
Crosstalk between Different DNA Repair Pathways Contributes to Neurodegenerative Diseases


Abstract
:Simple Summary
Abstract
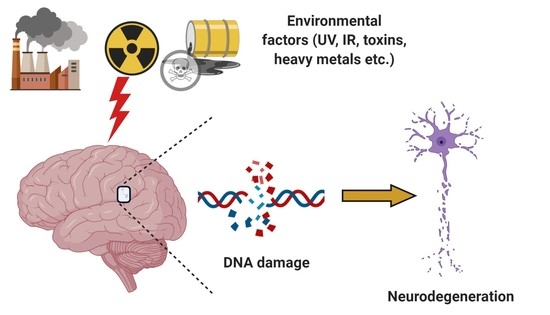
1. Introduction
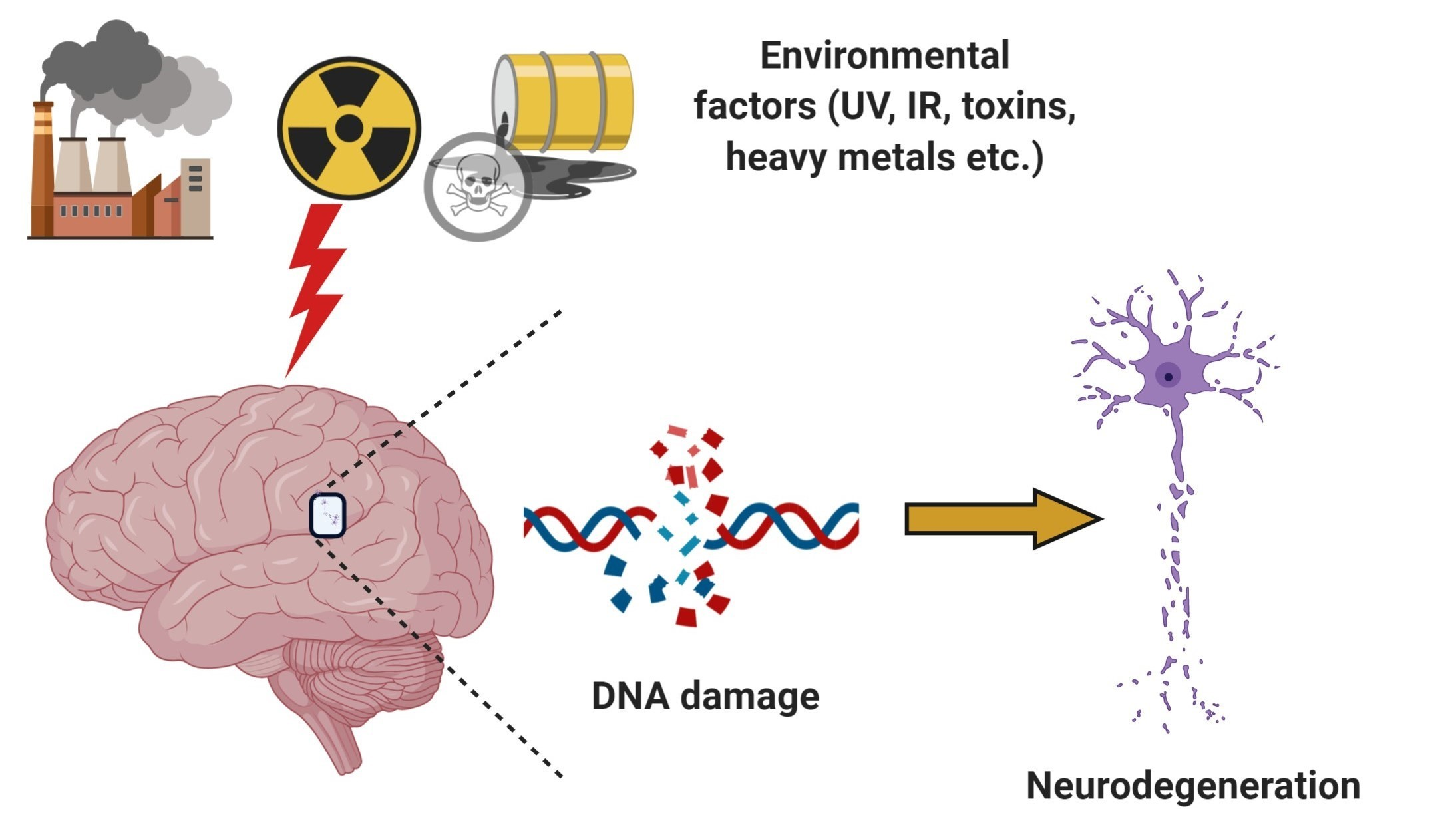
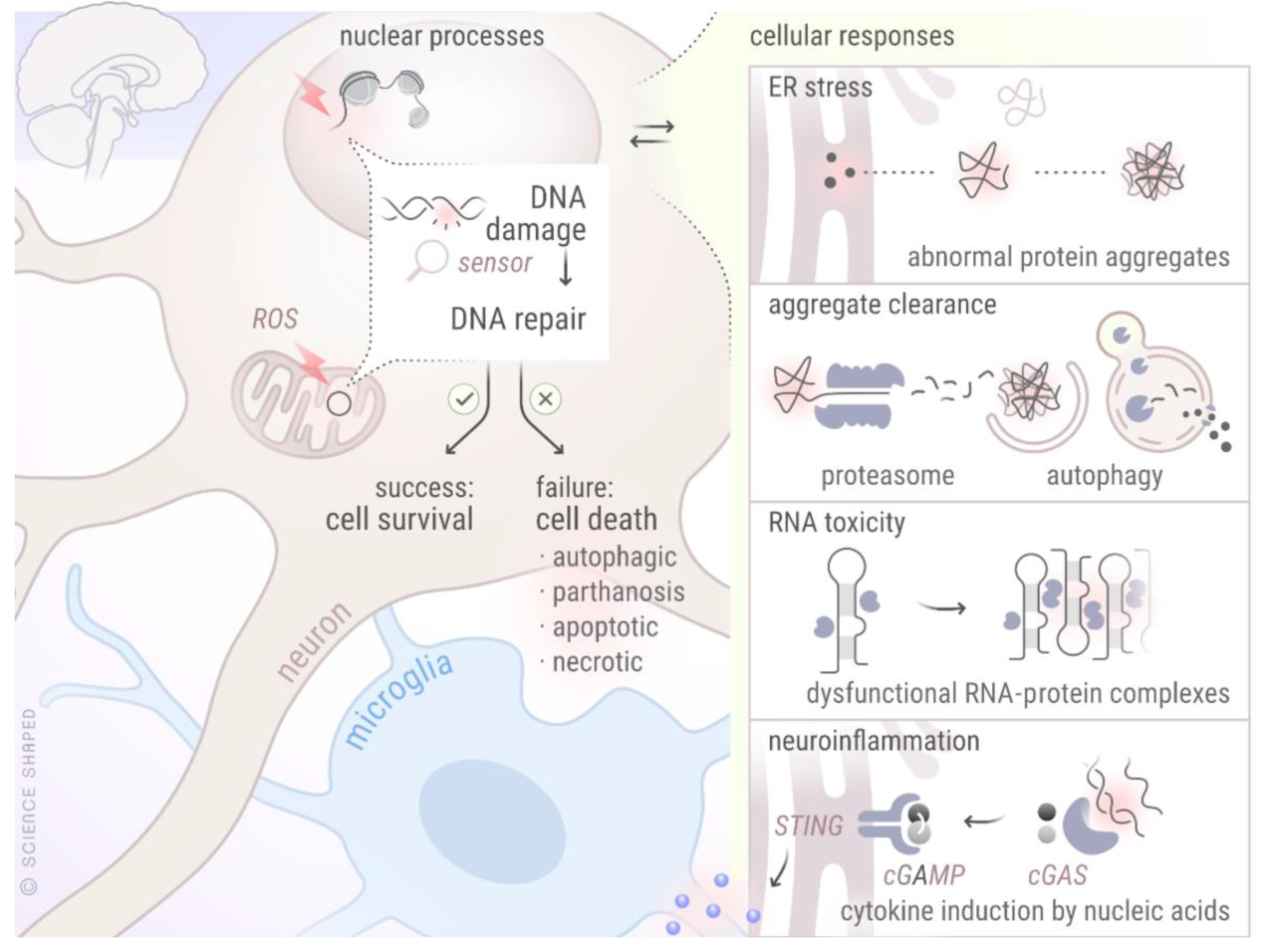
2. DNA Damage Response and Repair Mechanisms
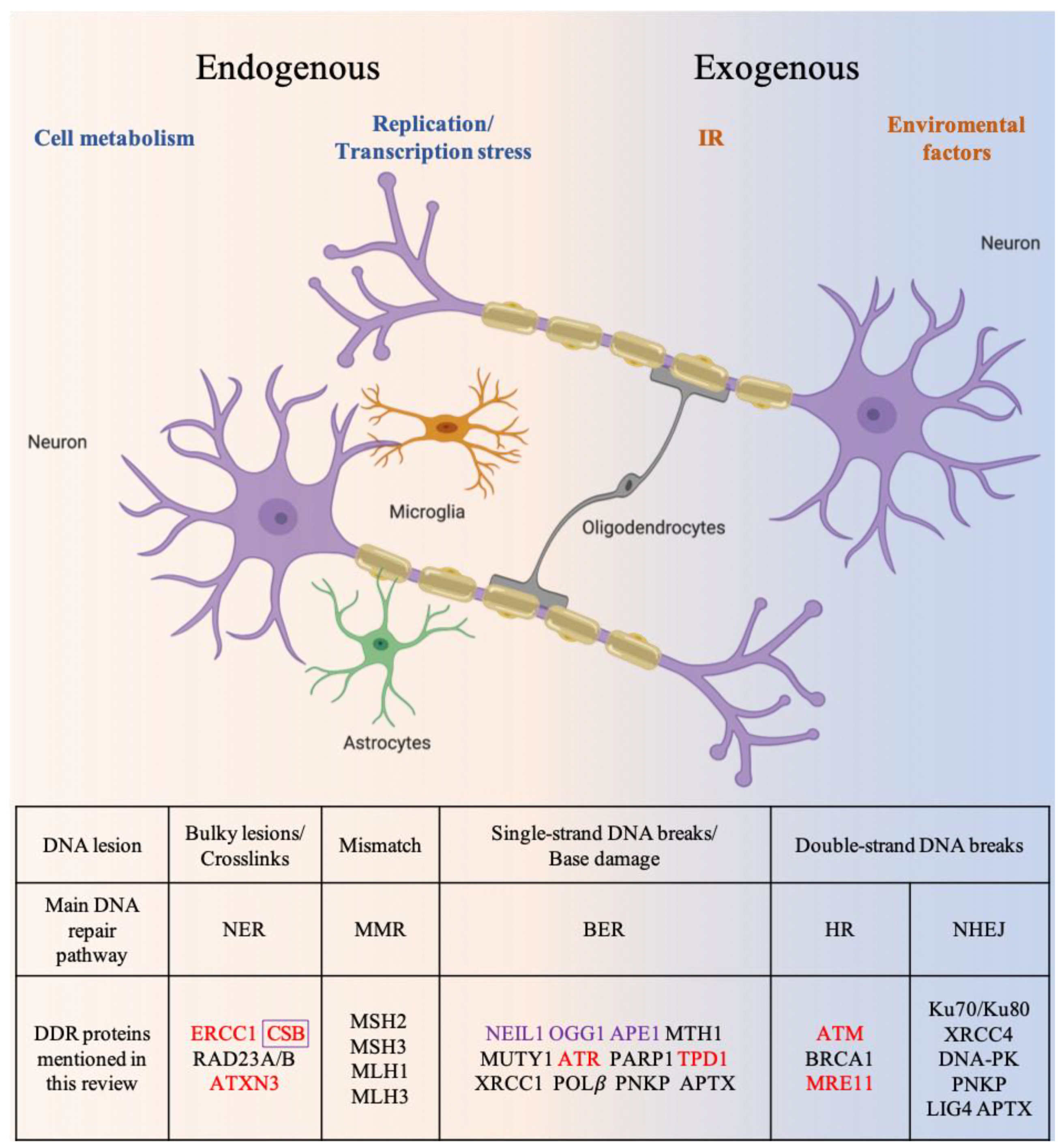
3. NDDs Related to the Defective DNA Repair Mechanisms
3.1. Alzheimer’s Disease
3.2. Parkinson’s Disease
3.3. Amyotrophic Lateral Sclerosis
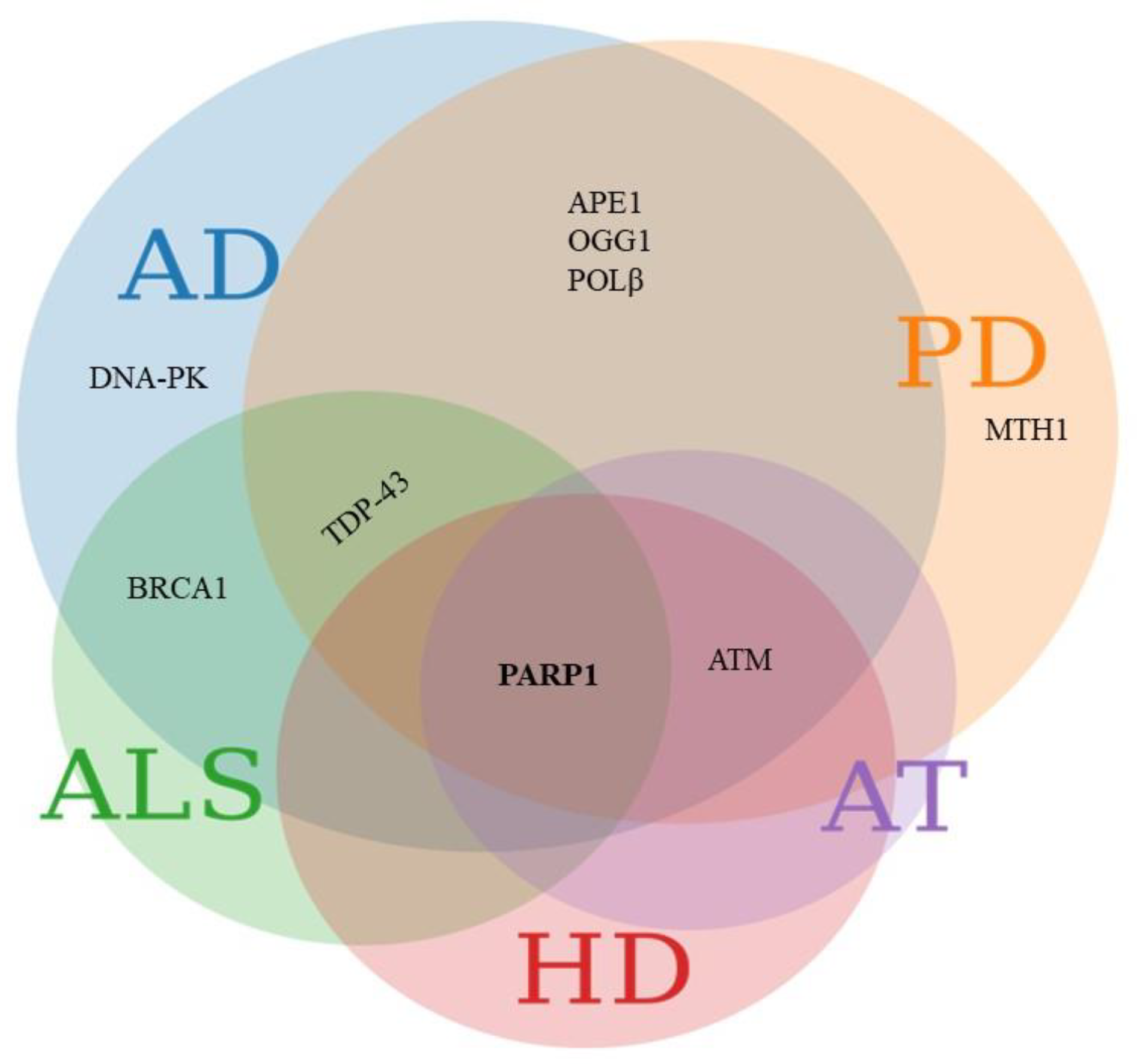
4. Crosstalk between DDR and Age-Related Neurodegenerative Diseases
5. Crosstalk between DDR and Other Cellular Processes Known to Be Disturbed in NDDs
5.1. DDR and Autophagy
5.2. DDR and Neuroinflammation
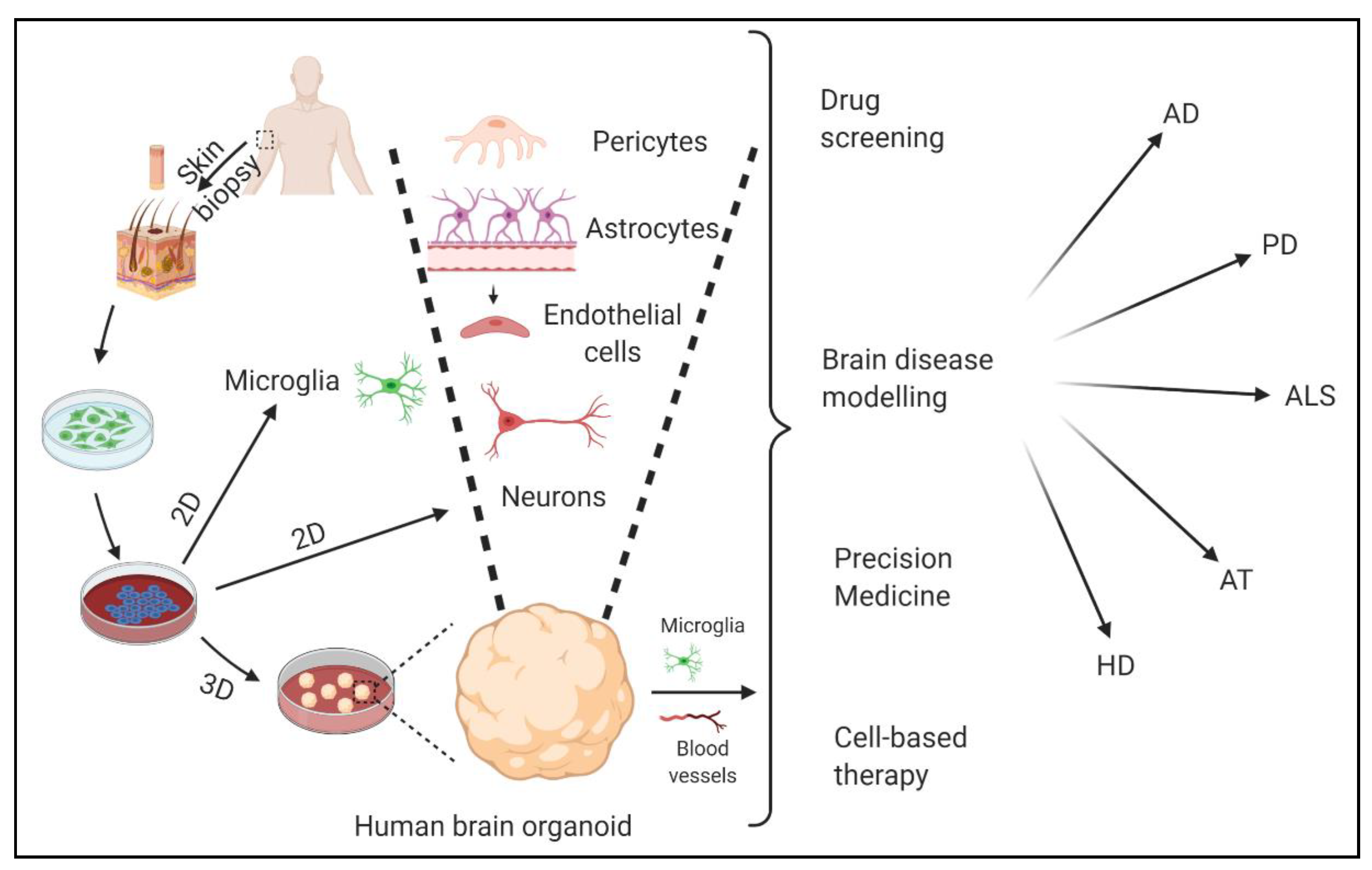
6. Model Systems to Study NDDs and Therapeutics
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delaney, S.; Jarem, D.A.; Volle, C.B.; Yennie, C.J. Chemical and biological consequences of oxidatively damaged guanine in DNA. Free Radic. Res. 2012, 46, 420–441. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.; Oka, S.; Tsuchimoto, D.; Abolhassani, N.; Nomaru, H.; Sakumi, K.; Yamada, H.; Nakabeppu, Y. 8-Oxoguanine causes neurodegeneration during MUTYH-mediated DNA base excision repair. J. Clin. Investig. 2012, 122, 4344–4361. [Google Scholar] [CrossRef] [Green Version]
- Krokan, H.E.; Drablos, F.; Slupphaug, G. Uracil in DNA-Occurrence, consequences and repair. Oncogene 2002, 21, 8935–8948. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, T.; Nyberg, B. Rate of depurination of native deoxyribonucleic acid. Biochemistry 1972, 11, 3610–3618. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Ramachandiran, S.; Hansen, J.M.; Jones, D.P.; Richardson, J.R.; Miller, G.W. Divergent mechanisms of paraquat, MPP+, and rotenone toxicity: Oxidation of thioredoxin and caspase-3 activation. Toxicol. Sci. 2007, 95, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [Green Version]
- Trist, B.G.; Davies, K.M.; Cottam, V.; Genoud, S.; Ortega, R.; Roudeau, S.; Carmona, A.; De Silva, K.; Wasinger, V.; Lewis, S.J.G.; et al. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain. Acta Neuropathol. 2017, 134, 113–127. [Google Scholar] [CrossRef]
- Rulten, S.L.; Caldecott, K.W. DNA strand break repair and neurodegeneration. DNA Repair 2013, 12, 558–567. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Ferguson, D.; Song, H.; Bassing, C.; Eckersdorff, M.; Alt, F.W.; Xu, Y. Functional interaction of H2AX, NBS1, and p53 in ATM-dependent DNA damage responses and tumor suppression. Mol. Cell Biol. 2005, 25, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, J.; Ford, B.N.; Brackley, M.E.; Glickman, B.W. Human DNA repair systems: An overview. Environ. Mol. Mutagen. 1999, 33, 3–20. [Google Scholar] [CrossRef]
- Nouspikel, T. DNA repair in mammalian cells: Nucleotide excision repair: Variations on versatility. Cell Mol. Life Sci. 2009, 66, 994–1009. [Google Scholar] [CrossRef]
- Nilsen, H.; Krokan, H.E. Base excision repair in a network of defence and tolerance. Carcinogenesis 2001, 22, 987–998. [Google Scholar] [CrossRef] [Green Version]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Fontana, G.A.; Gahlon, H.L. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fang, H.; Chi, Z.; Wu, Z.; Wei, D.; Mo, D.; Niu, K.; Balajee, A.S.; Hei, T.K.; Nie, L.; et al. XPD localizes in mitochondria and protects the mitochondrial genome from oxidative DNA damage. Nucleic Acids Res. 2015, 43, 5476–5488. [Google Scholar] [CrossRef] [PubMed]
- Aamann, M.D.; Sorensen, M.M.; Hvitby, C.; Berquist, B.R.; Muftuoglu, M.; Tian, J.; de Souza-Pinto, N.C.; Scheibye-Knudsen, M.; Wilson, D.M., 3rd; Stevnsner, T.; et al. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J. 2010, 24, 2334–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Sumberaz, P. Mitochondrial DNA Damage: Prevalence, Biological Consequence, and Emerging Pathways. Chem. Res. Toxicol. 2020, 33, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandsma, I.; Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef]
- Nair, N.; Shoaib, M.; Sorensen, C.S. Chromatin Dynamics in Genome Stability: Roles in Suppressing Endogenous DNA Damage and Facilitating DNA Repair. Int. J. Mol. Sci. 2017, 18, 1486. [Google Scholar] [CrossRef] [Green Version]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef]
- Bassing, C.H.; Suh, H.; Ferguson, D.O.; Chua, K.F.; Manis, J.; Eckersdorff, M.; Gleason, M.; Bronson, R.; Lee, C.; Alt, F.W. Histone H2AX: A dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003, 114, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Ditch, S.; Paull, T.T. The ATM protein kinase and cellular redox signaling: Beyond the DNA damage response. Trends Biochem. Sci. 2012, 37, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Pizzamiglio, L.; Focchi, E.; Antonucci, F. ATM Protein Kinase: Old and New Implications in Neuronal Pathways and Brain Circuitry. Cells 2020, 9, 1969. [Google Scholar] [CrossRef] [PubMed]
- Anisenko, A.; Kan, M.; Shadrina, O.; Brattseva, A.; Gottikh, M. Phosphorylation Targets of DNA-PK and Their Role in HIV-1 Replication. Cells 2020, 9, 1907. [Google Scholar] [CrossRef]
- Alt, F.W.; Zhang, Y.; Meng, F.L.; Guo, C.; Schwer, B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 2013, 152, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Woodbine, L.; Neal, J.A.; Sasi, N.K.; Shimada, M.; Deem, K.; Coleman, H.; Dobyns, W.B.; Ogi, T.; Meek, K.; Davies, E.G.; et al. PRKDC mutations in a SCID patient with profound neurological abnormalities. J. Clin. Investig. 2013, 123, 2969–2980. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Crowe, J.L.; Liu, X.; Nakajima, S.; Wang, Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X.; et al. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol. Cell 2015, 58, 172–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.J.; Baltimore, D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003, 17, 615–628. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, A.; Wilson, D.M., 3rd. The involvement of DNA-damage and -repair defects in neurological dysfunction. Am. J. Hum. Genet. 2008, 82, 539–566. [Google Scholar] [CrossRef] [Green Version]
- Coon, E.A.; Benarroch, E.E. DNA damage response: Selected review and neurologic implications. Neurology 2018, 90, 367–376. [Google Scholar] [CrossRef]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Wheeler, V.C.; Chao, M.J.; Vonsattel, J.P.; Pinto, R.M.; Lucente, D.; Abu-Elneel, K.; Ramos, E.M.; Mysore, J.S.; Gillis, T.; et al. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 2015, 162, 516–526. [Google Scholar] [CrossRef] [Green Version]
- McMurray, C.T. Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet. 2010, 11, 786–799. [Google Scholar] [CrossRef] [Green Version]
- Panigrahi, G.B.; Lau, R.; Montgomery, S.E.; Leonard, M.R.; Pearson, C.E. Slipped (CTG)*(CAG) repeats can be correctly repaired, escape repair or undergo error-prone repair. Nat. Struct. Mol. Biol. 2005, 12, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Massey, T.H.; Jones, L. The central role of DNA damage and repair in CAG repeat diseases. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, T.; Mocle, A.J.; Hung, C.L.; Xia, J.; van Roon-Mom, W.M.; Truant, R. Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum. Mol. Genet. 2017, 26, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, R.; Liu, Y.; Silva-Fernandes, A.; Fang, X.; Paulucci-Holthauzen, A.; Chatterjee, A.; Zhang, H.L.; Matsuura, T.; Choudhary, S.; Ashizawa, T.; et al. Inactivation of PNKP by mutant ATXN3 triggers apoptosis by activating the DNA damage-response pathway in SCA3. PLoS Genet. 2015, 11, e1004834. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Kannan, A.; Bhatia, K.; Branzei, D.; Gangwani, L. Combined deficiency of Senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res. 2018, 46, 8326–8346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, A.; Jiang, X.; He, L.; Ahmad, S.; Gangwani, L. ZPR1 prevents R-loop accumulation, upregulates SMN2 expression and rescues spinal muscular atrophy. Brain 2020, 143, 69–93. [Google Scholar] [CrossRef]
- Barker, W.W.; Luis, C.A.; Kashuba, A.; Luis, M.; Harwood, D.G.; Loewenstein, D.; Waters, C.; Jimison, P.; Shepherd, E.; Sevush, S.; et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 2002, 16, 203–212. [Google Scholar] [CrossRef]
- Griffin, W.S. Inflammation and neurodegenerative diseases. Am. J. Clin. Nutr. 2006, 83, 470S–474S. [Google Scholar] [CrossRef] [Green Version]
- Castellani, R.J.; Rolston, R.K.; Smith, M.A. Alzheimer disease. Dis. Mon. 2010, 56, 484–546. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Dosunmu, R.; Wu, J.; Basha, M.R.; Zawia, N.H. Environmental and dietary risk factors in Alzheimer’s disease. Expert Rev. Neurother. 2007, 7, 887–900. [Google Scholar] [CrossRef]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Muller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef]
- Wingo, T.S.; Lah, J.J.; Levey, A.I.; Cutler, D.J. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch. Neurol 2012, 69, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, L.; Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J. Clin. Investig. 2005, 115, 1449–1457. [Google Scholar] [CrossRef] [Green Version]
- Raber, J.; Huang, Y.; Ashford, J.W. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging 2004, 25, 641–650. [Google Scholar] [CrossRef]
- Adamec, E.; Vonsattel, J.P.; Nixon, R.A. DNA strand breaks in Alzheimer’s disease. Brain Res. 1999, 849, 67–77. [Google Scholar] [CrossRef]
- Myung, N.H.; Zhu, X.; Kruman, I.I.; Castellani, R.J.; Petersen, R.B.; Siedlak, S.L.; Perry, G.; Smith, M.A.; Lee, H.G. Evidence of DNA damage in Alzheimer disease: Phosphorylation of histone H2AX in astrocytes. Age 2008, 30, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Shanbhag, N.M.; Evans, M.D.; Mao, W.; Nana, A.L.; Seeley, W.W.; Adame, A.; Rissman, R.A.; Masliah, E.; Mucke, L. Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 1998, 71, 2034–2040. [Google Scholar] [CrossRef] [Green Version]
- Lyras, L.; Cairns, N.J.; Jenner, A.; Jenner, P.; Halliwell, B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J. Neurochem. 1997, 68, 2061–2069. [Google Scholar] [CrossRef]
- Shen, X.; Chen, J.; Li, J.; Kofler, J.; Herrup, K. Neurons in Vulnerable Regions of the Alzheimer’s Disease Brain Display Reduced ATM Signaling. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Suberbielle, E.; Djukic, B.; Evans, M.; Kim, D.H.; Taneja, P.; Wang, X.; Finucane, M.; Knox, J.; Ho, K.; Devidze, N.; et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 2015, 6, 8897. [Google Scholar] [CrossRef] [PubMed]
- Kanungo, J. DNA-PK Deficiency in Alzheimer’s Disease. J. Neurol. Neuromedicine 2016, 1, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Res. 2000, 855, 116–123. [Google Scholar] [CrossRef]
- Weissman, L.; Jo, D.G.; Sorensen, M.M.; de Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007, 35, 5545–5555. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Shamanna, R.A.; Croteau, D.L.; Bohr, V.A. Base excision DNA repair levels in mitochondrial lysates of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1293–1300. [Google Scholar] [CrossRef] [Green Version]
- Rao, K.S.; Annapurna, V.V.; Raji, N.S. DNA polymerase-beta may be the main player for defective DNA repair in aging rat neurons. Ann. N. Y. Acad. Sci. 2001, 928, 113–120. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Smoczer, C.; Pace, B.; Patterson, D.; Cress Cabelof, D. Loss of DNA polymerase beta induces cellular senescence. Environ. Mol. Mutagen. 2018, 59, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Misiak, M.; Wang, Y.; Ghosh, S.; Leandro, G.S.; Liu, D.; Tian, J.; Baptiste, B.A.; Cong, W.N.; Brenerman, B.M.; et al. DNA polymerase beta deficiency leads to neurodegeneration and exacerbates Alzheimer disease phenotypes. Nucleic Acids Res. 2015, 43, 943–959. [Google Scholar] [CrossRef]
- Gkikas, I.; Palikaras, K.; Tavernarakis, N. The Role of Mitophagy in Innate Immunity. Front. Immunol. 2018, 9, 1283. [Google Scholar] [CrossRef]
- Tan, M.M.X.; Malek, N.; Lawton, M.A.; Hubbard, L.; Pittman, A.M.; Joseph, T.; Hehir, J.; Swallow, D.M.A.; Grosset, K.A.; Marrinan, S.L.; et al. Genetic analysis of Mendelian mutations in a large UK population-based Parkinson’s disease study. Brain 2019, 142, 2828–2844. [Google Scholar] [CrossRef]
- Gaare, J.J.; Nido, G.; Dolle, C.; Sztromwasser, P.; Alves, G.; Tysnes, O.B.; Haugarvoll, K.; Tzoulis, C. Meta-analysis of whole-exome sequencing data from two independent cohorts finds no evidence for rare variant enrichment in Parkinson disease associated loci. PLoS ONE 2020, 15, e0239824. [Google Scholar] [CrossRef] [PubMed]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 1997, 69, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Gupta, V.B.; Anitha, M.; Harikrishna, T.; Shankar, S.K.; Muthane, U.; Subba Rao, K.; Jagannatha Rao, K.S. Studies on genomic DNA topology and stability in brain regions of Parkinson’s disease. Arch. Biochem. Biophys. 2006, 449, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, T.; Hara, K.; Sethi, K.D.; Morgan, J.C.; Borlongan, C.V. Increased 8-OHdG levels in the urine, serum, and substantia nigra of hemiparkinsonian rats. Brain Res. 2007, 1133, 49–52. [Google Scholar] [CrossRef]
- Sepe, S.; Milanese, C.; Gabriels, S.; Derks, K.W.; Payan-Gomez, C.; van, I.W.F.; Rijksen, Y.M.; Nigg, A.L.; Moreno, S.; Cerri, S.; et al. Inefficient DNA Repair Is an Aging-Related Modifier of Parkinson’s Disease. Cell Rep. 2016, 15, 1866–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Fukae, J.; Hatano, T.; Kubo, S.; Ohtsubo, T.; Nakabeppu, Y.; Mori, H.; Mizuno, Y.; Hattori, N. Up-regulation of hMUTYH, a DNA repair enzyme, in the mitochondria of substantia nigra in Parkinson’s disease. Acta Neuropathol. 2006, 112, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef] [PubMed]
- Fukae, J.; Takanashi, M.; Kubo, S.; Nishioka, K.; Nakabeppu, Y.; Mori, H.; Mizuno, Y.; Hattori, N. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson’s disease and related neurodegenerative disorders. Acta Neuropathol. 2005, 109, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Cardozo-Pelaez, F.; Sanchez-Contreras, M.; Nevin, A.B. Ogg1 null mice exhibit age-associated loss of the nigrostriatal pathway and increased sensitivity to MPTP. Neurochem. Int. 2012, 61, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Scott, T.L.; Wicker, C.A.; Suganya, R.; Dhar, B.; Pittman, T.; Horbinski, C.; Izumi, T. Polyubiquitination of apurinic/apyrimidinic endonuclease 1 by Parkin. Mol. Carcinog. 2017, 56, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Sanders, L.H.; McCoy, J.; Hu, X.; Mastroberardino, P.G.; Dickinson, B.C.; Chang, C.J.; Chu, C.T.; Van Houten, B.; Greenamyre, J.T. Mitochondrial DNA damage: Molecular marker of vulnerable nigral neurons in Parkinson’s disease. Neurobiol. Dis. 2014, 70, 214–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gencer, M.; Dasdemir, S.; Cakmakoglu, B.; Cetinkaya, Y.; Varlibas, F.; Tireli, H.; Kucukali, C.I.; Ozkok, E.; Aydin, M. DNA repair genes in Parkinson’s disease. Genet. Test. Mol. Biomark. 2012, 16, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.H.; Paul, K.C.; Howlett, E.H.; Lawal, H.; Boppana, S.; Bronstein, J.M.; Ritz, B.; Greenamyre, J.T. Editor’s Highlight: Base Excision Repair Variants and Pesticide Exposure Increase Parkinson’s Disease Risk. Toxicol. Sci. 2017, 158, 188–198. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrimsdottir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Kerchner, G.A.; Ayalon, G.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Vasquez, V.; Mitra, J.; Hegde, P.M.; Pandey, A.; Sengupta, S.; Mitra, S.; Rao, K.S.; Hegde, M.L. Chromatin-Bound Oxidized alpha-Synuclein Causes Strand Breaks in Neuronal Genomes in in vitro Models of Parkinson’s Disease. J. Alzheimers Dis. 2017, 60, S133–S150. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.I.; Mao, X.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragagnin, A.M.G.; Shadfar, S.; Vidal, M.; Jamali, M.S.; Atkin, J.D. Motor Neuron Susceptibility in ALS/FTD. Front. Neurosci. 2019, 13, 532. [Google Scholar] [CrossRef] [Green Version]
- Ajroud-Driss, S.; Siddique, T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim. Biophys. Acta 2015, 1852, 679–684. [Google Scholar] [CrossRef] [Green Version]
- Peters, O.M.; Ghasemi, M.; Brown, R.H., Jr. Emerging mechanisms of molecular pathology in ALS. J. Clin. Investig. 2015, 125, 1767–1779. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, B.; Izaurieta, P.; Fritz, E.; Alvarez, F.J. Early pathogenesis in the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J. Cell Biochem. 2012, 113, 3301–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, W.G.; Krasin, F. A new hypothesis of the etiology of amyotrophic lateral sclerosis. The DNA hypothesis. Arch. Neurol. 1982, 39, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol. Commun. 2020, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Brenner, D.; Muller, K.; Wieland, T.; Weydt, P.; Bohm, S.; Lule, D.; Hubers, A.; Neuwirth, C.; Weber, M.; Borck, G.; et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 2016, 139, e28. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, C.F.; Riley, D.J.; Chen, P.L. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle 2011, 10, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higelin, J.; Catanese, A.; Semelink-Sedlacek, L.L.; Oeztuerk, S.; Lutz, A.K.; Bausinger, J.; Barbi, G.; Speit, G.; Andersen, P.M.; Ludolph, A.C.; et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res. 2018, 30, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, A.Y.; Martin, L.J. DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in the brain and spinal cord of individuals with amyotrophic lateral sclerosis. Neuromol. Med. 2002, 2, 47–60. [Google Scholar] [CrossRef]
- Murakami, T.; Nagai, M.; Miyazaki, K.; Morimoto, N.; Ohta, Y.; Kurata, T.; Takehisa, Y.; Kamiya, T.; Abe, K. Early decrease of mitochondrial DNA repair enzymes in spinal motor neurons of presymptomatic transgenic mice carrying a mutant SOD1 gene. Brain Res. 2007, 1150, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.; Gomez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014, 42, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef]
- Andrade, N.S.; Ramic, M.; Esanov, R.; Liu, W.; Rybin, M.J.; Gaidosh, G.; Abdallah, A.; Del’Olio, S.; Huff, T.C.; Chee, N.T.; et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol. Neurodegener. 2020, 15, 13. [Google Scholar] [CrossRef] [Green Version]
- Farg, M.A.; Konopka, A.; Soo, K.Y.; Ito, D.; Atkin, J.D. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017, 26, 2882–2896. [Google Scholar] [CrossRef]
- Petersen, A.J.; Rimkus, S.A.; Wassarman, D.A. ATM kinase inhibition in glial cells activates the innate immune response and causes neurodegeneration in Drosophila. Proc. Natl. Acad. Sci. USA 2012, 109, E656–E664. [Google Scholar] [CrossRef] [Green Version]
- Illuzzi, J.; Yerkes, S.; Parekh-Olmedo, H.; Kmiec, E.B. DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J. Neurosci. Res. 2009, 87, 733–747. [Google Scholar] [CrossRef]
- Lu, X.H.; Mattis, V.B.; Wang, N.; Al-Ramahi, I.; van den Berg, N.; Fratantoni, S.A.; Waldvogel, H.; Greiner, E.; Osmand, A.; Elzein, K.; et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci. Transl. Med. 2014, 6, 268ra178. [Google Scholar] [CrossRef] [PubMed]
- Milanese, C.; Cerri, S.; Ulusoy, A.; Gornati, S.V.; Plat, A.; Gabriels, S.; Blandini, F.; Di Monte, D.A.; Hoeijmakers, J.H.; Mastroberardino, P.G. Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis. 2018, 9, 818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mano, T.; Nagata, K.; Nonaka, T.; Tarutani, A.; Imamura, T.; Hashimoto, T.; Bannai, T.; Koshi-Mano, K.; Tsuchida, T.; Ohtomo, R.; et al. Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E9645–E9654. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Reddy, P.H. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: Implications for early intervention and therapeutics. Biochim. Biophys. Acta 2011, 1812, 1359–1370. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, M.; Mano, T.; Saito, Y.; Murayama, S.; Toda, T.; Iwata, A. Colocalization of BRCA1 with Tau Aggregates in Human Tauopathies. Brain Sci. 2019, 10, 7. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Kaneko, S.; Dickson, D.W.; Kusaka, H. Aberrant Accumulation of BRCA1 in Alzheimer Disease and Other Tauopathies. J. Neuropathol. Exp. Neurol. 2020, 79, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Wezyk, M.; Szybinska, A.; Wojsiat, J.; Szczerba, M.; Day, K.; Ronnholm, H.; Kele, M.; Berdynski, M.; Peplonska, B.; Fichna, J.P.; et al. Overactive BRCA1 Affects Presenilin 1 in Induced Pluripotent Stem Cell-Derived Neurons in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 175–202. [Google Scholar] [CrossRef] [PubMed]
- Noristani, H.N.; Sabourin, J.C.; Gerber, Y.N.; Teigell, M.; Sommacal, A.; Vivanco, M.; Weber, M.; Perrin, F.E. Brca1 is expressed in human microglia and is dysregulated in human and animal model of ALS. Mol. Neurodegener. 2015, 10, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, P.J. DNA repair in neural cells: Basic science and clinical implications. Mutat. Res. 2002, 509, 93–108. [Google Scholar] [CrossRef]
- Shackelford, D.A. DNA end joining activity is reduced in Alzheimer’s disease. Neurobiol. Aging 2006, 27, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Davydov, V.; Hansen, L.A.; Shackelford, D.A. Is DNA repair compromised in Alzheimer’s disease? Neurobiol. Aging 2003, 24, 953–968. [Google Scholar] [CrossRef]
- Cardinale, A.; Racaniello, M.; Saladini, S.; De Chiara, G.; Mollinari, C.; de Stefano, M.C.; Pocchiari, M.; Garaci, E.; Merlo, D. Sublethal doses of beta-amyloid peptide abrogate DNA-dependent protein kinase activity. J. Biol. Chem. 2012, 287, 2618–2631. [Google Scholar] [CrossRef] [Green Version]
- Altmeyer, M.; Messner, S.; Hassa, P.O.; Fey, M.; Hottiger, M.O. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009, 37, 3723–3738. [Google Scholar] [CrossRef] [Green Version]
- Aredia, F.; Scovassi, A.I. Involvement of PARPs in cell death. Front. Biosci. 2014, 6, 308–317. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef] [Green Version]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)—Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front. Mol. Biosci. 2020, 7, 191. [Google Scholar] [CrossRef]
- Beneke, S. Regulation of chromatin structure by poly(ADP-ribosyl)ation. Front. Genet. 2012, 3, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkle, A.; Virag, L. Poly(ADP-ribose): PARadigms and PARadoxes. Mol. Asp. Med. 2013, 34, 1046–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef]
- Croteau, D.L.; Fang, E.F.; Nilsen, H.; Bohr, V.A. NAD(+) in DNA repair and mitochondrial maintenance. Cell Cycle 2017, 16, 491–492. [Google Scholar] [CrossRef] [Green Version]
- Love, S.; Barber, R.; Wilcock, G.K. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain 1999, 122 Pt 2, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, T.M.; Suh, S.W.; Higashi, Y.; Berman, A.E.; Escartin, C.; Won, S.J.; Wang, C.; Cho, S.H.; Gan, L.; Swanson, R.A. Poly(ADP-ribose)polymerase-1 modulates microglial responses to amyloid beta. J. Neuroinflammation 2011, 8, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martire, S.; Fuso, A.; Rotili, D.; Tempera, I.; Giordano, C.; De Zottis, I.; Muzi, A.; Vernole, P.; Graziani, G.; Lococo, E.; et al. PARP-1 modulates amyloid beta peptide-induced neuronal damage. PLoS ONE 2013, 8, e72169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Mandir, A.S.; Przedborski, S.; Jackson-Lewis, V.; Wang, Z.Q.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Hoffman, B.E.; Guastella, D.B.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc. Natl. Acad. Sci. USA 1999, 96, 5774–5779. [Google Scholar] [CrossRef] [Green Version]
- Shibata, N.; Kakita, A.; Takahashi, H.; Ihara, Y.; Nobukuni, K.; Fujimura, H.; Sakoda, S.; Sasaki, S.; Yamamoto, T.; Kobayashi, M. Persistent cleavage and nuclear translocation of apoptosis-inducing factor in motor neurons in the spinal cord of sporadic amyotrophic lateral sclerosis patients. Acta Neuropathol. 2009, 118, 755–762. [Google Scholar] [CrossRef] [PubMed]
- McGurk, L.; Mojsilovic-Petrovic, J.; Van Deerlin, V.M.; Shorter, J.; Kalb, R.G.; Lee, V.M.; Trojanowski, J.Q.; Lee, E.B.; Bonini, N.M. Nuclear poly(ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2018, 6, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.K.; Shin, K.S.; Kang, S.J. AIF translocates to the nucleus in the spinal motor neurons in a mouse model of ALS. Neurosci. Lett. 2006, 406, 205–210. [Google Scholar] [CrossRef]
- Schondorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [Green Version]
- Fifita, J.A.; Williams, K.L.; Sundaramoorthy, V.; McCann, E.P.; Nicholson, G.A.; Atkin, J.D.; Blair, I.P. A novel amyotrophic lateral sclerosis mutation in OPTN induces ER stress and Golgi fragmentation in vitro. Amyotroph. Lateral Scler. Frontotemporal Degener. 2017, 18, 126–133. [Google Scholar] [CrossRef]
- Korac, J.; Schaeffer, V.; Kovacevic, I.; Clement, A.M.; Jungblut, B.; Behl, C.; Terzic, J.; Dikic, I. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell Sci. 2013, 126, 580–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goode, A.; Butler, K.; Long, J.; Cavey, J.; Scott, D.; Shaw, B.; Sollenberger, J.; Gell, C.; Johansen, T.; Oldham, N.J.; et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 2016, 12, 1094–1104. [Google Scholar] [CrossRef] [Green Version]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef]
- Orlotti, N.I.; Cimino-Reale, G.; Borghini, E.; Pennati, M.; Sissi, C.; Perrone, F.; Palumbo, M.; Daidone, M.G.; Folini, M.; Zaffaroni, N. Autophagy acts as a safeguard mechanism against G-quadruplex ligand-mediated DNA damage. Autophagy 2012, 8, 1185–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eapen, V.V.; Haber, J.E. DNA damage signaling triggers the cytoplasm-to-vacuole pathway of autophagy to regulate cell cycle progression. Autophagy 2013, 9, 440–441. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.; Kim, J.; Walker, C.L. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010, 6, 672–673. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Tse, K.H.; Chow, H.M.; Gan, Y.; Song, X.; Ma, F.; Qian, Y.X.Y.; She, W.; Herrup, K. ATM loss disrupts the autophagy-lysosomal pathway. Autophagy 2020, 1–13. [Google Scholar] [CrossRef]
- Rodriguez-Vargas, J.M.; Ruiz-Magana, M.J.; Ruiz-Ruiz, C.; Majuelos-Melguizo, J.; Peralta-Leal, A.; Rodriguez, M.I.; Munoz-Gamez, J.A.; de Almodovar, M.R.; Siles, E.; Rivas, A.L.; et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 2012, 22, 1181–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Fang, Y.; Yan, L.; Xu, L.; Zhang, S.; Cao, Y.; Xu, L.; Zhang, X.; Xie, J.; Jiang, G.; et al. Nuclear localization of Beclin 1 promotes radiation-induced DNA damage repair independent of autophagy. Sci. Rep. 2017, 7, 45385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SenGupta, T.; Torgersen, M.L.; Kassahun, H.; Vellai, T.; Simonsen, A.; Nilsen, H. Base excision repair AP endonucleases and mismatch repair act together to induce checkpoint-mediated autophagy. Nat. Commun. 2013, 4, 2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, G.; Carroll, B.; Sarallah, R.; Correia-Melo, C.; Ogrodnik, M.; Nelson, G.; Otten, E.G.; Manni, D.; Antrobus, R.; Morgan, B.A.; et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 2016, 12, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Ni, D.; Ghozalli, I.; Pirooz, S.D.; Ma, B.; Liang, C. UVRAG: At the crossroad of autophagy and genomic stability. Autophagy 2012, 8, 1392–1393. [Google Scholar] [CrossRef]
- Dan, X.; Babbar, M.; Moore, A.; Wechter, N.; Tian, J.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. DNA damage invokes mitophagy through a pathway involving Spata18. Nucleic Acids Res. 2020, 48, 6611–6623. [Google Scholar] [CrossRef]
- Chaurasia, M.; Gupta, S.; Das, A.; Dwarakanath, B.S.; Simonsen, A.; Sharma, K. Radiation induces EIF2AK3/PERK and ERN1/IRE1 mediated pro-survival autophagy. Autophagy 2019, 15, 1391–1406. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [Green Version]
- Mikhed, Y.; Gorlach, A.; Knaus, U.G.; Daiber, A. Redox regulation of genome stability by effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol. 2015, 5, 275–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moritz, E.; Pauly, K.; Bravard, A.; Hall, J.; Radicella, J.P.; Epe, B. hOGG1-Cys326 variant cells are hypersensitive to DNA repair inhibition by nitric oxide. Carcinogenesis 2014, 35, 1426–1433. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Chin, A.C. Neuroinflammation and the cGAS-STING pathway. J. Neurophysiol. 2019, 121, 1087–1091. [Google Scholar] [CrossRef]
- Quek, H.; Luff, J.; Cheung, K.; Kozlov, S.; Gatei, M.; Lee, C.S.; Bellingham, M.C.; Noakes, P.G.; Lim, Y.C.; Barnett, N.L.; et al. A rat model of ataxia-telangiectasia: Evidence for a neurodegenerative phenotype. Hum. Mol. Genet. 2017, 26, 109–123. [Google Scholar] [CrossRef]
- Xu, Q.; Xu, W.; Cheng, H.; Yuan, H.; Tan, X. Efficacy and mechanism of cGAMP to suppress Alzheimer’s disease by elevating TREM2. Brain Behav. Immun. 2019, 81, 495–508. [Google Scholar] [CrossRef]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; Konig, I.R.; Delcambre, S.; Petrucci, S.; Balck, A.; Bruggemann, N.; Zimprich, A.; Wasner, K.; Pereira, S.L.; Avenali, M.; et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 2020, 143, 3041–3051. [Google Scholar] [CrossRef]
- Liachko, N.F.; Guthrie, C.R.; Kraemer, B.C. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci. 2010, 30, 16208–16219. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [Green Version]
- Ciura, S.; Lattante, S.; Le Ber, I.; Latouche, M.; Tostivint, H.; Brice, A.; Kabashi, E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 180–187. [Google Scholar] [CrossRef]
- Imamura, S.; Kishi, S. Molecular cloning and functional characterization of zebrafish ATM. Int. J. Biochem. Cell Biol. 2005, 37, 1105–1116. [Google Scholar] [CrossRef]
- Lakso, M.; Vartiainen, S.; Moilanen, A.M.; Sirvio, J.; Thomas, J.H.; Nass, R.; Blakely, R.D.; Wong, G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J. Neurochem. 2003, 86, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.; Plasterk, R.H.; Nollen, E.A. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008, 4, e1000027. [Google Scholar] [CrossRef]
- McColl, G.; Roberts, B.R.; Pukala, T.L.; Kenche, V.B.; Roberts, C.M.; Link, C.D.; Ryan, T.M.; Masters, C.L.; Barnham, K.J.; Bush, A.I.; et al. Utility of an improved model of amyloid-beta (Abeta(1)(-)(4)(2)) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease. Mol. Neurodegener. 2012, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Wu, Z.; Butko, P.; Christen, Y.; Lambert, M.P.; Klein, W.L.; Link, C.D.; Luo, Y. Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J. Neurosci. 2006, 26, 13102–13113. [Google Scholar] [CrossRef]
- Shen, L.; Wang, C.; Chen, L.; Leung, K.L.; Lo, E.; Lakso, M.; Wong, G. TDP-1/TDP-43 potentiates human alpha-Synuclein (HASN) neurodegeneration in Caenorhabditis elegans. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165876. [Google Scholar] [CrossRef]
- Muqit, M.M.; Feany, M.B. Modelling neurodegenerative diseases in Drosophila: A fruitful approach? Nat. Rev. Neurosci. 2002, 3, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Pitchai, A.; Rajaretinam, R.K.; Freeman, J.L. Zebrafish as an Emerging Model for Bioassay-Guided Natural Product Drug Discovery for Neurological Disorders. Medicines 2019, 6, 61. [Google Scholar] [CrossRef] [Green Version]
- Saleem, S.; Kannan, R.R. Zebrafish: An emerging real-time model system to study Alzheimer’s disease and neurospecific drug discovery. Cell Death Discov. 2018, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Xi, Y.; Noble, S.; Ekker, M. Modeling neurodegeneration in zebrafish. Curr. Neurol. Neurosci. Rep. 2011, 11, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Abugable, A.A.; Morris, J.L.M.; Palminha, N.M.; Zaksauskaite, R.; Ray, S.; El-Khamisy, S.F. DNA repair and neurological disease: From molecular understanding to the development of diagnostics and model organisms. DNA Repair 2019, 81, 102669. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef]
- Kimura, J.; Shimizu, K.; Kajima, K.; Yokosuka, A.; Mimaki, Y.; Oku, N.; Ohizumi, Y. Nobiletin Reduces Intracellular and Extracellular beta-Amyloid in iPS Cell-Derived Alzheimer’s Disease Model Neurons. Biol. Pharm. Bull. 2018, 41, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic Correction of SOD1 Mutant iPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cell Rep. 2017, 8, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Gunther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef]
- Seminary, E.R.; Sison, S.L.; Ebert, A.D. Modeling Protein Aggregation and the Heat Shock Response in ALS iPSC-Derived Motor Neurons. Front. Neurosci. 2018, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, D.; Magotani, H.; Kikuchi, T.; Ikeda, M.; Hiramatsu, S.; Yoshida, K.; Amano, N.; Nomura, M.; Umekage, M.; Morizane, A.; et al. Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson’s disease. Nat. Commun. 2020, 11, 3369. [Google Scholar] [CrossRef]
- Lopez de Maturana, R.; Lang, V.; Zubiarrain, A.; Sousa, A.; Vazquez, N.; Gorostidi, A.; Aguila, J.; Lopez de Munain, A.; Rodriguez, M.; Sanchez-Pernaute, R. Mutations in LRRK2 impair NF-kappaB pathway in iPSC-derived neurons. J. Neuroinflammation 2016, 13, 295. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1294. [Google Scholar] [CrossRef] [Green Version]
- Dutta, D.; Heo, I.; Clevers, H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol. Med. 2017, 23, 393–410. [Google Scholar] [CrossRef]
- Ormel, P.R.; Vieira de Sa, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef]
- Yan, Y.; Song, L.; Bejoy, J.; Zhao, J.; Kanekiyo, T.; Bu, G.; Zhou, Y.; Li, Y. Modeling Neurodegenerative Microenvironment Using Cortical Organoids Derived from Human Stem Cells. Tissue Eng. Part A 2018, 24, 1125–1137. [Google Scholar] [CrossRef]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Penney, J.; Ralvenius, W.T.; Tsai, L.H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K.; Kim, J. Modeling G2019S-LRRK2 Sporadic Parkinson’s Disease in 3D Midbrain Organoids. Stem Cell Rep. 2019, 12, 518–531. [Google Scholar] [CrossRef] [Green Version]
- Monzel, A.S.; Smits, L.M.; Hemmer, K.; Hachi, S.; Moreno, E.L.; van Wuellen, T.; Jarazo, J.; Walter, J.; Bruggemann, I.; Boussaad, I.; et al. Derivation of Human Midbrain-Specific Organoids from Neuroepithelial Stem Cells. Stem Cell Rep. 2017, 8, 1144–1154. [Google Scholar] [CrossRef]
- Smits, L.M.; Reinhardt, L.; Reinhardt, P.; Glatza, M.; Monzel, A.S.; Stanslowsky, N.; Rosato-Siri, M.D.; Zanon, A.; Antony, P.M.; Bellmann, J.; et al. Modeling Parkinson’s disease in midbrain-like organoids. NPJ Parkinsons Dis. 2019, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osaki, T.; Sivathanu, V.; Kamm, R.D. Engineered 3D vascular and neuronal networks in a microfluidic platform. Sci. Rep. 2018, 8, 5168. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, F.K.; Al-Janabi, T.; Hardy, J.; Karmiloff-Smith, A.; Nizetic, D.; Tybulewicz, V.L.; Fisher, E.M.; Strydom, A. A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 2015, 16, 564–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandmann, O.; Davis, M.B.; Marsden, C.D.; Harding, A.E. Sequence of the superoxide dismutase 1 (SOD 1) gene in familial Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1995, 59, 90–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Rees, H.D.; Weintraub, S.T.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative modifications and aggregation of Cu,Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J. Biol. Chem. 2005, 280, 11648–11655. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | DDR Genes | NDD | References |
|---|---|---|---|
| C. elegans | PARP1 | AD | [146] |
| TARDBP | ALS | [183] | |
| PARP1 | AT | [140] | |
| XPA | XP | [184] | |
| CSB | CS | [184] | |
| ATM | AT | [184] | |
| D. melanogaster | ATM | AT | [72] |
| AD | [120] | ||
| HD | [122] | ||
| D. rerio | C9ORF72 | ALS | [185] |
| ATM | AT | [186] | |
| M. musculus | ATM | HD | [122] |
| AD | [72] | ||
| BRCA1 | ALS | [129] | |
| AD | [73] | ||
| POLβ | AD | [80] | |
| OGG1 | PD | [91] | |
| ALS | [112] | ||
| ERCC1 | PD | [87] | |
| PARP1 | AD | [145] | |
| PD | [147] | ||
| ALS | [150] | ||
| R. norvegicus | ATM | PD | [123] |
| iPSCs | BRCA1 | AD | [128] |
| NEK1 | ALS | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, S.; You, P.; SenGupta, T.; Nilsen, H.; Sharma, K. Crosstalk between Different DNA Repair Pathways Contributes to Neurodegenerative Diseases. Biology 2021, 10, 163. https://doi.org/10.3390/biology10020163
Gupta S, You P, SenGupta T, Nilsen H, Sharma K. Crosstalk between Different DNA Repair Pathways Contributes to Neurodegenerative Diseases. Biology. 2021; 10(2):163. https://doi.org/10.3390/biology10020163
Chicago/Turabian StyleGupta, Swapnil, Panpan You, Tanima SenGupta, Hilde Nilsen, and Kulbhushan Sharma. 2021. "Crosstalk between Different DNA Repair Pathways Contributes to Neurodegenerative Diseases" Biology 10, no. 2: 163. https://doi.org/10.3390/biology10020163
APA StyleGupta, S., You, P., SenGupta, T., Nilsen, H., & Sharma, K. (2021). Crosstalk between Different DNA Repair Pathways Contributes to Neurodegenerative Diseases. Biology, 10(2), 163. https://doi.org/10.3390/biology10020163