Transcription Factors
4. Epigenetic Regulation
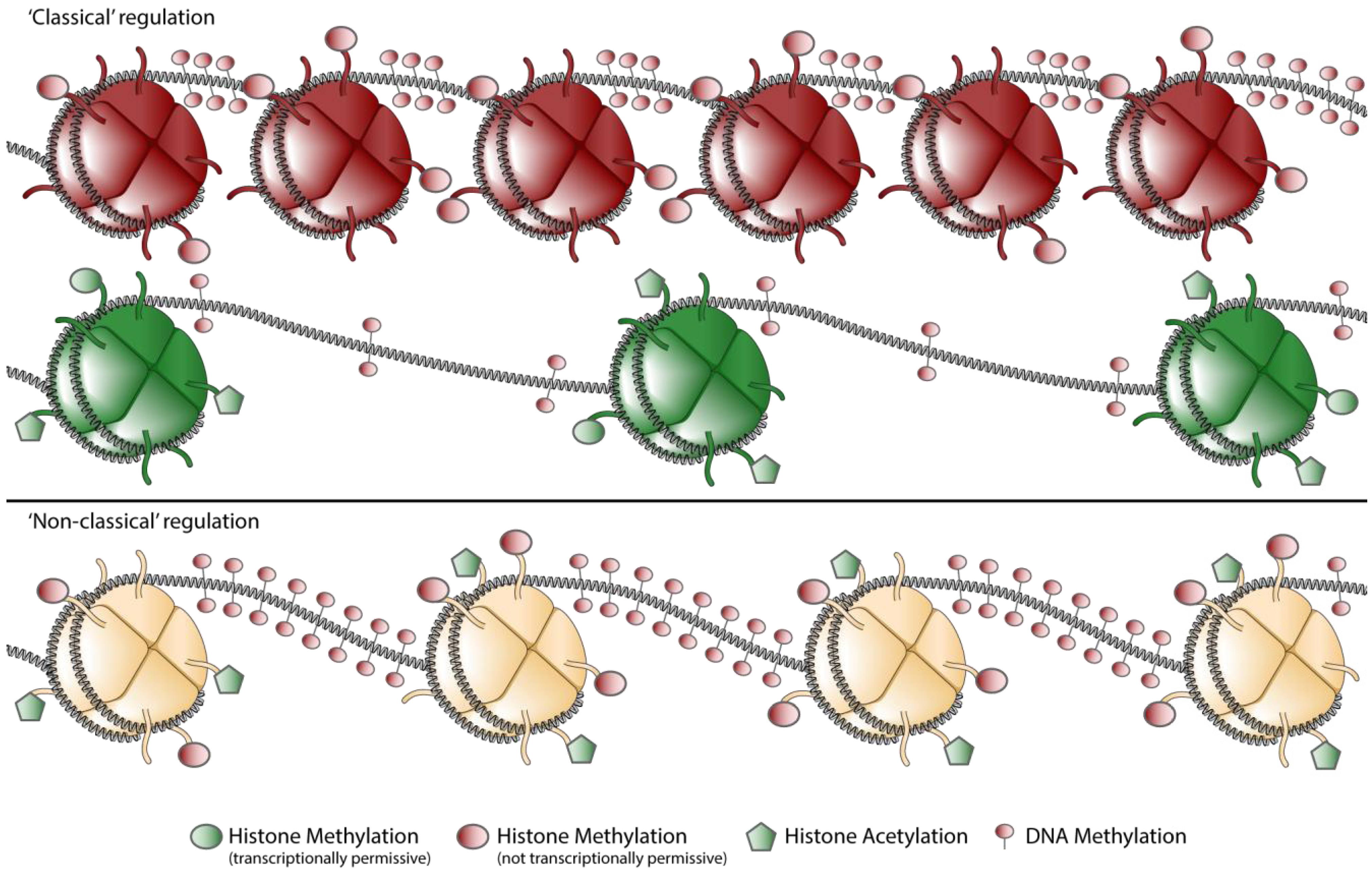
4.1. “Classical” Epigenetic Regulation

4.2. “Non-classical” Epigenetic Regulation
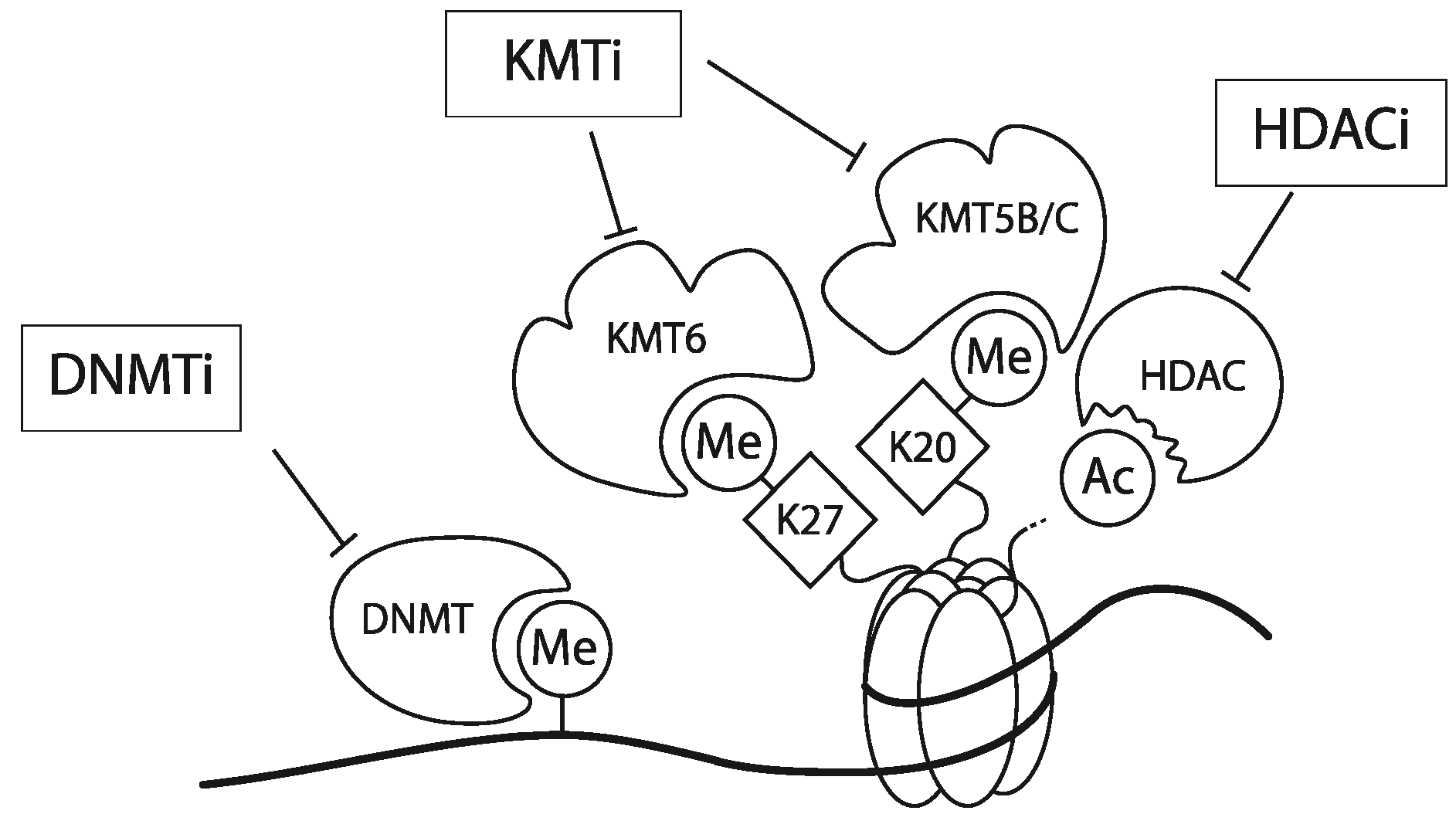
4.3. Epigenetic Intervention

5. Conclusions
Acknowledgments
References and Notes
- Samson, M.; Labbe, O.; Mollereau, C.; Vassart, G.; Parmentier, M. Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry 1996, 35, 3362–3367. [Google Scholar] [CrossRef]
- Combadiere, C.; Ahuja, S.K.; Tiffany, H.L.; Murphy, P.M. Cloning and functional expression of CC CKR5, a human monocyte CC chemokine receptor selective for MIP-1(alpha), MIP-1(beta), and RANTS. J. Leukoc. Biol. 1996, 60, 147–152. [Google Scholar]
- Raport, C.J.; Gosling, J.; Schweickart, V.L.; Gray, P.W.; Charo, I.F. Molecular cloning and functional characterization of a novel human CC chemokine receptor (CCR5) for RANTES, MIP-1beta, and MIP-1alpha. J. Biol. Chem. 1996, 271, 17161–17166. [Google Scholar]
- Bursill, C.A.; Channon, K.M.; Greaves, D.R. The role of chemokines in atherosclerosis: Recent evidence from experimental models and population genetics. Curr. Opin. Lipidol. 2004, 15, 145–149. [Google Scholar] [CrossRef]
- Ribeiro, S.; Horuk, R. The clinical potential of chemokine receptor antagonists. Pharmacol. Ther. 2005, 107, 44–58. [Google Scholar] [CrossRef]
- Biber, K.; Zuurman, M.W.; Dijkstra, I.M.; Boddeke, H.W. Chemokines in the brain: Neuroimmunology and beyond. Curr. Opin. Pharmacol. 2002, 2, 63–68. [Google Scholar] [CrossRef]
- Schober, A. Chemokines in vascular dysfunction and remodeling. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1950–1959. [Google Scholar] [CrossRef]
- Zernecke, A.; Liehn, E.A.; Gao, J.-L.; Kuziel, W.A.; Murphy, P.M.; Weber, C. Deficiency in CCR5 but not CCR1 protects against neointima formation in atherosclerosis-prone mice: Involvement of IL-10. Blood 2006, 107, 4240–4243. [Google Scholar] [CrossRef]
- Zernecke, A.; Shagdarsuren, E.; Weber, C. Chemokines in atherosclerosis: An update. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1897–1908. [Google Scholar] [CrossRef]
- Kraaijeveld, A.O.; de Jager, S.C.; de Jager, W.J.; Prakken, B.J.; McColl, S.R.; Haspels, I.; Putter, H.; van Berkel, T.J.; Nagelkerken, L.; Jukema, J.W.; et al. CC chemokine ligand-5 (CCL5/RANTES) and CC chemokine ligand-18 (CCL18/PARC) are specific markers of refractory unstable angina pectoris and are transiently raised during severe ischemic symptoms. Circulation 2007, 116, 1931–1941. [Google Scholar] [CrossRef]
- Wu, L.; Paxton, W.A.; Kassam, N.; Ruffing, N.; Rottman, J.B.; Sullivan, N.; Choe, H.; Sodroski, J.; Newman, W.; Koup, R.A.; et al. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J. Exp. Med. 1997, 185, 1681–1691. [Google Scholar] [CrossRef]
- Oswald-Richter, K.; Grill, S.M.; Leelawong, M.; Tseng, M.; Kalams, S.A.; Hulgan, T.; Haas, D.W.; Unutmaz, D. Identification of a CCR5-expressing T cell subset that is resistant to R5-tropic HIV infection. PLoS Pathog. 2007, 3, e58. [Google Scholar] [CrossRef]
- Carrington, M.; Dean, M.; Martin, M.P.; O’Brien, S.J. Genetics of HIV-1 infection: Chemokine receptor CCR5 polymorphism and its consequences. Hum. Mol. Genet. 1999, 8, 1939–1945. [Google Scholar]
- Ebert, L.M.; McColl, S.R. Up-regulation of CCR5 and CCR6 on distinct subpopulations of antigen-activated CD4+ T lymphocytes. J. Immunol. 2002, 168, 65–72. [Google Scholar]
- Mummidi, S.; Adams, L.M.; VanCompernolle, S.E.; Kalkonde, M.; Camargo, J.F.; Kulkarni, H.; Bellinger, A.S.; Bonello, G.; Tagoh, H.; Ahuja, S.S.; et al. Production of specific mRNA transcripts, usage of an alternate promoter, and octamer-binding transcription factors influence the surface expression levels of the HIV coreceptor CCR5 on primary T cells. J. Immunol. 2007, 178, 5668–5681. [Google Scholar]
- Mummidi, S.; Ahuja, S.S.; McDaniel, B.L.; Ahuja, S.K. The human CC chemokine receptor 5 (CCR5) gene. Multiple transcripts with 5'-end heterogeneity, dual promoter usage, and evidence for polymorphisms within the regulatory regions and noncoding exons. J. Biol. Chem. 1997, 272, 30662–30671. [Google Scholar]
- Bleul, C.C.; Wu, L.; Hoxie, J.A.; Springer, T.A.; Mackay, C.R. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 1925–1930. [Google Scholar] [CrossRef]
- Guignard, F.; Combadiere, C.; Tiffany, H.L.; Murphy, P.M. Gene organization and promoter function for CC chemokine receptor 5 (CCR5). J. Immunol. 1998, 160, 985–992. [Google Scholar]
- Van der Merwe, P.A.; Davis, S.J. The immunological synapse—A multitasking system. Science 2002, 295, 1479–1480. [Google Scholar] [CrossRef]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse: A molecular machine controlling T cell activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef]
- Serbina, N.V.; Jia, T.; Hohl, T.M.; Pamer, E.G. Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 2008, 26, 421–452. [Google Scholar] [CrossRef]
- Schmitz, G.; Leuthauser-Jaschinski, K.; Orso, E. Are circulating monocytes as microglia orthologues appropriate biomarker targets for neuronal diseases? Cent. Nerv. Syst. Agents Med. Chem. 2009, 9, 307–330. [Google Scholar] [CrossRef]
- Chan, W.Y.; Kohsaka, S.; Rezaie, P. The origin and cell lineage of microglia: New concepts. Brain Res. Rev. 2007, 53, 344–354. [Google Scholar] [CrossRef]
- Hansson, G.K.; Robertson, A.K.L.; Soderberg-Naucler, C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef]
- Fox, R.J.; Kivisakk, P.; Fisher, E.; Tucky, B.; Lee, J.C.; Rudick, R.A.; Ransohoff, R.M. Multiple sclerosis: Chemokine receptor expression on circulating lymphocytes in correlation with radiographic measures of tissue injury. Mult. Scler. 2008, 14, 1036–1043. [Google Scholar] [CrossRef]
- Trebst, C.; Konig, F.; Ransohoff, R.; Bruck, W.; Stangel, M. CCR5 expression on macrophages/microglia is associated with early remyelination in multiple sclerosis lesions. Mult. Scler. 2008, 14, 728–733. [Google Scholar] [CrossRef]
- Mummidi, S.; Bamshad, M.; Ahuja, S.S.; Gonzalez, E.; Feuillet, P.M.; Begum, K.; Galvis, M.C.; Kostecki, V.; Valente, A.J.; Murthy, K.K.; et al. Evolution of human and non-human primate CC chemokine receptor 5 gene and mRNA. Potential roles for haplotype and mRNA diversity, differential haplotype-specific transcriptional activity, and altered transcription factor binding to polymorphic nucleotides in the pathogenesis of HIV-1 and simian immunodeficiency virus. J. Biol. Chem. 2000, 275, 18946–18961. [Google Scholar]
- Wierda, R.J.; Kuipers, H.F.; van Eggermond, M.C.J.A.; Benard, A.; van Leeuwen, J.C.; Carluccio, S.; Geutskens, S.B.; Jukema, J.W.; Marquez, V.E.; Quax, P.H.A.; et al. Epigenetic control of CCR5 transcript levels in immune cells and modulation by small molecules inhibitors. J. Cell. Mol. Med. 2012, 16, 1866–1877. [Google Scholar] [CrossRef]
- Liu, R.; Zhao, X.; Gurney, T.A.; Landau, N.R. Functional analysis of the proximal CCR5 promoter. AIDS Res. Hum. Retroviruses 1998, 14, 1509–1519. [Google Scholar] [CrossRef]
- Kuipers, H.F.; Biesta, P.J.; Montagne, L.J.; van Haastert, E.S.; van der Valk, P.; van den Elsen, P.J. CC chemokine receptor 5 gene promoter activation by the cyclic AMP response element binding transcription factor. Blood 2008, 112, 1610–1619. [Google Scholar] [CrossRef]
- Banerjee, A.; Pirrone, V.; Wigdahl, B.; Nonnemacher, M.R. Transcriptional regulation of the chemokine co-receptor CCR5 by the cAMP/PKA/CREB pathway. Biomed. Pharmacother. 2011, 65, 293–297. [Google Scholar] [CrossRef]
- Jin, Q.; Agrawal, L.; Meyer, L.; Tubiana, R.; Theodorou, I.; Alkhatib, G. CCR5Delta32 59537-G/A promoter polymorphism is associated with low translational efficiency and the loss of CCR5Delta32 protective effects. J. Virol. 2008, 82, 2418–2426. [Google Scholar] [CrossRef]
- Moriuchi, H.; Moriuchi, M.; Fauci, A.S. Cloning and analysis of the promoter region of CCR5, a coreceptor for HIV-1 entry. J. Immunol. 1997, 159, 5441–5449. [Google Scholar]
- Moriuchi, M.; Moriuchi, H. Octamer transcription factors up-regulate the expression of CCR5, a coreceptor for HIV-1 entry. J. Biol. Chem. 2001, 276, 8639–8642. [Google Scholar]
- Moriuchi, M.; Moriuchi, H. YY1 transcription factor down-regulates expression of CCR5, a major coreceptor for HIV-1. J. Biol. Chem. 2003, 278, 13003–13007. [Google Scholar] [CrossRef]
- Moriuchi, M.; Moriuchi, H.; Fauci, A.S. GATA-1 transcription factor transactivates the promoter for CCR5, a coreceptor for human immunodeficiency virus type 1 entry. Blood 1999, 93, 1433–1435. [Google Scholar]
- Richardson, M.W.; Jadlowsky, J.; Didigu, C.A.; Doms, R.W.; Riley, J.L. Kruppel-like Factor 2 Modulates CCR5 Expression and Susceptibility to HIV-1 Infection. J. Immunol. 2012. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745. [Google Scholar] [CrossRef]
- Larsen, F.; Gundersen, G.; Lopez, R.; Prydz, H. CpG islands as gene markers in the human genome. Genomics 1992, 13, 1095–1107. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef]
- Rice, J.C.; Briggs, S.D.; Ueberheide, B.; Barber, C.M.; Shabanowitz, J.; Hunt, D.F.; Shinkai, Y.; Allis, C.D. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol. Cell 2003, 12, 1591–1598. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 2004, 14, 155–164. [Google Scholar] [CrossRef]
- Schotta, G.; Lachner, M.; Sarma, K.; Ebert, A.; Sengupta, R.; Reuter, G.; Reinberg, D.; Jenuwein, T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004, 18, 1251–1262. [Google Scholar] [CrossRef]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef]
- Wang, P.; Lin, C.; Smith, E.R.; Guo, H.; Sanderson, B.W.; Wu, M.; Gogol, M.; Alexander, T.; Seidel, C.; Wiedemann, L.M.; et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 2009, 29, 6074–6085. [Google Scholar]
- Yan, C.; Boyd, D.D. Histone H3 acetylation and H3 K4 methylation define distinct chromatin regions permissive for transgene expression. Mol. Cell. Biol. 2006, 26, 6357–6371. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Vaissiere, T.; Sawan, C.; Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 2008, 659, 40–48. [Google Scholar] [CrossRef]
- Lamond, A.I.; Earnshaw, W.C. Structure and function in the nucleus. Science 1998, 280, 547–553. [Google Scholar] [CrossRef]
- Van Steensel, B. Chromatin: Constructing the big picture. EMBO J. 2011, 30, 1885–1895. [Google Scholar] [CrossRef]
- Schwartz, Y.B.; Pirrotta, V. Polycomb complexes and epigenetic states. Curr. Opin. Cell Biol. 2008, 20, 266–273. [Google Scholar] [CrossRef]
- Bender, J. Chromatin-based silencing mechanisms. Curr. Opin. Plant Biol. 2004, 7, 521–526. [Google Scholar] [CrossRef]
- Ball, D.J.; Gross, D.S.; Garrard, W.T. 5-methylcytosine is localized in nucleosomes that contain histone H1. Proc. Natl. Acad. Sci. USA 1983, 80, 5490–5494. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Bapat, S.A.; Jin, V.; Berry, N.; Balch, C.; Sharma, N.; Kurrey, N.; Zhang, S.; Fang, F.; Lan, X.; Li, M.; et al. Multivalent epigenetic marks confer microenvironment-responsive epigenetic plasticity to ovarian cancer cells. Epigenetics 2010, 5, 716–729. [Google Scholar] [CrossRef]
- De Gobbi, M.; Garrick, D.; Lynch, M.; Vernimmen, D.; Hughes, J.R.; Goardon, N.; Luc, S.; Lower, K.M.; Sloane-Stanley, J.A.; Pina, C.; et al. Generation of bivalent chromatin domains during cell fate decisions. Epigenet. Chromatin 2011, 4, 9. [Google Scholar] [CrossRef]
- Saito, A.; Yamashita, T.; Mariko, Y.; Nosaka, Y.; Tsuchiya, K.; Ando, T.; Suzuki, T.; Tsuruo, T.; Nakanishi, O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 4592–4597. [Google Scholar]
- Hu, E.; Dul, E.; Sung, C.-M.; Chen, Z.; Kirkpatrick, R.; Zhang, G.-F.; Johanson, K.; Liu, R.; Lago, A.; Hofmann, G.; et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther. 2003, 307, 720–728. [Google Scholar] [CrossRef]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; de Francesco, R.; Gallinari, P.; et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar]
- Matalon, S.; Palmer, B.E.; Nold, M.F.; Furlan, A.; Kassu, A.; Fossati, G.; Mascagni, P.; Dinarello, C.A. The histone deacetylase inhibitor ITF2357 decreases surface CXCR4 and CCR5 expression on CD4(+) T-cells and monocytes and is superior to valproic acid for latent HIV-1 expression in vitro. J. Acquir. Immune Defic. Syndr. 2010, 54, 1–9. [Google Scholar]
- Gerstner, T.; Bell, N.; Konig, S. Oral valproic acid for epilepsy—Long-term experience in therapy and side effects. Expert. Opin. Pharmacother. 2008, 9, 285–292. [Google Scholar] [CrossRef]
- Henry, T.R. The history of valproate in clinical neuroscience. Psychopharmacol. Bull. 2003, 37, 5–16. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wierda, R.J.; Van den Elsen, P.J. Genetic and Epigenetic Regulation of CCR5 Transcription. Biology 2012, 1, 869-879. https://doi.org/10.3390/biology1030869
Wierda RJ, Van den Elsen PJ. Genetic and Epigenetic Regulation of CCR5 Transcription. Biology. 2012; 1(3):869-879. https://doi.org/10.3390/biology1030869
Chicago/Turabian StyleWierda, Rutger J., and Peter J. Van den Elsen. 2012. "Genetic and Epigenetic Regulation of CCR5 Transcription" Biology 1, no. 3: 869-879. https://doi.org/10.3390/biology1030869
APA StyleWierda, R. J., & Van den Elsen, P. J. (2012). Genetic and Epigenetic Regulation of CCR5 Transcription. Biology, 1(3), 869-879. https://doi.org/10.3390/biology1030869