
Carfilzomib in Combination with Bortezomib Enhances Apoptotic Cell Death in B16-F1 Melanoma Cells

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
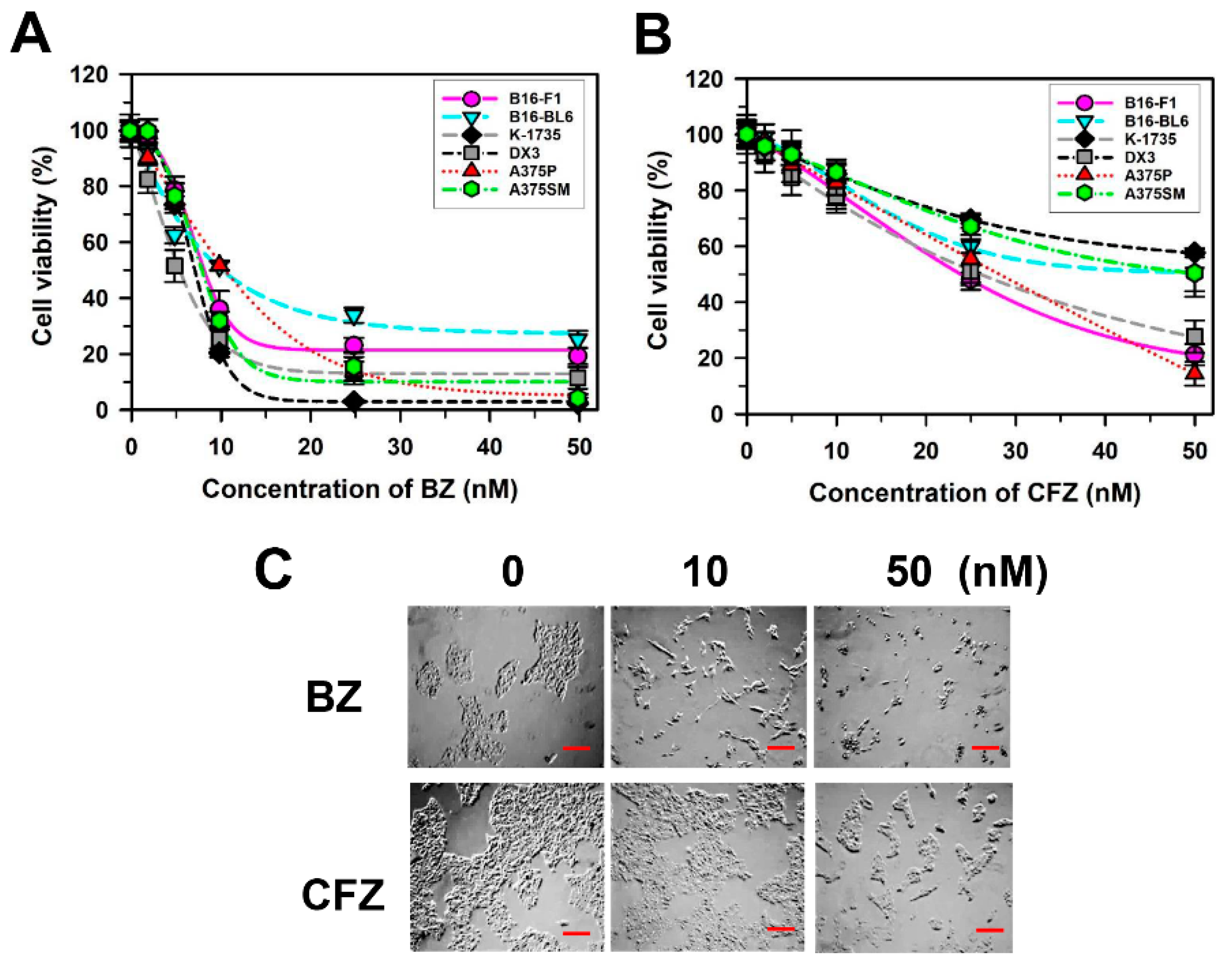
2.1. BZ and CFZ Induce Apoptosis in Various Types of Melanoma Cells Including B16-F1 Cells
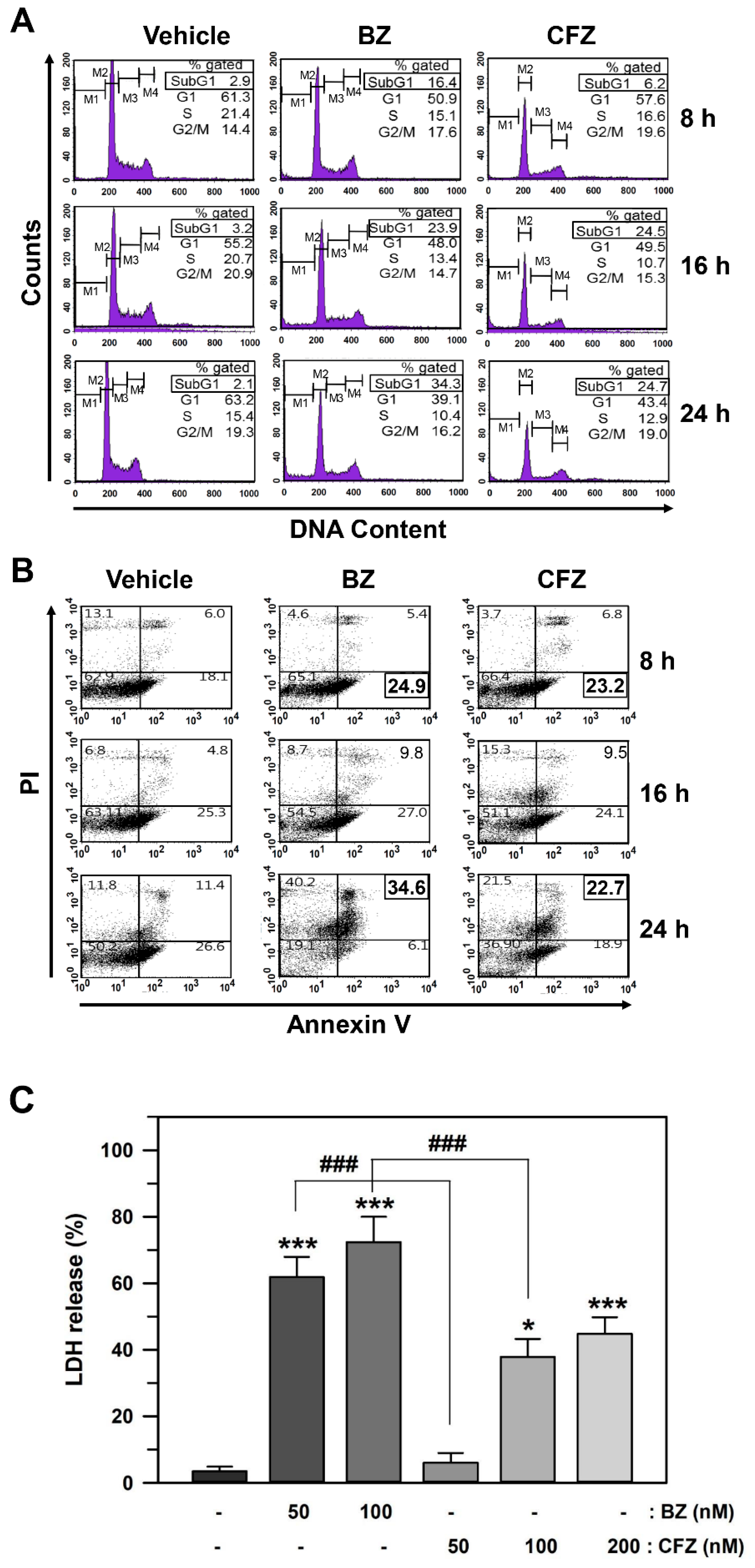
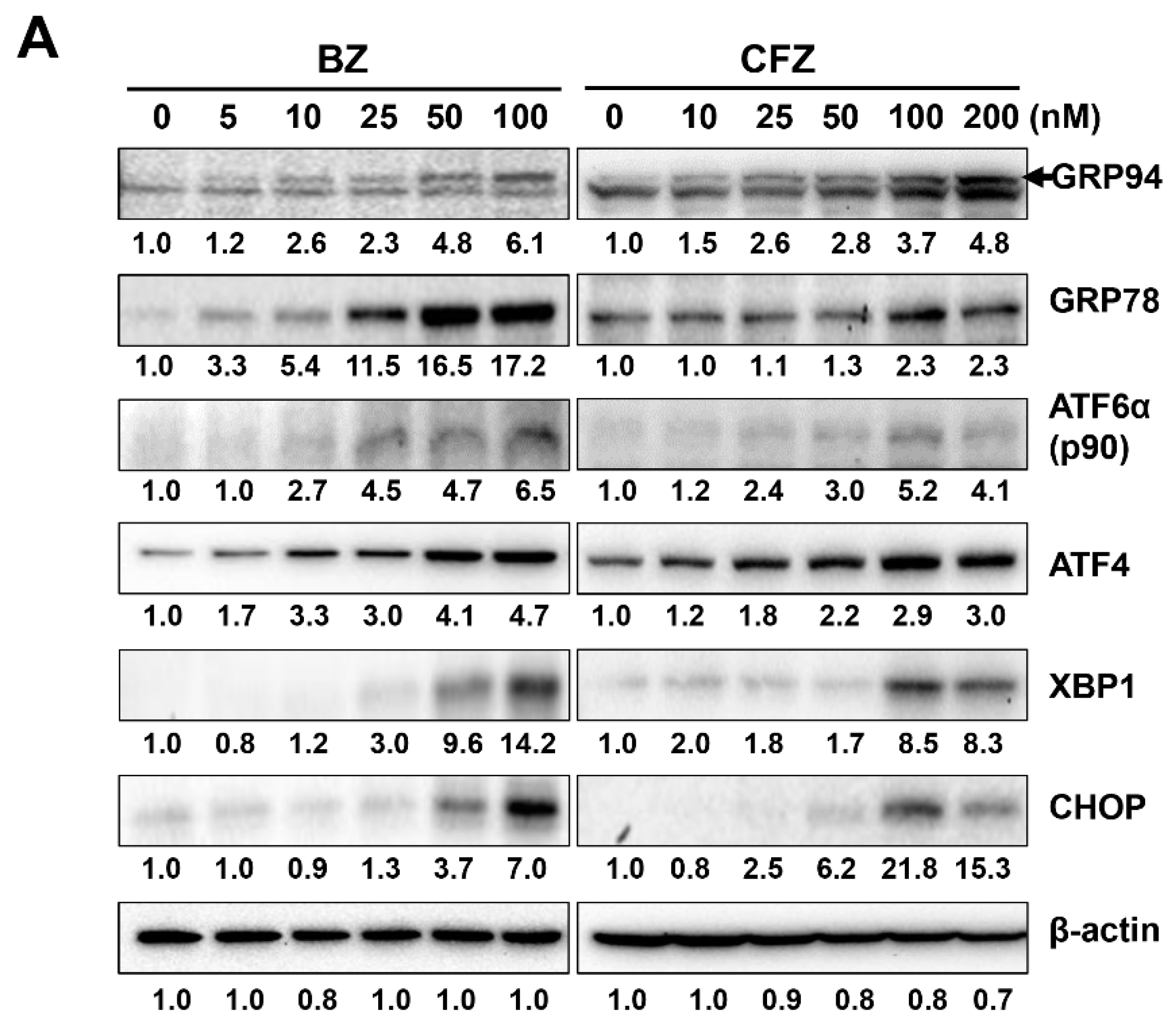
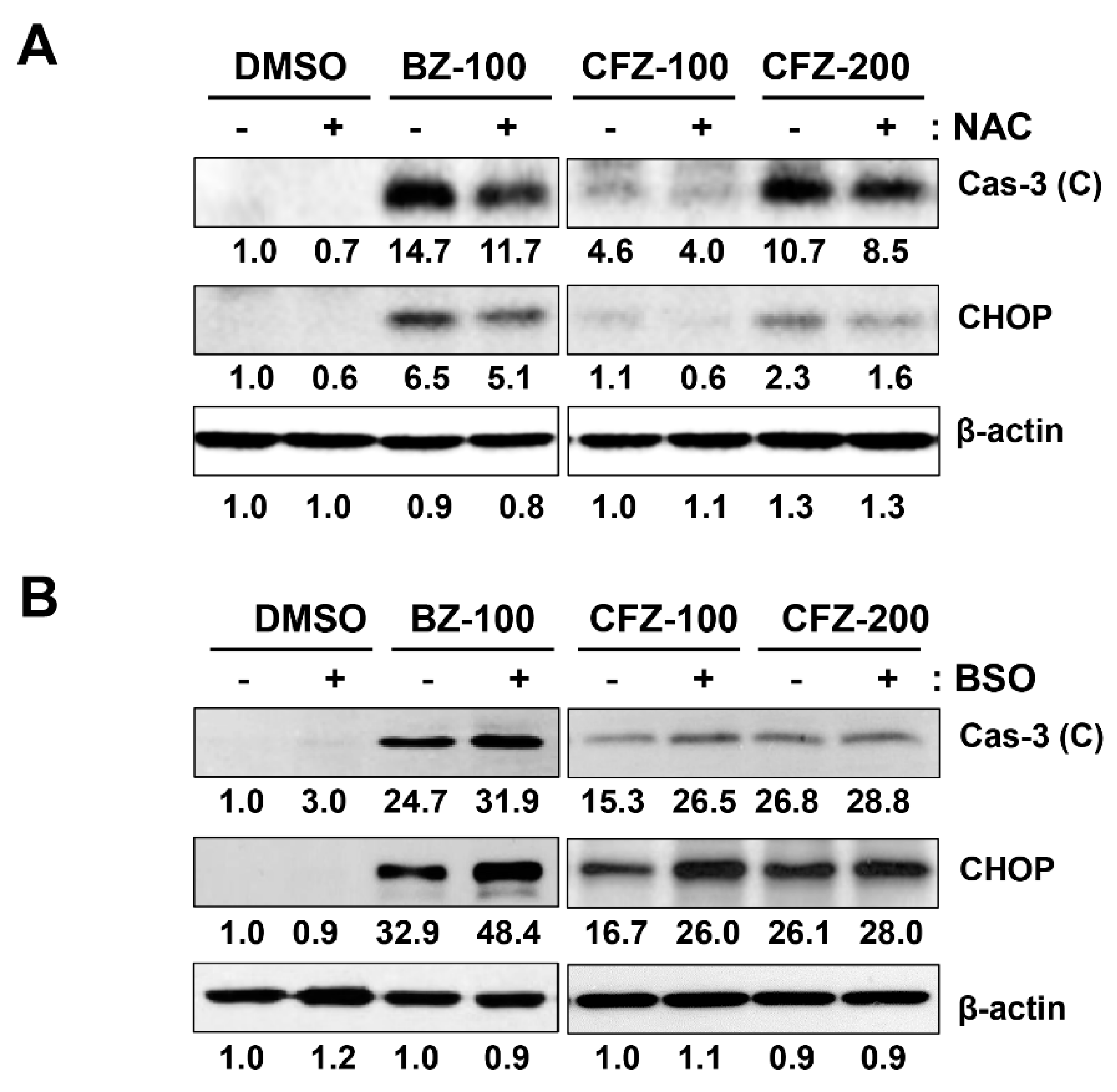
2.2. BZ and CFZ Induce Apoptosis via ER Stress in B16-F1 Cells
2.3. BZ and CFZ Increase ROS Accumulation and Mitochondrial Membrane Potential Loss in B16-F1 Cells
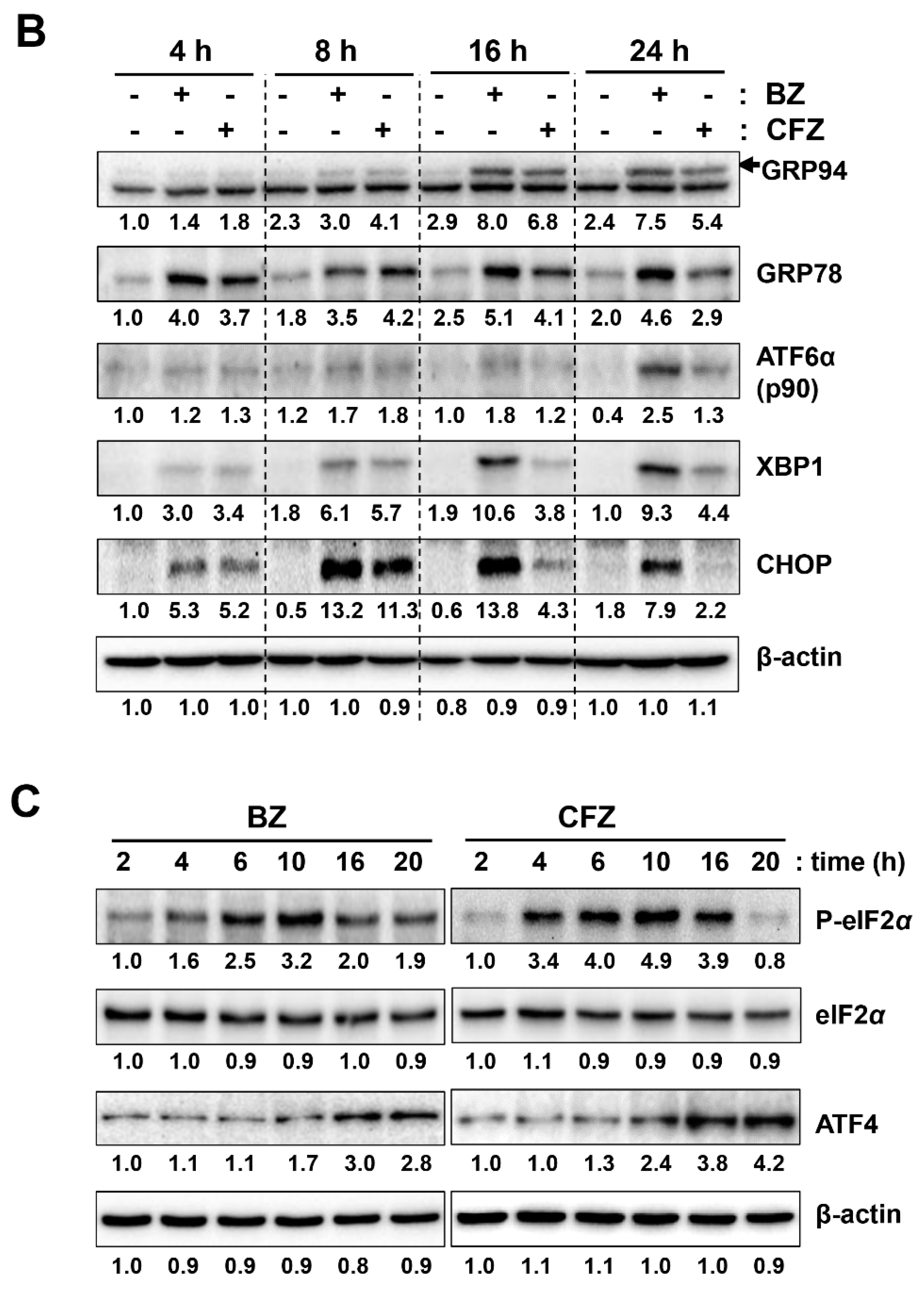
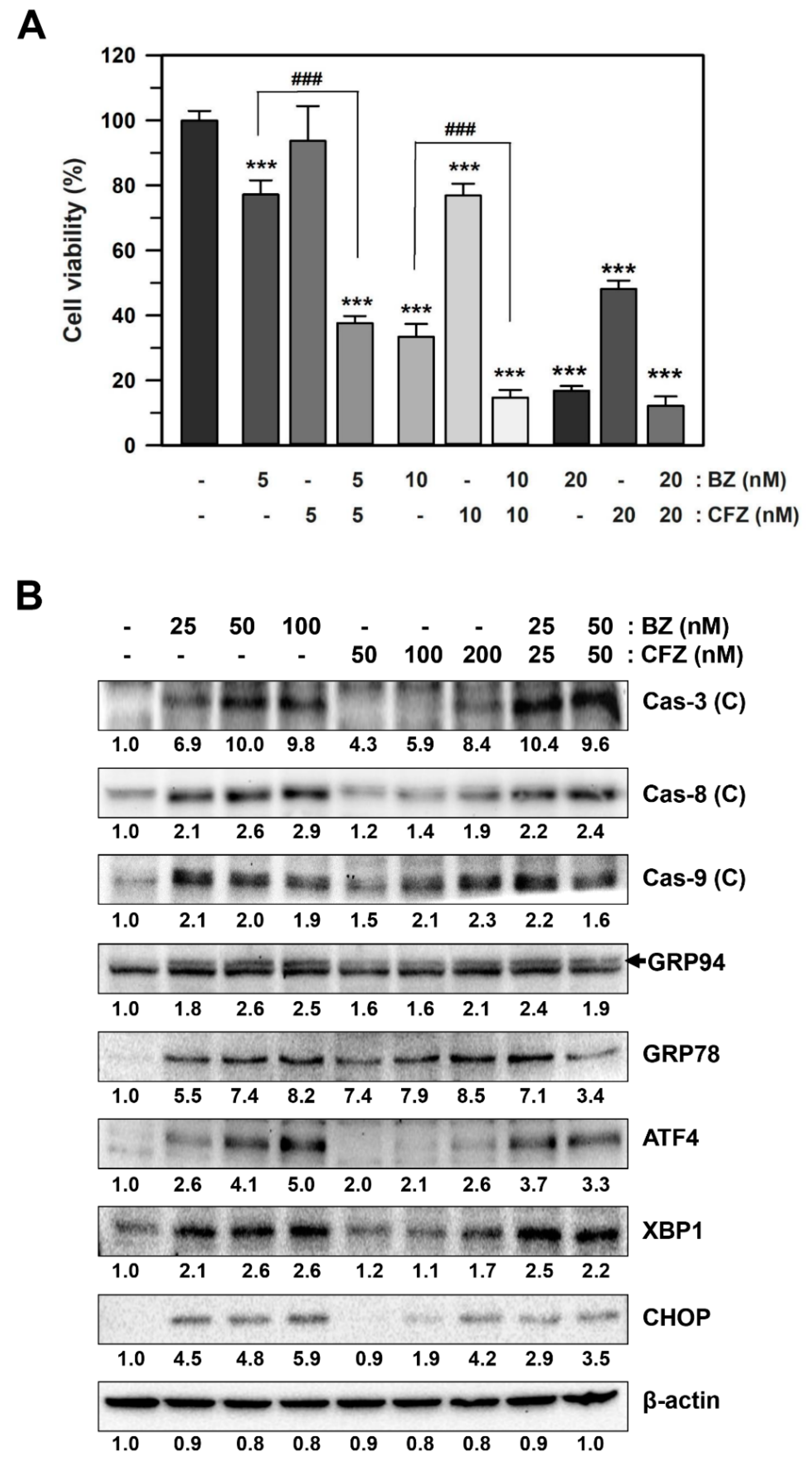
2.4. BZ and CFZ Synergistically Induce ER Stress and Apoptosis in B16-F1 Cells
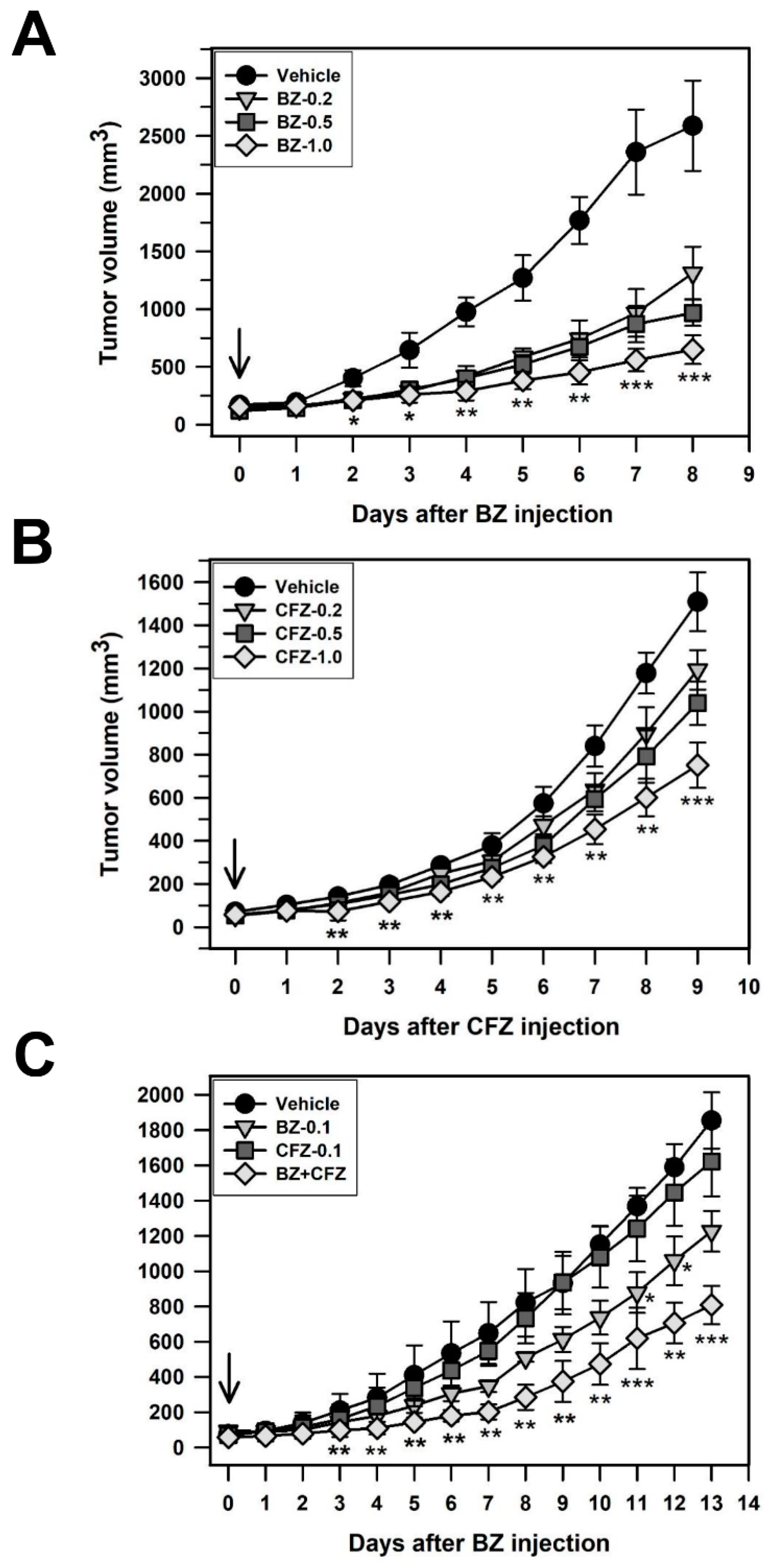
2.5. BZ and CFZ Synergistically Reduce Tumor Growth in a Xenograft Model Mouse C57BL/6
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. MTT Assay
4.4. Lactate Dehydrogenase Release Assay for Cytotoxicity Detection
4.5. Flow Cytometry for DNA Content Analysis and Apoptosis Detection
4.6. Western Blot Analysis
4.7. Measurement of Reactive Oxygen Species Production
4.8. Measurement of Mitochondrial Membrane Potential
4.9. Xenograft Model Experiment
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [Green Version]
- Lilenbaum, R.C.; Herndon, J.E., 2nd; List, M.A.; Desch, C.; Watson, D.M.; Miller, A.A.; Graziano, S.L.; Perry, M.C.; Saville, W.; Chahinian, P.; et al. Single-agent versus combination chemotherapy in advanced non-small-cell lung cancer: The cancer and leukemia group B (study 9730). J. Clin. Oncol. 2005, 23, 190–196. [Google Scholar] [CrossRef]
- Ali, Z.; Yousaf, N.; Larkin, J. Melanoma epidemiology, biology and prognosis. Eur. J. Cancer Suppl. 2013, 11, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghi, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kempen, L.C.; Redpath, M.; Robert, C.; Spatz, A. Molecular pathology of cutaneous melanoma. Melanoma Manag. 2014, 1, 151–164. [Google Scholar] [CrossRef]
- Apalla, Z.; Lallas, A.; Sotiriou, E.; Lazaridou, E.; Ioannides, D. Epidemiological trends in skin cancer. Dermatol. Pr. Concept. 2017, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.T.; Zmijewski, M.A.; Plonka, P.M.; Szaflarski, J.P.; Paus, R. How UV Light Touches the Brain and Endocrine System Through Skin, and Why. Endocrinology 2018, 159, 1992–2007. [Google Scholar] [CrossRef] [Green Version]
- Hawryluk, E.B.; Tsao, H. Melanoma: Clinical Features and Genomic Insights. Cold Spring Harb. Perspect. Med. 2014, 4, a015388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garman, B.; Anastopoulos, I.N.; Krepler, C.; Brafford, P.; Sproesser, K.; Jiang, Y.; Wubbenhorst, B.; Amaravadi, R.; Bennett, J.; Beqiri, M.; et al. Genetic and Genomic Characterization of 462 Melanoma Patient-Derived Xenografts, Tumor Biopsies, and Cell Lines. Cell Rep. 2017, 21, 1936–1952. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, V.O.; Bolshakov, S.V.; Walker, C.; Ananthaswamy, H.N. Genomic alterations in spontaneous and carcinogen-induced murine melanoma cell lines. Oncogene 2004, 23, 2347–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Cancer Society Survival Rates for Melanoma. Available online: https://www.cancer.org/cancer/melanoma-skin-cancer/about/key-statistics.html (accessed on 4 January 2018).
- National Cancer Institute Drugs Approved for Melanoma. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/melanoma (accessed on 13 June 2018).
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600–Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [Green Version]
- King, A.J.; Arnone, M.R.; Bleam, M.R.; Moss, K.G.; Yang, J.; Fedorowicz, K.E.; Smitheman, K.N.; Erhardt, J.A.; Hughes-Earle, A.; Kane-Carson, L.S.; et al. Dabrafenib; Preclinical Characterization, Increased Efficacy when Combined with Trametinib, while BRAF/MEK Tool Combination Reduced Skin Lesions. PLoS ONE 2013, 8, e67583. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.B.; Kefford, R.; Pavlick, A.C.; Infante, J.R.; Ribas, A.; Sosman, J.A.; Fecher, L.A.; Millward, M.; McArthur, G.A.; Hwu, P.; et al. Phase II Study of the MEK1/MEK2 Inhibitor Trametinib in Patients With Metastatic BRAF-Mutant Cutaneous Melanoma Previously Treated With or Without a BRAF Inhibitor. J. Clin. Oncol. 2013, 31, 482–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-H.; Ahn, J.-H.; Lee, M. Upregulation of MicroRNA-1246 Is Associated with BRAF Inhibitor Resistance in Melanoma Cells with Mutant BRAF. Cancer Res. Treat. 2017, 49, 947–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, R.M. Patient-Derived Orthotopic Xenograft (PDOX) Models of Melanoma. Int. J. Mol. Sci. 2017, 18, 1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Piva, M.; Sun, L.; Hong, A.; Moriceau, G.; Kong, X.; Zhang, H.; Lomeli, S.; Qian, J.; Yu, C.C.; et al. Recurrent Tumor Cell–Intrinsic and –Extrinsic Alterations during MAPKi-Induced Melanoma Regression and Early Adaptation. Cancer Discov. 2017, 7, 1248–1265. [Google Scholar] [CrossRef] [Green Version]
- Milijasevic, B.; Stefanovic, D.; Lalić-Popović, M.; Tomic, Z.; Kolarović, J.; Lalošević, D.; Mikov, M. Acute toxic effects of single dose dacarbazine: hematological and histological changes in an animal model. Biotech. Histochem. 2014, 89, 583–590. [Google Scholar] [CrossRef]
- Finley, D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Adams, J. The proteasome: A suitable antineoplastic target. Nat. Rev. Cancer 2004, 4, 349–360. [Google Scholar] [CrossRef]
- Hoeller, D.; Dikic, I. Targeting the ubiquitin system in cancer therapy. Nat. Cell Biol. 2009, 458, 438–444. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [Green Version]
- Kane, R.C.; Farrell, A.T.; Sridhara, R.; Pazdur, R. United States Food and Drug Administration Approval Summary: Bortezomib for the Treatment of Progressive Multiple Myeloma after One Prior Therapy. Clin. Cancer Res. 2006, 12, 2955–2960. [Google Scholar] [CrossRef] [Green Version]
- San Miguel, J.F.; Schlag, R.; Khuageva, N.K.; Dimopoulos, M.A.; Shpilberg, O.; Kropff, M.; Spicka, I.; Petrucci, M.T.; Palumbo, A.; Samoilova, O.S.; et al. Bortezomib plus Melphalan and Prednisone for Initial Treatment of Multiple Myeloma. New Engl. J. Med. 2008, 359, 906–917. [Google Scholar] [CrossRef] [Green Version]
- Teicher, B.A.; Anderson, K.C. CCR 20th Anniversary Commentary: In the Beginning, There Was PS-341. Clin. Cancer Res. 2015, 21, 939–941. [Google Scholar] [CrossRef] [Green Version]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal Structure of the Boronic Acid-Based Proteasome Inhibitor Bortezomib in Complex with the Yeast 20S Proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, J.; Henneberg, F.; Mata, R.A.; Tittmann, K.; Schneider, T.R.; Stark, H.; Bourenkov, G.; Chari, A. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pancheri, E.; Guglielmi, V.; Wilczynski, G.M.; Malatesta, M.; Tonin, P.; Tomelleri, G.; Nowis, D.; Vattemi, G. Non-Hematologic Toxicity of Bortezomib in Multiple Myeloma: The Neuromuscular and Cardiovascular Adverse Effects. Cancers 2020, 12, 2540. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Callander, N.S.; Hillengass, J.; Liedtke, M.; Baljevic, M.; Campagnaro, E.; Castillo, J.J.; Chandler, J.C.; Cornell, R.F.; Costello, C.; et al. NCCN Guidelines Insights: Multiple Myeloma, Version 1.2020. J. Natl. Compr. Cancer Netw. 2019, 17, 1154–1165. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hájek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef] [Green Version]
- Bose, P.; Batalo, M.S.; Holkova, B.; Grant, S. Bortezomib for the treatment of non-Hodgkin’s lymphoma. Expert Opin. Pharmacother. 2014, 15, 2443–2459. [Google Scholar] [CrossRef]
- Sayar, H.; Cripe, L.D.; Saliba, A.N.; Abu Zaid, M.; Konig, H.; Boswell, H.S. Combination of sorafenib, vorinostat and bortezomib for the treatment of poor-risk AML: Report of two consecutive clinical trials. Leuk. Res. 2019, 77, 30–33. [Google Scholar] [CrossRef] [Green Version]
- Kondagunta, G.V.; Drucker, B.J.; Schwartz, L.H.; Bacik, J.; Marion, S.; Russo, P.; Mazumdar, M.; Motzer, R.J. Phase II Trial of Bortezomib for Patients With Advanced Renal Cell Carcinoma. J. Clin. Oncol. 2004, 22, 3720–3725. [Google Scholar] [CrossRef]
- Shahshahan, M.A.; Beckley, M.N.; Jazirehi, A.R. Potential usage of proteasome inhibitor bortezomib (Velcade, PS-341) in the treatment of metastatic melanoma: Basic and clinical aspects. Am. J. Cancer Res. 2011, 1, 913–924. [Google Scholar]
- Herndon, T.M.; Deisseroth, A.; Kaminskas, E.; Kane, R.C.; Koti, K.M.; Rothmann, M.D.; Habtemariam, B.; Bullock, J.; Bray, J.D.; Hawes, J.; et al. Food and Drug Administration Approval: Carfilzomib for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 4559–4563. [Google Scholar] [CrossRef] [Green Version]
- Kortuem, K.M.; Stewart, A.K. Carfilzomib. Blood 2013, 121, 893–897. [Google Scholar] [CrossRef]
- Thompson, J.L. Carfilzomib: A Second-Generation Proteasome Inhibitor for the Treatment of Relapsed and Refractory Multiple Myeloma. Ann. Pharmacother. 2013, 47, 56–62. [Google Scholar] [CrossRef]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, Lenalidomide, and Dexamethasone for Relapsed Multiple Myeloma. New Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor Activity of PR-171, a Novel Irreversible Inhibitor of the Proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef] [Green Version]
- Parlati, F.; Lee, S.J.; Aujay, M.; Suzuki, E.; Levitsky, K.; Lorens, J.B.; Micklem, D.R.; Ruurs, P.; Sylvain, C.; Lu, Y.; et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009, 114, 3439–3447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harshbarger, W.; Miller, C.; Diedrich, C.; Sacchettini, J. Crystal Structure of the Human 20S Proteasome in Complex with Carfilzomib. Structure 2015, 23, 418–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.E.; Miller, Z.; Jun, Y.; Lee, W.; Kim, K.B. Next-generation proteasome inhibitors for cancer therapy. Transl. Res. 2018, 198, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Goldschmidt, H.; Niesvizky, R.; Joshua, D.; Chng, W.-J.; Oriol, A.; Orlowski, R.Z.; Ludwig, H.; Facon, T.; Hajek, R.; et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): An interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1327–1337. [Google Scholar] [CrossRef]
- Danhof, S.; Schreder, M.; Rasche, L.; Strifler, S.; Einsele, H.; Knop, S. ‘Real-life’ experience of preapproval carfilzomib-based therapy in myeloma - analysis of cardiac toxicity and predisposing factors. Eur. J. Haematol. 2015, 97, 25–32. [Google Scholar] [CrossRef]
- Efentakis, P.; Kremastiotis, G.; Varela, A.; Nikolaou, P.-E.; Papanagnou, E.-D.; Davos, C.H.; Tsoumani, M.; Agrogiannis, G.; Konstantinidou, A.; Kastritis, E.; et al. Molecular mechanisms of carfilzomib-induced cardiotoxicity in mice and the emerging cardioprotective role of metformin. Blood 2019, 133, 710–723. [Google Scholar] [CrossRef] [Green Version]
- Fotiou, D.; Roussou, M.; Gakiopoulou, C.; Psimenou, E.; Gavriatopoulou, M.; Migkou, M.; Kanellias, N.; Dialoupi, I.; Eleutherakis-Papaiakovou, E.; Giannouli, S.; et al. Carfilzomib-associated renal toxicity is common and unpredictable: A comprehensive analysis of 114 multiple myeloma patients. Blood Cancer J. 2020, 10, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.F.; Hanke, N.T.; Sands, B.J.; Carbajal, L.; Anderl, J.L.; Garland, L.L. Carfilzomib demonstrates broad anti-tumor activity in pre-clinical non-small cell and small cell lung cancer models. J. Exp. Clin. Cancer Res. 2014, 33, 111. [Google Scholar] [CrossRef] [Green Version]
- Park, J.E.; Park, J.; Jun, Y.; Oh, Y.; Ryoo, G.; Jeong, Y.-S.; Gadalla, H.H.; Min, J.S.; Jo, J.H.; Song, M.G.; et al. Expanding therapeutic utility of carfilzomib for breast cancer therapy by novel albumin-coated nanocrystal formulation. J. Control. Release 2019, 302, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-I.; Jeong, Y.J.; Yu, A.-R.; Kwak, H.J.; Cha, J.-Y.; Kang, I.; Yeo, E.-J. Carfilzomib enhances cisplatin-induced apoptosis in SK-N-BE(2)-M17 human neuroblastoma cells. Sci. Rep. 2019, 9, 5039. [Google Scholar] [CrossRef]
- Lee, A.S.; Hendershot, L.M. ER stress and cancer. Cancer Biol. Ther. 2006, 5, 721–722. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Chen, A.W.; Varner, J.D. A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 2011, 108, 2777–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selimovic, D.; Porzig, B.B.; El-Khattouti, A.; Badura, H.E.; Ahmad, M.; Ghanjati, F.; Santourlidis, S.; Haikel, Y.; Hassan, M. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell. Signal. 2013, 25, 308–318. [Google Scholar] [CrossRef]
- Li, Z.W.; Chen, H.; Campbell, R.A.; Bonavida, B.; Berenson, J.R. NF-kappaB in the pathogenesis and treatment of multiple myeloma. Curr. Opin. Hematol. 2008, 15, 391–399. [Google Scholar]
- Hideshima, T.; Mitsiades, C.; Akiyama, M.; Hayashi, T.; Chauhan, D.; Richardson, P.; Schlossman, R.; Podar, K.; Munshi, N.C.; Mitsiades, N.; et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood 2003, 101, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, S.V.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. Proteasome Inhibition as a Novel Therapeutic Target in Human Cancer. J. Clin. Oncol. 2005, 23, 630–639. [Google Scholar] [CrossRef]
- Meeth, K.; Wang, J.X.; Micevic, G.; Damsky, W.; Bosenberg, M.W. The YUMM lines: A series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment. Cell Melanoma Res. 2016, 29, 590–597. [Google Scholar] [CrossRef]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef]
- Choi, J.H.; Lee, J.Y.; Choi, A.Y.; Hwang, K.Y.; Choe, W.; Yoon, K.S.; Ha, J.; Yeo, E.J.; Kang, I. Apicidin induces endoplasmic reticulum stress- and mitochondrial dysfunction-associated apoptosis via phospholipase Cgamma1- and Ca(2+)-dependent pathway in mouse Neuro-2a neuroblastoma cells. Apoptosis 2012, 17, 1340–1358. [Google Scholar] [CrossRef]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free. Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef]
- Goy, A.; Bernstein, S.H.; Kahl, B.S.; Djulbegovic, B.; Robertson, M.J.; De Vos, S.; Epner, E.; Krishnan, A.; Leonard, J.P.; Lonial, S.; et al. Bortezomib in patients with relapsed or refractory mantle cell lymphoma: updated time-to-event analyses of the multicenter phase 2 PINNACLE study. Ann. Oncol. 2008, 20, 520–525. [Google Scholar] [CrossRef]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Zhang, N.; Fu, J.-N.; Chou, T.-C. Synergistic combination of microtubule targeting anticancer fludelone with cytoprotective panaxytriol derived from panax ginseng against MX-1 cells in vitro: Experimental design and data analysis using the combination index method. Am. J. Cancer Res. 2015, 6, 97–104. [Google Scholar] [PubMed]
- Tallarida, R.J. Quantitative Methods for Assessing Drug Synergism. Genes Cancer 2011, 2, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Amiri, K.I.; Horton, L.W.; LaFleur, B.J.; Sosman, J.A.; Richmond, A. Augmenting chemosensitivity of malignant melanoma tumors via proteasome inhibition: Implication for bortezomib (VELCADE, PS-341) as a therapeutic agent for malignant melanoma. Cancer Res. 2004, 64. [Google Scholar] [CrossRef] [Green Version]
- Freudlsperger, C.; Thies, A.; Pfüller, U.; Schumacher, U. The proteasome inhibitor bortezomib augments anti-proliferative effects of mistletoe lectin-I and the PPAR-gamma agonist rosiglitazone in human melanoma cells. Anticancer. Res. 2007, 27, 207–213. [Google Scholar]
- Qin, J.-Z.; Ziffra, J.; Stennett, L.; Bodner, B.; Bonish, B.K.; Chaturvedi, V.; Bennett, F.; Pollock, P.M.; Trent, J.M.; Hendrix, M.J.C.; et al. Proteasome Inhibitors Trigger NOXA-Mediated Apoptosis in Melanoma and Myeloma Cells. Cancer Res. 2005, 65, 6282–6293. [Google Scholar] [CrossRef] [Green Version]
- Sorolla, A.; Yeramian, A.; Dolcet, X.; Perez De Santos, A.; Llobet, D.; Schoenenberger, J.; Casanova, J.; Soria, X.; Egido, R.; Llombart, A.; et al. Effect of proteasome inhibitors on proliferation and apoptosis of human cutaneous melanoma-derived cell lines. Br. J. Dermatol. 2008, 158, 496–504. [Google Scholar] [CrossRef]
- Grayschopfer, V.C.; Wellbrock, C.; Marais, R. Melanoma biology and new targeted therapy. Nat. Cell Biol. 2007, 445, 851–857. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Montano, X.; Shamsher, M.; Whitehead, P.; Dawson, K.; Newton, J. Analysis of p53 in human cutaneous melanoma cell lines. Oncogene 1994, 9, 1455–1459. [Google Scholar]
- Easty, D.J.; Ganz, S.E.; Farr, C.J.; Lai, C.; Herlyn, M.; Bennett, D.C. Novel and Known Protein Tyrosine Kinases and Their Abnormal Expression in Human Melanoma. J. Investig. Dermatol. 1993, 101, 679–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, D.T.; Demenais, F.; Goldstein, A.M.; Bergman, W.; Bishop, J.N.; Bressac-de Paillerets, B.; Chompret, A.; Ghiorzo, P.; Gruis, N.; Hansson, J.; et al. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J. Natl. Cancer Inst. 2002, 94, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monzon, J.; Liu, L.; Brill, H.; Goldstein, A.M.; Tucker, M.A.; From, L.; McLaughlin, J.; Hogg, D.; Lassam, N.J. CDKN2A mutations in multiple primary melanomas. N. Engl. J. Med. 1998, 338, 879–887. [Google Scholar] [CrossRef]
- Zang, Y.; Thomas, S.M.; Chan, E.T.; Kirk, C.J.; Freilino, M.L.; DeLancey, H.M.; Grandis, J.R.; Li, C.; Johnson, D.E. Carfilzomib and ONX 0912 inhibit cell survival and tumor growth of head and neck cancer and their activities are enhanced by suppression of Mcl-1 or autophagy. Clin. Cancer Res. 2012, 18, 5639–5649. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Pham, L.V.; Newberry, K.J.; Ou, Z.; Liang, R.; Qian, J.; Sun, L.; Blonska, M.; You, Y.; Yang, J.; et al. In Vitro and In Vivo Therapeutic Efficacy of Carfilomib in Mantle Cell Lymphoma: Targeting the Immunoproteasome. Mol. Cancer Ther. 2013, 12, 2494–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravitz, L. Chemoprevention: First line of defence. Nat. Cell Biol. 2011, 471, S5–S7. [Google Scholar] [CrossRef]
- Atay, C.; Kwak, T.; Lavilla-Alonso, S.; Donthireddy, L.; Richards, A.D.; Moberg, V.; Pilon-Thomas, S.; Schell, M.J.; Messina, J.L.; Rebecca, V.W.; et al. BRAF Targeting Sensitizes Resistant Melanoma to Cytotoxic T Cells. Clin. Cancer Res. 2019, 25, 2783–2794. [Google Scholar] [CrossRef] [Green Version]
- Cosenza, M.; Civallero, M.; Pozzi, S.; Marcheselli, L.; Bari, A.; Sacchi, S. The combination of bortezomib with enzastaurin or lenalidomide enhances cytotoxicity in follicular and mantle cell lymphoma cell lines. Hematol. Oncol. 2014, 33, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Ji, C.; Mayfield, J.E.; Goel, A.; Xiao, J.; Dixon, J.E.; Guo, X. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. USA 2018, 115, 8155–8160. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin Pigmentation in Mammalian Skin and Its Hormonal Regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nat. Cell Biol. 2007, 445, 843–850. [Google Scholar] [CrossRef]
- Slominski, R.M.; Zmijewski, M.A.; Slominski, A.T. The role of melanin pigment in melanoma. Exp. Dermatol. 2015, 24, 258–259. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.T.; Lim, S.H.; Lee, B.; Kang, I.; Yeo, E.-J. Compound C Inhibits B16-F1 Tumor Growth in a Syngeneic Mouse Model Via the Blockage of Cell Cycle Progression and Angiogenesis. Cancers 2019, 11, 823. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.J.; Kim, G.-E.; Lee, Y.T.; Jeong, M.-S.; Kang, I.; Yang, D.; Yeo, E.-J. Inhibition of platelet-derived growth factor receptor tyrosine kinase and downstream signaling pathways by Compound C. Cell. Signal. 2013, 25, 883–897. [Google Scholar] [CrossRef]
- Christensen, M.E.; Jansen, E.S.; Sanchez, W.; Waterhouse, N.J. Flow cytometry based assays for the measurement of apoptosis-associated mitochondrial membrane depolarisation and cytochrome c release. Methods 2013, 61, 138–145. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Melanoma Cells | IC50 (nM) * | |
|---|---|---|
| BZ | CFZ | |
| B16-F1 (murine) | 8.1 | 23.9 |
| B16-BL6 (murine, higher metastatic) | 10.1 | 43.4 |
| K-1735 (murine, carcinogen-induced) | 5.4 | 26.1 |
| DX3 (human) | 7.1 | 57.6 |
| A375P (human, low metastatic) | 10.4 | 28.2 |
| A375SM (human, highly metastatic) | 7.9 | 51.0 |
| Drug | Cell Viability (Fa) a | Parameters b | CI c | DRI d | Comments | ||||
|---|---|---|---|---|---|---|---|---|---|
| BZ (nM) | CFZ (nM) | m | Dm | r | BZ | CFZ | |||
| 5 | - | 0.772 | –2.029 | 8.397 | –0.979 | ||||
| 10 | - | 0.334 | |||||||
| 20 | - | 0.169 | |||||||
| - | 5 | 0.937 | –2.002 | 18.91 | –0.999 | ||||
| - | 10 | 0.769 | |||||||
| - | 20 | 0.481 | |||||||
| 5 | 5 | 0.376 | –1.058 | 5.263 | –0.926 | 0.669 | 2.16 | 4.87 | Moderate synergism, favorable dose-reduction |
| 10 | 10 | 0.147 | 0.720 | 2.00 | 4.55 | ||||
| 20 | 20 | 0.122 | 1.295 | 1.11 | 2.53 | Antagonism, no dose- reduction for BZ but favorable for CFZ | |||
| Simulation | |||||||||
| 0.05 | 1.703 | 0.84 | 1.94 | Antagonism No or not favorable dose-reduction for BZ but favorable for CFZ | |||||
| 0.10 | 1.216 | 1.18 | 2.70 | ||||||
| 0.25 | 0.742 | 1.94 | 4.40 | Slight synergism, favorable dose-reduction | |||||
| 0.50 | 0.453 | 3.19 | 7.19 | Moderate synergism, favorable dose-reduction | |||||
| 0.75 | 0.276 | 5.24 | 11.7 | High synergism, favorable dose-reduction | |||||
| 0.90 | 0.168 | 8.62 | 19.1 | ||||||
| 0.95 | 0.120 | 12.1 | 26.7 | ||||||
| Days | N | Chi-Square (χ2) | Degree of Freedom (df) | Significance (p) |
|---|---|---|---|---|
| 0 | 20 | 6.020 | 3 | 0.111 |
| 1 | 20 | 3.160 | 3 | 0.368 |
| 2 | 20 | 10.768 | 3 | 0.013 * |
| 3 | 20 | 11.366 | 3 | 0.010 * |
| 4 | 20 | 14.154 | 3 | 0.003 ** |
| 5 | 20 | 15.274 | 3 | 0.002 ** |
| 6 | 20 | 15.332 | 3 | 0.002 ** |
| 7 | 20 | 15.754 | 3 | 0.001 ** |
| 8 | 20 | 17.583 | 3 | 0.001 ** |
| Days | N | Chi-Square (χ2) | Degree of Freedom (df) | Significance (p) |
|---|---|---|---|---|
| 0 | 20 | 3.909 | 3 | 0.271 |
| 1 | 20 | 6.020 | 3 | 0.111 |
| 2 | 20 | 13.118 | 3 | 0.004 ** |
| 3 | 20 | 13.674 | 3 | 0.003 ** |
| 4 | 20 | 14.842 | 3 | 0.002 ** |
| 5 | 20 | 12.389 | 3 | 0.006 ** |
| 6 | 20 | 16.419 | 3 | 0.001 ** |
| 7 | 20 | 15.642 | 3 | 0.001 ** |
| 8 | 20 | 15.915 | 3 | 0.001 ** |
| 9 | 20 | 17.010 | 3 | 0.001 ** |
| Days | N | Chi-Square (χ2) | Degree of Freedom (df) | Significance (p) |
|---|---|---|---|---|
| 0 | 24 | 6.111 | 3 | 0.106 |
| 1 | 24 | 4.419 | 3 | 0.220 |
| 2 | 24 | 6.099 | 3 | 0.107 |
| 3 | 24 | 11.346 | 3 | 0.010 * |
| 4 | 24 | 13.614 | 3 | 0.003 ** |
| 5 | 24 | 17.394 | 3 | 0.001 ** |
| 6 | 24 | 18.257 | 3 | 0.000 *** |
| 7 | 24 | 19.641 | 3 | 0.000 *** |
| 8 | 24 | 19.667 | 3 | 0.000 *** |
| 9 | 24 | 19.238 | 3 | 0.000 *** |
| 10 | 24 | 19.297 | 3 | 0.000 *** |
| 11 | 24 | 19.262 | 3 | 0.000 *** |
| 12 | 24 | 19.873 | 3 | 0.000 *** |
| 13 | 24 | 20.193 | 3 | 0.000 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.S.; Lim, S.H.; Yu, A.-R.; Hwang, C.Y.; Kang, I.; Yeo, E.-J. Carfilzomib in Combination with Bortezomib Enhances Apoptotic Cell Death in B16-F1 Melanoma Cells. Biology 2021, 10, 153. https://doi.org/10.3390/biology10020153
Lee MS, Lim SH, Yu A-R, Hwang CY, Kang I, Yeo E-J. Carfilzomib in Combination with Bortezomib Enhances Apoptotic Cell Death in B16-F1 Melanoma Cells. Biology. 2021; 10(2):153. https://doi.org/10.3390/biology10020153
Chicago/Turabian StyleLee, Min Seung, So Hyun Lim, Ah-Ran Yu, Chi Yeon Hwang, Insug Kang, and Eui-Ju Yeo. 2021. "Carfilzomib in Combination with Bortezomib Enhances Apoptotic Cell Death in B16-F1 Melanoma Cells" Biology 10, no. 2: 153. https://doi.org/10.3390/biology10020153
APA StyleLee, M. S., Lim, S. H., Yu, A. -R., Hwang, C. Y., Kang, I., & Yeo, E. -J. (2021). Carfilzomib in Combination with Bortezomib Enhances Apoptotic Cell Death in B16-F1 Melanoma Cells. Biology, 10(2), 153. https://doi.org/10.3390/biology10020153