
TERT Promoter Revertant Mutation Inhibits Melanoma Growth through Intrinsic Apoptosis

Abstract
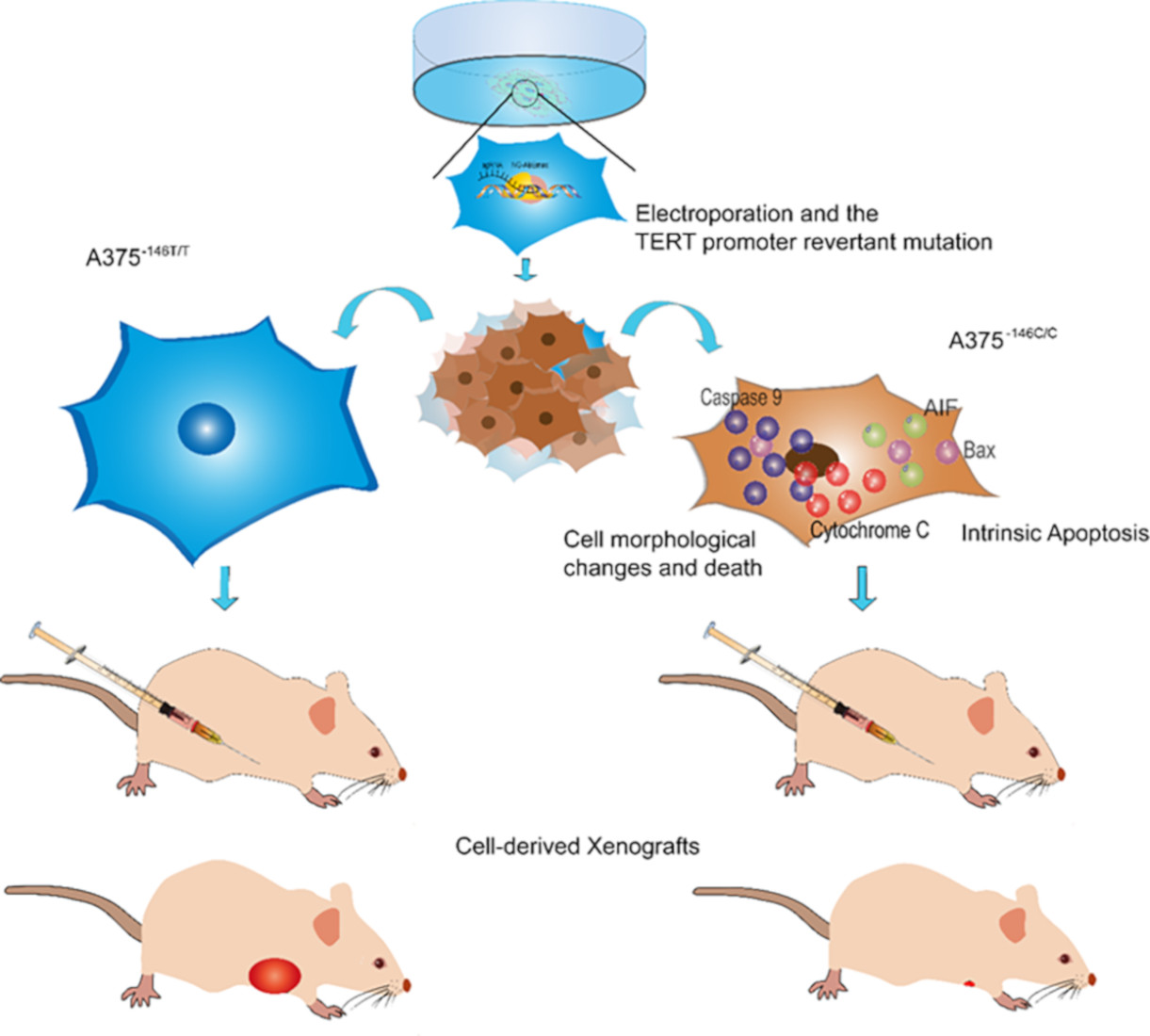
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines and Cell Culture Conditions
2.3. Plasmid Construction
2.4. Electroporation, Selection of A375 and PCR
2.5. Real-Time Q-TRAP
2.6. Telomere Length Measurements
2.7. Transwell Migration and Invasion Assays
2.8. Off-Target Analysis
2.9. Reverse Transcriptase-PCR
2.10. Western Blotting
2.11. Cell Proliferation Assays
2.12. Tumor Xenografts
2.13. DNA Damage Assay (γ-H2AX Immunofluorescence Assay)
2.14. Statistics
3. Results
3.1. TERT Promoter Revertant Mutation Inhibits TERT Expression
3.2. TERT Promoter Revertant Mutation Reduces Telomere Length, Migration and Invasion, and Induces Cellular Deformation
3.3. Off-Target Analysis
3.4. TERT Promoter Revertant Mutation Inhibits Melanoma Growth In Vitro and In Vivo
3.5. TERT Promoter Revertant Mutation and Wnt/β-Catenin Signaling
3.6. TERT Promoter Revertant Mutation Activates the Intrinsic Apoptosis Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Vos, T.; Abajobir, A.A.; Abate, K.H.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abdulkader, R.S.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef]
- Gardner, L.J.; Strunck, J.L.; Wu, Y.P.; Grossman, D. Current controversies in early-stage melanoma: Questions on incidence, screening, and histologic regression. J. Am. Acad. Dermatol. 2019, 80, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Alqathama, A. BRAF in malignant melanoma progression and metastasis: Potentials and challenges. Am. J. Cancer Res. 2020, 10, 1103–1114. [Google Scholar] [PubMed]
- Peters, G. Tumor suppression for ARFicionados: The relative contributions of p16INK4a and p14ARF in melanoma. J. Natl. Cancer Inst. 2008, 100, 757–759. [Google Scholar] [CrossRef]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef]
- Shaughnessy, M.; Njauw, C.N.; Artomov, M.; Tsao, H. Classifying Melanoma by TERT Promoter Mutational Status. J. Investig. Dermatol. 2020, 140, 390–394.e391. [Google Scholar] [CrossRef]
- Jenkins, N.C.; Jung, J.; Liu, T.; Wilde, M.; Holmen, S.L.; Grossman, D. Familial melanoma-associated mutations in p16 uncouple its tumor-suppressor functions. J. Investig. Dermatol. 2013, 133, 1043–1051. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Wang, Y.; Sušac, L.; Feigon, J. Structural Biology of Telomerase. Cold Spring Harb. Perspect. Biol. 2019, 11, a032383. [Google Scholar] [CrossRef]
- Li, X.; Qian, X.; Wang, B.; Xia, Y.; Zheng, Y.; Du, L.; Xu, D.; Xing, D.; DePinho, R.A.; Lu, Z. Programmable base editing of mutated TERT promoter inhibits brain tumour growth. Nat. Cell Biol. 2020, 22, 282–288. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, T.; Zhu, G.; Xing, M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat. Commun. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Mu, N.; Wang, N.; Strååt, K.; Sofiadis, A.; Guo, Y.; Stenman, A.; Li, K.; Cheng, G.; Zhang, L.; et al. GABPA inhibits invasion/metastasis in papillary thyroid carcinoma by regulating DICER1 expression. Oncogene 2019, 38, 965–979. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yuan, X.; Li, K.; Dai, M.; Zhang, L.; Wu, Y.; Sun, C.; Chen, Y.; Cheng, G.; Liu, C.; et al. GABPA is a master regulator of luminal identity and restrains aggressive diseases in bladder cancer. Cell Death Differ. 2020, 27, 1862–1877. [Google Scholar] [CrossRef]
- Bao, Y.; Prescott, J.; Yuan, C.; Zhang, M.; Kraft, P.; Babic, A.; Morales-Oyarvide, V.; Qian, Z.R.; Buring, J.E.; Cochrane, B.B.; et al. Leucocyte telomere length, genetic variants at the TERT gene region and risk of pancreatic cancer. Gut 2017, 66, 1116–1122. [Google Scholar] [CrossRef]
- Hoffmeyer, K.; Raggioli, A.; Rudloff, S.; Anton, R.; Hierholzer, A.; Del Valle, I.; Hein, K.; Vogt, R.; Kemler, R. Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science 2012, 336, 1549–1554. [Google Scholar] [CrossRef]
- Madan, B.; Harmston, N.; Nallan, G.; Montoya, A.; Faull, P.; Petretto, E.; Virshup, D.M. Temporal dynamics of Wnt-dependent transcriptome reveal an oncogenic Wnt/MYC/ribosome axis. J. Clin. Investig. 2018, 128, 5620–5633. [Google Scholar] [CrossRef]
- Celeghin, A.; Giunco, S.; Freguja, R.; Zangrossi, M.; Nalio, S.; Dolcetti, R.; De Rossi, A. Short-term inhibition of TERT induces telomere length-independent cell cycle arrest and apoptotic response in EBV-immortalized and transformed B cells. Cell Death Dis. 2016, 7, e2562. [Google Scholar] [CrossRef]
- Chen, P.; Gu, W.L.; Gong, M.Z.; Wang, J.; Li, D.Q. shRNA-mediated silencing of hTERT suppresses proliferation and promotes apoptosis in osteosarcoma cells. Cancer Gene Ther. 2017, 24, 325–332. [Google Scholar] [CrossRef]
- Mattson, M.P.; Klapper, W. Emerging roles for telomerase in neuronal development and apoptosis. J. Neurosci. Res. 2001, 63, 1–9. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Benimetskaya, L.; Ayyanar, K.; Kornblum, N.; Castanotto, D.; Rossi, J.; Wu, S.; Lai, J.; Brown, B.D.; Popova, N.; Miller, P.; et al. Bcl-2 protein in 518A2 melanoma cells in vivo and in vitro. Clin. Cancer Res. 2006, 12, 4940–4948. [Google Scholar] [CrossRef]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef]
- Pattingre, S.; Levine, B. Bcl-2 inhibition of autophagy: A new route to cancer? Cancer Res. 2006, 66, 2885–2888. [Google Scholar] [CrossRef]
- Wen, L.; Zhao, C.; Song, J.; Ma, L.; Ruan, J.; Xia, X.; Chen, Y.E.; Zhang, J.; Ma, P.X.; Xu, J. CRISPR/Cas9-Mediated TERT Disruption in Cancer Cells. Int. J. Mol. Sci. 2020, 21, 653. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, X.; Li, G.; Huang, S.; Sun, W.; Sun, Q.; Li, L.; Huang, X.; Liu, J.; Wang, L. Editing Properties of Base Editors with SpCas9-NG in Discarded Human Tripronuclear Zygotes. CRISPR J. 2021, 4, 710–727. [Google Scholar] [CrossRef]
- Fellmann, C.; Gowen, B.G.; Lin, P.C.; Doudna, J.A.; Corn, J.E. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat. Rev. Drug Discov. 2017, 16, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Mao, A.; Xu, M.; Weng, Q.; Mao, J.; Ji, J. CRISPR-Cas9 for cancer therapy: Opportunities and challenges. Cancer Lett. 2019, 447, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Savić, N.; Schwank, G. Advances in therapeutic CRISPR/Cas9 genome editing. Transl. Res. 2016, 168, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.M.; Fu, X.; Zhu, J.; Katrekar, D.; Shih, Y.V.; Marlett, J.; Cabotaje, J.; Tat, J.; Naughton, J.; Lisowski, L.; et al. In Situ Gene Therapy via AAV-CRISPR-Cas9-Mediated Targeted Gene Regulation. Mol. Ther. 2018, 26, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, F.; Gao, G. CRISPR-Based Therapeutic Genome Editing: Strategies and In Vivo Delivery by AAV Vectors. Cell 2020, 181, 136–150. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Y.; He, L.; Pu, W.; Yu, W.; Li, Y.; Wu, Y.T.; Xu, C.; Wei, Y.; Ding, Q.; et al. In Vivo AAV-CRISPR/Cas9-Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation 2020, 141, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Wu, Y.; Gemberling, M.P.; Oliver, M.L.; Waller, M.A.; Bohning, J.D.; Robinson-Hamm, J.N.; Bulaklak, K.; Castellanos Rivera, R.M.; Collier, J.H.; et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 427–432. [Google Scholar] [CrossRef]
- Min, Y.L.; Bassel-Duby, R.; Olson, E.N. CRISPR Correction of Duchenne Muscular Dystrophy. Annu. Rev. Med. 2019, 70, 239–255. [Google Scholar] [CrossRef]
- Liu, Z.; Shan, H.; Chen, S.; Chen, M.; Song, Y.; Lai, L.; Li, Z. Highly efficient base editing with expanded targeting scope using SpCas9-NG in rabbits. FASEB J. 2020, 34, 588–596. [Google Scholar] [CrossRef]
- Herbert, B.S.; Hochreiter, A.E.; Wright, W.E.; Shay, J.W. Nonradioactive detection of telomerase activity using the telomeric repeat amplification protocol. Nat. Protoc. 2006, 1, 1583–1590. [Google Scholar] [CrossRef]
- Werner, C.M.; Hecksteden, A.; Morsch, A.; Zundler, J.; Wegmann, M.; Kratzsch, J.; Thiery, J.; Hohl, M.; Bittenbring, J.T.; Neumann, F.; et al. Differential effects of endurance, interval, and resistance training on telomerase activity and telomere length in a randomized, controlled study. Eur. Heart J. 2019, 40, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Cawthon, R.M. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002, 30, e47. [Google Scholar] [CrossRef]
- Simon, M.; Hosen, I.; Gousias, K.; Rachakonda, S.; Heidenreich, B.; Gessi, M.; Schramm, J.; Hemminki, K.; Waha, A.; Kumar, R. TERT promoter mutations: A novel independent prognostic factor in primary glioblastomas. Neuro-Oncology 2015, 17, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Vallarelli, A.F.; Rachakonda, P.S.; André, J.; Heidenreich, B.; Riffaud, L.; Bensussan, A.; Kumar, R.; Dumaz, N. TERT promoter mutations in melanoma render TERT expression dependent on MAPK pathway activation. Oncotarget 2016, 7, 53127–53136. [Google Scholar] [CrossRef]
- Van Cruchten, S.; Van Den Broeck, W. Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat. Histol. Embryol. 2002, 31, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Li, M.; Tang, X.; You, W.; Wang, Y.; Chen, Y.; Liu, Y.; Yuan, H.; Gao, C.; Chen, X.; Xiao, Z.; et al. HMEJ-mediated site-specific integration of a myostatin inhibitor increases skeletal muscle mass in porcine. Mol. Ther. Nucleic Acids 2021, 26, 49–62. [Google Scholar] [CrossRef]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of β-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef]
- Sun, Y.; Yu, M.; Qu, M.; Ma, Y.; Zheng, D.; Yue, Y.; Guo, S.; Tang, L.; Li, G.; Zheng, W.; et al. Hepatitis B virus-triggered PTEN/β-catenin/c-Myc signaling enhances PD-L1 expression to promote immune evasion. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G162–G173. [Google Scholar] [CrossRef]
- Nayak, G.; Odaka, Y.; Prasad, V.; Solano, A.F.; Yeo, E.J.; Vemaraju, S.; Molkentin, J.D.; Trumpp, A.; Williams, B.; Rao, S.; et al. Developmental vascular regression is regulated by a Wnt/β-catenin, MYC and CDKN1A pathway that controls cell proliferation and cell death. Development 2018, 145, dev154898. [Google Scholar] [CrossRef]
- Lee, E.J.; Seo, E.; Kim, J.W.; Nam, S.A.; Lee, J.Y.; Jun, J.; Oh, S.; Park, M.; Jho, E.H.; Yoo, K.H.; et al. TAZ/Wnt-β-catenin/c-MYC axis regulates cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2020, 117, 29001–29012. [Google Scholar] [CrossRef]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef]
- Arantes, L.; Cruvinel-Carloni, A.; de Carvalho, A.C.; Sorroche, B.P.; Carvalho, A.L.; Scapulatempo-Neto, C.; Reis, R.M. TERT Promoter Mutation C228T Increases Risk for Tumor Recurrence and Death in Head and Neck Cancer Patients. Front. Oncol. 2020, 10, 1275. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, K.; Yekula, A.; Small, J.L.; Rosh, Z.S.; Kang, K.M.; Wang, L.; Lau, S.; Zhang, H.; Lee, H.; Bettegowda, C.; et al. TERT Promoter Mutation Analysis for Blood-Based Diagnosis and Monitoring of Gliomas. Clin. Cancer Res. 2021, 27, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Vinagre, J.; Almeida, A.; Pópulo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef] [PubMed]
- García-Sáez, A.J. The secrets of the Bcl-2 family. Cell Death Differ. 2012, 19, 1733–1740. [Google Scholar] [CrossRef]
- Thompson, E.B. The many roles of c-Myc in apoptosis. Annu. Rev. Physiol. 1998, 60, 575–600. [Google Scholar] [CrossRef] [PubMed]
- Belling, J.N.; Heidenreich, L.K.; Tian, Z.; Mendoza, A.M.; Chiou, T.T.; Gong, Y.; Chen, N.Y.; Young, T.D.; Wattanatorn, N.; Park, J.H.; et al. Acoustofluidic sonoporation for gene delivery to human hematopoietic stem and progenitor cells. Proc. Natl. Acad. Sci. USA 2020, 117, 10976–10982. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Chen, Y.; Li, C.; Xiao, Z.; Yuan, H.; Zhang, Y.; Pang, D.; Tang, X.; Li, M.; Ouyang, H. TERT Promoter Revertant Mutation Inhibits Melanoma Growth through Intrinsic Apoptosis. Biology 2022, 11, 141. https://doi.org/10.3390/biology11010141
Wang Y, Chen Y, Li C, Xiao Z, Yuan H, Zhang Y, Pang D, Tang X, Li M, Ouyang H. TERT Promoter Revertant Mutation Inhibits Melanoma Growth through Intrinsic Apoptosis. Biology. 2022; 11(1):141. https://doi.org/10.3390/biology11010141
Chicago/Turabian StyleWang, Yanbing, Yiwu Chen, Chang Li, Zhiwei Xiao, Hongming Yuan, Yuanzhu Zhang, Daxin Pang, Xiaochun Tang, Mengjing Li, and Hongsheng Ouyang. 2022. "TERT Promoter Revertant Mutation Inhibits Melanoma Growth through Intrinsic Apoptosis" Biology 11, no. 1: 141. https://doi.org/10.3390/biology11010141
APA StyleWang, Y., Chen, Y., Li, C., Xiao, Z., Yuan, H., Zhang, Y., Pang, D., Tang, X., Li, M., & Ouyang, H. (2022). TERT Promoter Revertant Mutation Inhibits Melanoma Growth through Intrinsic Apoptosis. Biology, 11(1), 141. https://doi.org/10.3390/biology11010141