
Endothelial Cell GATA2 Modulates the Cardiomyocyte Stress Response through the Regulation of Two Long Non-Coding RNAs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Human Tissue and Serum Samples
2.3. Cell Culture
2.4. Plasmids and Adenoviruses
2.5. Co-Culture of Endothelial Cells and Cardiomyocytes
2.6. Stretch Assay
2.7. Luciferase Assay
2.8. RNA Isolation and Real-Time PCR
2.9. Agilent Microarray
2.10. Immunoblot Analysis
2.11. ChIP Assay
2.12. Histological Analysis
2.13. Statistical Analysis
3. Results
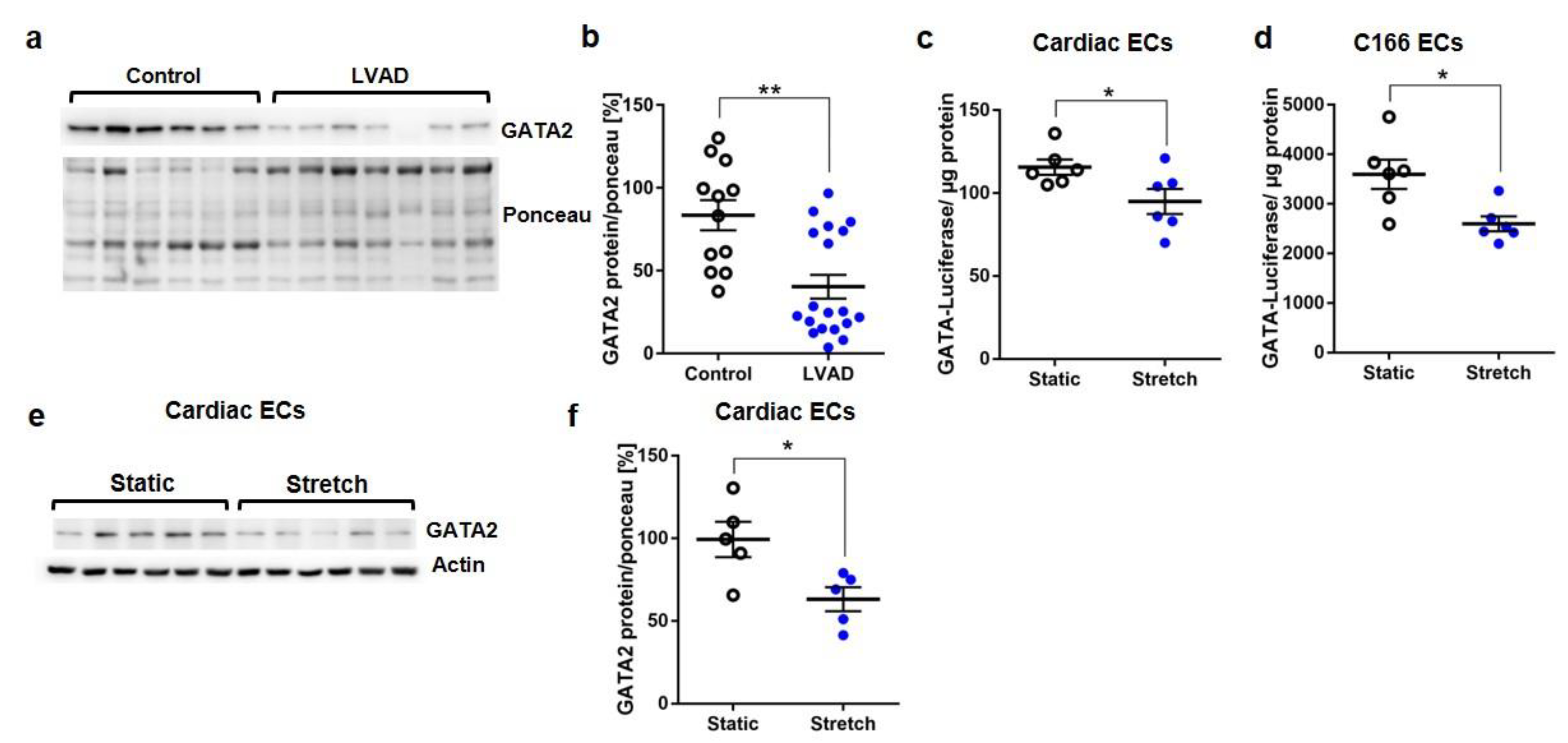
3.1. Reduced GATA Activation upon Mechanical Stretch in Endothelial Cells
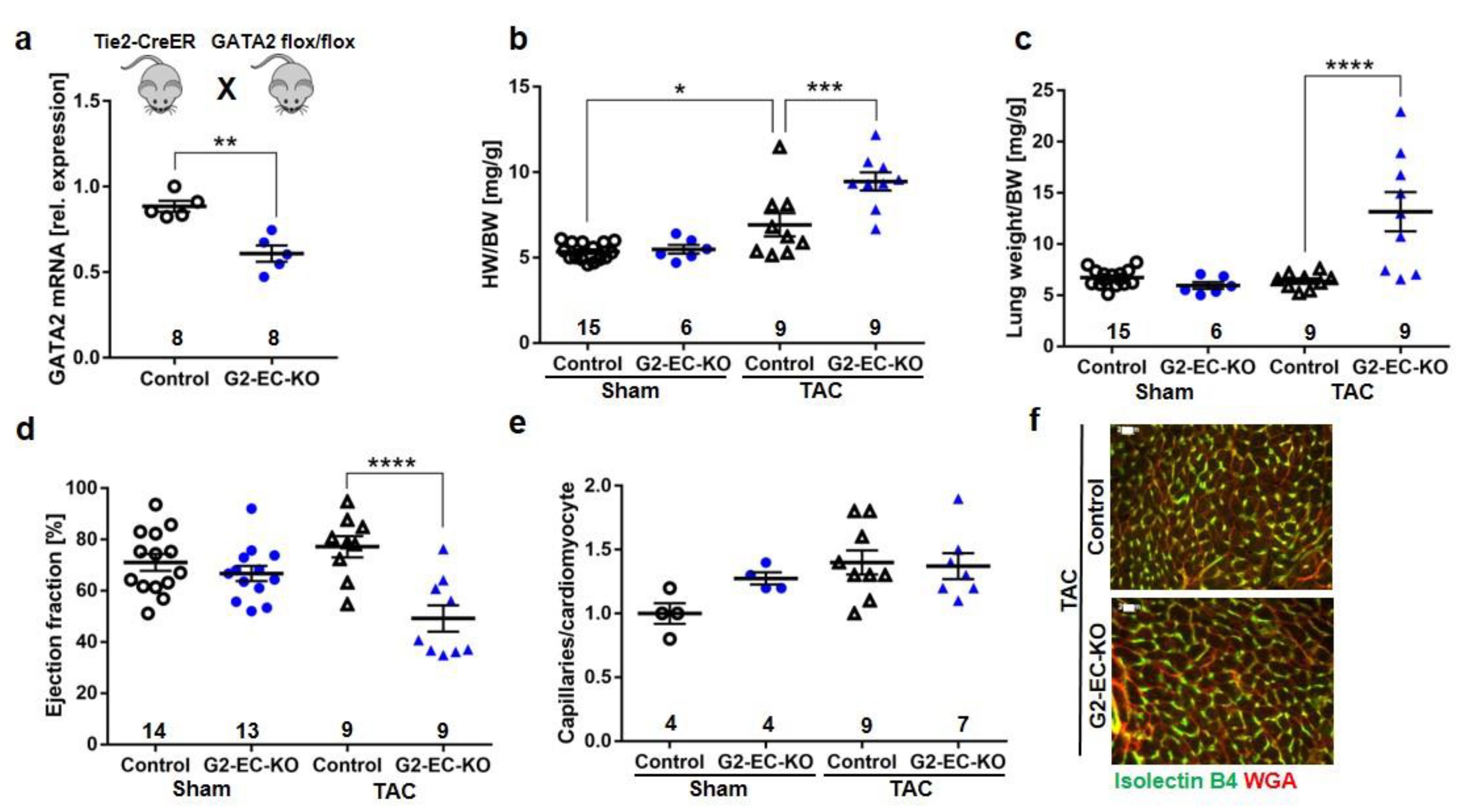
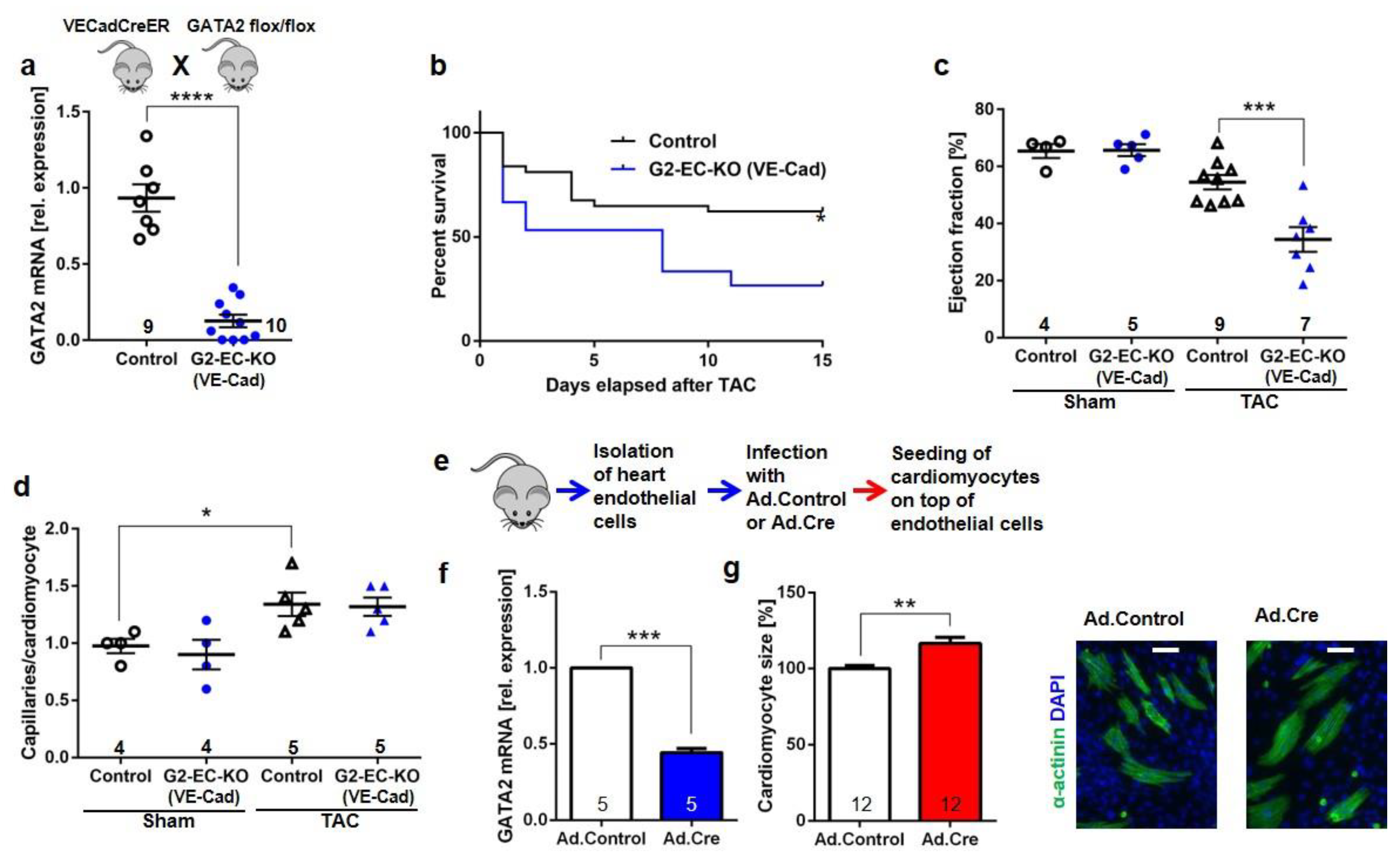
3.2. Endothelial Specific GATA2 Knock-Out Induces Cardiac Failure upon Pressure Overload
3.3. Disturbed Cardiomyocyte Stress Signaling in Endothelial Specific GATA2 Knock-Out Mice
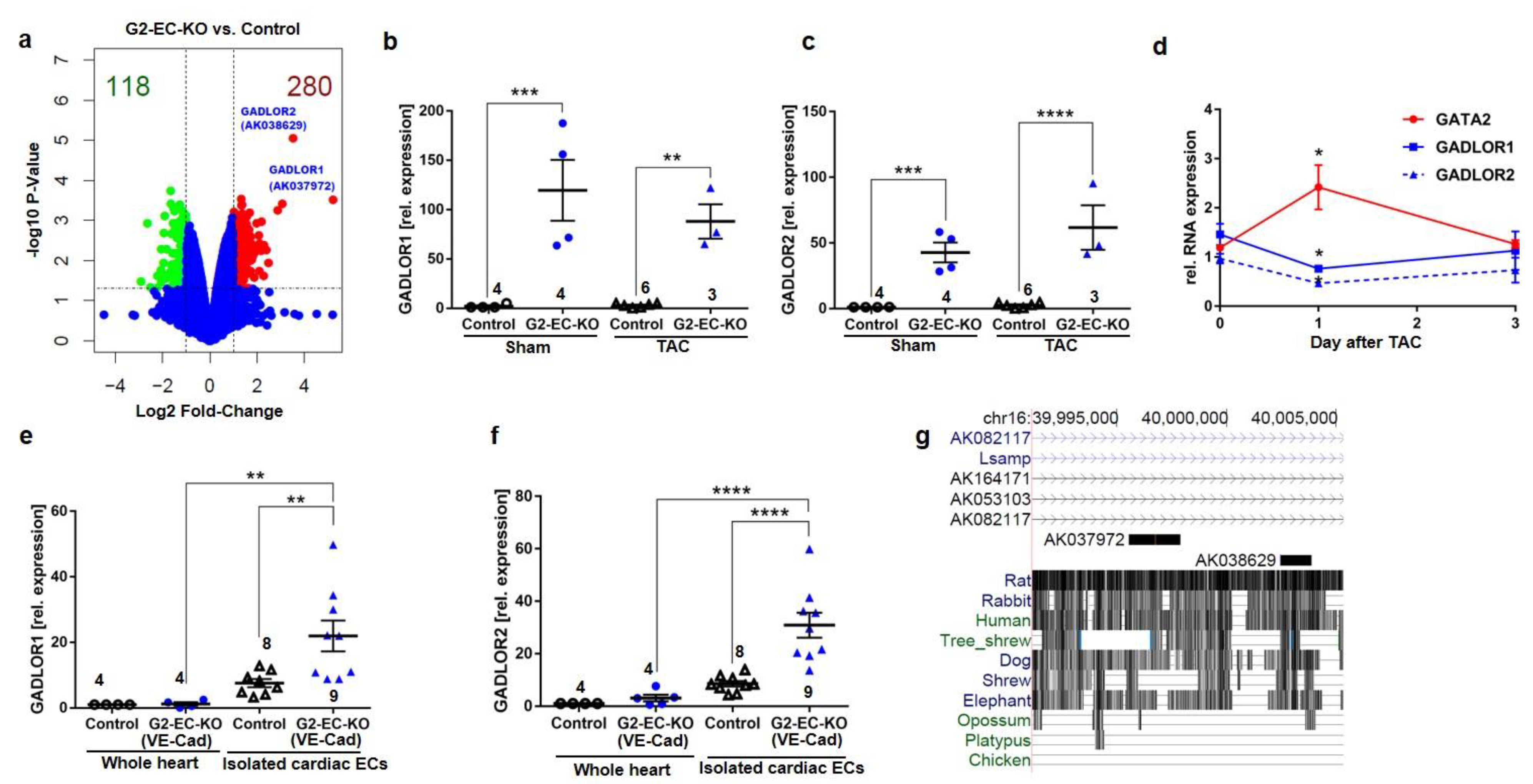
3.4. The Long Non-Coding RNAs GADLOR1 and GADLOR2 Are Released in Response to Reduced GATA2 Expression and during Heart Failure
3.5. Disturbed Cardiomyocyte Stress Signaling due to GADLOR Uptake
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar]
- Frantz, S.; Hundertmark, M.J.; Schulz-Menger, J.; Bengel, F.M.; Bauersachs, J. Left ventricular remodelling post-myocardial infarction: Pathophysiology, imaging, and novel therapies. Eur. Heart J. 2022, 43, 2549–2561. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Reboll, M.R.; Korf-Klingebiel, M.; Wollert, K.C. Angiogenesis after acute myocardial infarction. Cardiovasc. Res. 2021, 117, 1257–1273. [Google Scholar] [CrossRef] [PubMed]
- Heineke, J.; Auger-Messier, M.; Xu, J.; Oka, T.; Sargent, M.A.; York, A.; Klevitsky, R.; Vaikunth, S.; Duncan, S.A.; Aronow, B.J.; et al. Cardiomyocyte GATA4 functions as a stress-responsive regulator of angiogenesis in the murine heart. J. Clin. Investig. 2007, 117, 3198–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M.; et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007, 446, 444–448. [Google Scholar] [CrossRef]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Narmoneva, D.A.; Vukmirovic, R.; Davis, M.E.; Kamm, R.D.; Lee, R.T. Endothelial cells promote cardiac myocyte survival and spatial reorganization: Implications for cardiac regeneration. Circulation 2004, 110, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Izumiya, Y.; Shiojima, I.; Sato, K.; Sawyer, D.B.; Colucci, W.S.; Walsh, K. Vascular Endothelial Growth Factor Blockade Promotes the Transition from Compensatory Cardiac Hypertrophy to Failure in Response to Pressure Overload. Hypertension 2006, 47, 887–893. [Google Scholar] [CrossRef]
- Tirziu, D.; Chorianopoulos, E.; Moodie, K.L.; Palac, R.T.; Zhuang, Z.W.; Tjwa, M.; Roncal, C.; Eriksson, U.; Fu, Q.; Elfenbein, A.; et al. Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J. Clin. Investig. 2007, 117, 3188–3197. [Google Scholar] [CrossRef]
- Chen, J.; Yaniz-Galende, E.; Kagan, H.J.; Liang, L.; Hekmaty, S.; Giannarelli, C.; Hajjar, R.J. Abnormalities of capillary microarchitecture in a rat model of coronary ischemic congestive heart failure. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H830–H840. [Google Scholar] [CrossRef] [Green Version]
- Appari, M.; Breitbart, A.; Brandes, F.; Szaroszyk, M.; Froese, N.; Korf-Klingebiel, M.; Malek Mohammadi, M.; Grund, A.; Scharf, G.M.; Wang, H.; et al. C1q-TNF-Related Protein-9 Promotes Cardiac Hypertrophy and Failure. Circ Res. 2017, 120, 66–77. [Google Scholar] [CrossRef]
- Heineke, J. Wag the dog: How endothelial cells regulate cardiomyocyte growth. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 545–547. [Google Scholar] [CrossRef] [Green Version]
- Halkein, J.; Tabruyn, S.P.; Ricke-Hoch, M.; Haghikia, A.; Nguyen, N.Q.; Scherr, M.; Castermans, K.; Malvaux, L.; Lambert, V.; Thiry, M.; et al. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J. Clin. Investig. 2013, 123, 2143–2154. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, A.; Connor, K.M.; Mammoto, T.; Yung, C.W.; Huh, D.; Aderman, C.M.; Mostoslavsky, G.; Smith, L.E.; Ingber, D.E. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature 2009, 457, 1103–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnemann, A.K.; O’Geen, H.; Keles, S.; Farnham, P.J.; Bresnick, E.H. Genetic framework for GATA factor function in vascular biology. Proc. Natl. Acad. Sci. USA 2011, 108, 13641–13646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, F.Y.; Keller, G.; Kuo, F.C.; Weiss, M.; Chen, J.; Rosenblatt, M.; Alt, F.W.; Orkin, S.H. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature 1994, 371, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.D.; Hsu, A.P.; Ryu, M.J.; Wang, J.; Gao, X.; Boyer, M.E.; Liu, Y.; Lee, Y.; Calvo, K.R.; Keles, S.; et al. Cis-element mutated in GATA2-dependent immunodeficiency governs hematopoiesis and vascular integrity. J. Clin. Investig. 2012, 122, 3692–3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.C.; Hosoya, T.; Brandt, W.; Ku, C.J.; Hosoya-Ohmura, S.; Camper, S.A.; Yamamoto, M.; Engel, J.D. Conditional Gata2 inactivation results in HSC loss and lymphatic mispatterning. J. Clin. Investig. 2012, 122, 3705–3717. [Google Scholar] [CrossRef] [Green Version]
- Charles, M.A.; Saunders, T.L.; Wood, W.M.; Owens, K.; Parlow, A.F.; Camper, S.A.; Ridgway, E.C.; Gordon, D.F. Pituitary-specific Gata2 knockout: Effects on gonadotrope and thyrotrope function. Mol. Endocrinol. 2006, 20, 1366–1377. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.C.; Luscinskas, F.W. Isolation and culture of murine heart and lung endothelial cells for in vitro model systems. Methods Mol. Biol. 2006, 341, 141–154. [Google Scholar]
- Froese, N.; Cordero, J.; Abouissa, A.; Trogisch, F.A.; Grein, S.; Szaroszyk, M.; Wang, Y.; Gigina, A.; Korf-Klingebiel, M.; Bosnjak, B.; et al. Analysis of myocardial cellular gene expression during pressure overload reveals matrix based functional intercellular communication. iScience 2022, 25, 103965. [Google Scholar] [CrossRef]
- Wollert, K.C.; Taga, T.; Saito, M.; Narazaki, M.; Kishimoto, T.; Glembotski, C.C.; Vernallis, A.B.; Heath, J.K.; Pennica, D.; Wood, W.I.; et al. Cardiotrophin-1 activates a distinct form of cardiac muscle cell hypertrophy. Assembly of sarcomeric units in series VIA gp130/leukemia inhibitory factor receptor-dependent pathways. J. Biol. Chem. 1996, 271, 9535–9545. [Google Scholar] [CrossRef] [PubMed]
- Heineke, J.; Auger-Messier, M.; Correll, R.N.; Xu, J.; Benard, M.J.; Yuan, W.; Drexler, H.; Parise, L.V.; Molkentin, J.D. CIB1 is a regulator of pathological cardiac hypertrophy. Nat. Med. 2010, 16, 872–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froese, N.; Kattih, B.; Breitbart, A.; Grund, A.; Geffers, R.; Molkentin, J.D.; Kispert, A.; Wollert, K.C.; Drexler, H.; Heineke, J. GATA6 promotes angiogenic function and survival in endothelial cells by suppression of autocrine transforming growth factor beta/activin receptor-like kinase 5 signaling. J. Biol. Chem. 2011, 286, 5680–5690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maillet, M.; Davis, J.; Auger-Messier, M.; York, A.; Osinska, H.; Piquereau, J.; Lorenz, J.N.; Robbins, J.; Ventura-Clapier, R.; Molkentin, J.D. Heart-specific deletion of CnB1 reveals multiple mechanisms whereby calcineurin regulates cardiac growth and function. J. Biol. Chem. 2010, 285, 6716–6724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Rothermel, B.; Vega, R.B.; Frey, N.; McKinsey, T.A.; Olson, E.N.; Bassel-Duby, R.; Williams, R.S. Independent signals control expression of the calcineurin inhibitory proteins MCIP1 and MCIP2 in striated muscles. Circ. Res. 2000, 87, E61–E68. [Google Scholar] [CrossRef] [Green Version]
- Forde, A.; Constien, R.; Grone, H.J.; Hammerling, G.; Arnold, B. Temporal Cre-mediated recombination exclusively in endothelial cells using Tie2 regulatory elements. Genesis 2002, 33, 191–197. [Google Scholar] [CrossRef]
- Wang, Y.; Nakayama, M.; Pitulescu, M.E.; Schmidt, T.S.; Bochenek, M.L.; Sakakibara, A.; Adams, S.; Davy, A.; Deutsch, U.; Lüthi, U.; et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010, 465, 483–486. [Google Scholar] [CrossRef]
- Alam, T.; Medvedeva, Y.A.; Jia, H.; Brown, J.B.; Lipovich, L.; Bajic, V.B. Promoter analysis reveals globally differential regulation of human long non-coding RNA and protein-coding genes. PLoS ONE 2014, 9, e109443. [Google Scholar] [CrossRef] [Green Version]
- Kogure, T.; Yan, I.K.; Lin, W.L.; Patel, T. Extracellular Vesicle-Mediated Transfer of a Novel Long Noncoding RNA TUC339: A Mechanism of Intercellular Signaling in Human Hepatocellular Cancer. Genes Cancer 2014, 4, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yan, I.K.; Kogure, T.; Haga, H.; Patel, T. Extracellular vesicle-mediated transfer of long non-coding RNA ROR modulates chemosensitivity in human hepatocellular cancer. FEBS Open Bio 2014, 4, 458–467. [Google Scholar] [CrossRef] [Green Version]
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonrock, N.; Harvey, R.P.; Mattick, J.S. Long noncoding RNAs in cardiac development and pathophysiology. Circ. Res. 2012, 111, 1349–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.; Li, C.; Xing, Z.; Hu, Q.; Liang, K.; Han, L.; Wang, C.; Hawke, D.H.; Wang, S.; Zhang, Y.; et al. The LINK-A lncRNA activates normoxic HIF1α signalling in triple-negative breast cancer. Nat. Cell Biol. 2016, 18, 213–224. [Google Scholar] [CrossRef]
- Liu, X.; Xiao, Z.D.; Han, L.; Zhang, J.; Lee, S.W.; Wang, W.; Lee, H.; Zhuang, L.; Chen, J.; Lin, H.K.; et al. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat. Cell Biol. 2016, 18, 431–442. [Google Scholar] [CrossRef]
- DeBosch, B.; Treskov, I.; Lupu, T.S.; Weinheimer, C.; Kovacs, A.; Courtois, M.; Muslin, A.J. Akt1 is required for physiological cardiac growth. Circulation 2006, 113, 2097–2104. [Google Scholar] [CrossRef] [Green Version]
- Nishida, K.; Yamaguchi, O.; Hirotani, S.; Hikoso, S.; Higuchi, Y.; Watanabe, T.; Takeda, T.; Osuka, S.; Morita, T.; Kondoh, G.; et al. p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell Biol. 2004, 24, 10611–10620. [Google Scholar] [CrossRef] [Green Version]
- Braz, J.C.; Bueno, O.F.; Liang, Q.; Wilkins, B.J.; Dai, Y.S.; Parsons, S.; Braunwart, J.; Glascock, B.J.; Klevitsky, R.; Kimball, T.F.; et al. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J. Clin. Investig. 2003, 111, 1475–1486. [Google Scholar] [CrossRef] [Green Version]
- Molkentin, J.D.; Lu, J.R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Froese, N.; Szaroszyk, M.; Korf-Klingebiel, M.; Koch, K.; Schmitto, J.D.; Geffers, R.; Hilfiker-Kleiner, D.; Riehle, C.; Wollert, K.C.; Bauersachs, J.; et al. Endothelial Cell GATA2 Modulates the Cardiomyocyte Stress Response through the Regulation of Two Long Non-Coding RNAs. Biology 2022, 11, 1736. https://doi.org/10.3390/biology11121736
Froese N, Szaroszyk M, Korf-Klingebiel M, Koch K, Schmitto JD, Geffers R, Hilfiker-Kleiner D, Riehle C, Wollert KC, Bauersachs J, et al. Endothelial Cell GATA2 Modulates the Cardiomyocyte Stress Response through the Regulation of Two Long Non-Coding RNAs. Biology. 2022; 11(12):1736. https://doi.org/10.3390/biology11121736
Chicago/Turabian StyleFroese, Natali, Malgorzata Szaroszyk, Mortimer Korf-Klingebiel, Katrin Koch, Jan D. Schmitto, Robert Geffers, Denise Hilfiker-Kleiner, Christian Riehle, Kai C. Wollert, Johann Bauersachs, and et al. 2022. "Endothelial Cell GATA2 Modulates the Cardiomyocyte Stress Response through the Regulation of Two Long Non-Coding RNAs" Biology 11, no. 12: 1736. https://doi.org/10.3390/biology11121736
APA StyleFroese, N., Szaroszyk, M., Korf-Klingebiel, M., Koch, K., Schmitto, J. D., Geffers, R., Hilfiker-Kleiner, D., Riehle, C., Wollert, K. C., Bauersachs, J., & Heineke, J. (2022). Endothelial Cell GATA2 Modulates the Cardiomyocyte Stress Response through the Regulation of Two Long Non-Coding RNAs. Biology, 11(12), 1736. https://doi.org/10.3390/biology11121736