
Detecting Blood Methylation Signatures in Response to Childhood Cancer Radiotherapy via Machine Learning Methods



 and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
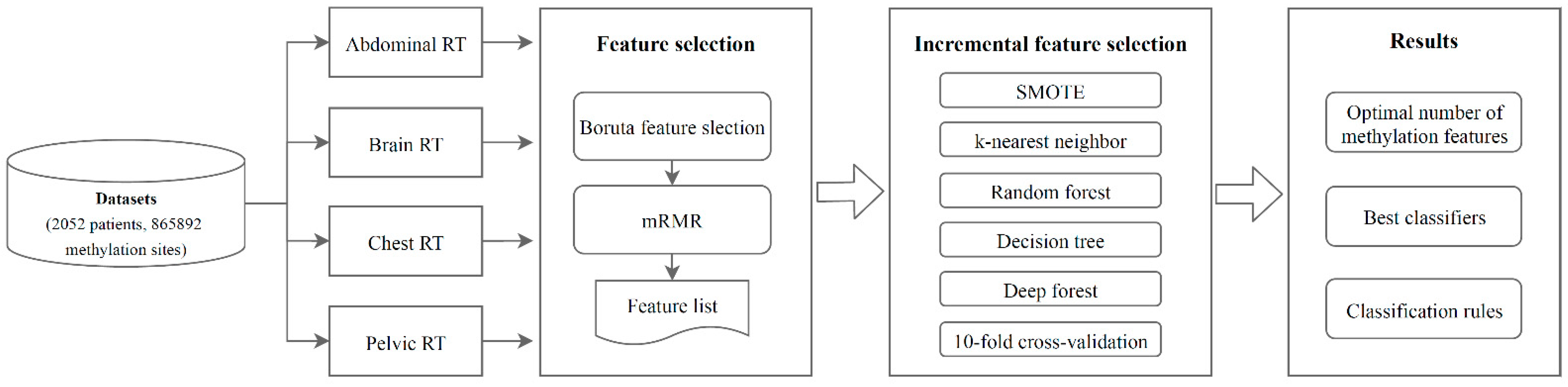
2. Materials and Methods
2.1. Datasets
2.2. Boruta Feature Selection
2.3. mRMR
2.4. IFS
2.5. Classification Algorithms
2.5.1. DF
2.5.2. kNN
2.5.3. RF
2.5.4. DT
2.6. SMOTE
2.7. Performance Measurement
3. Results
3.1. Results of Feature Selection of the Methylation Datasets via the Boruta and mRMR Methods
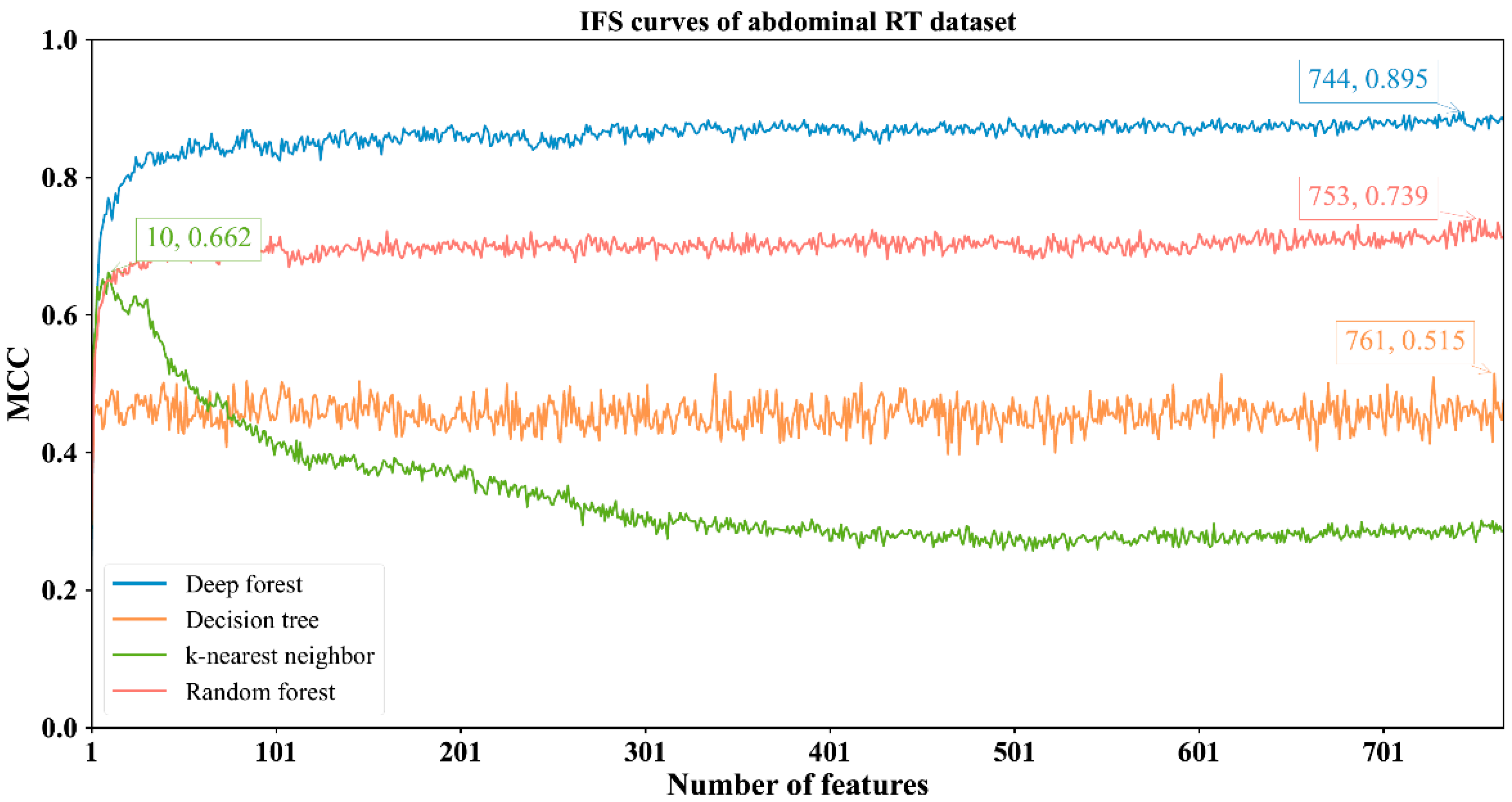
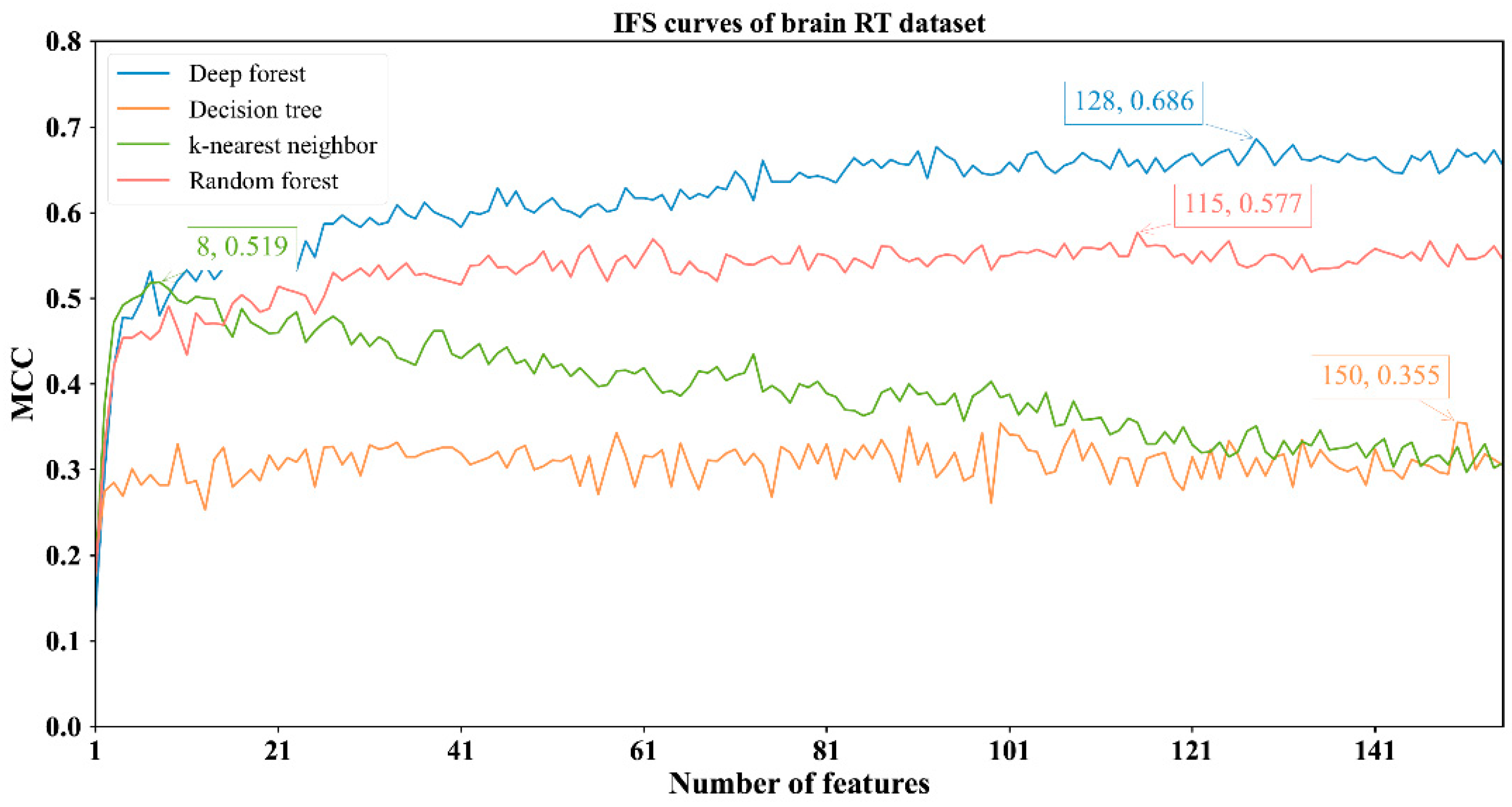
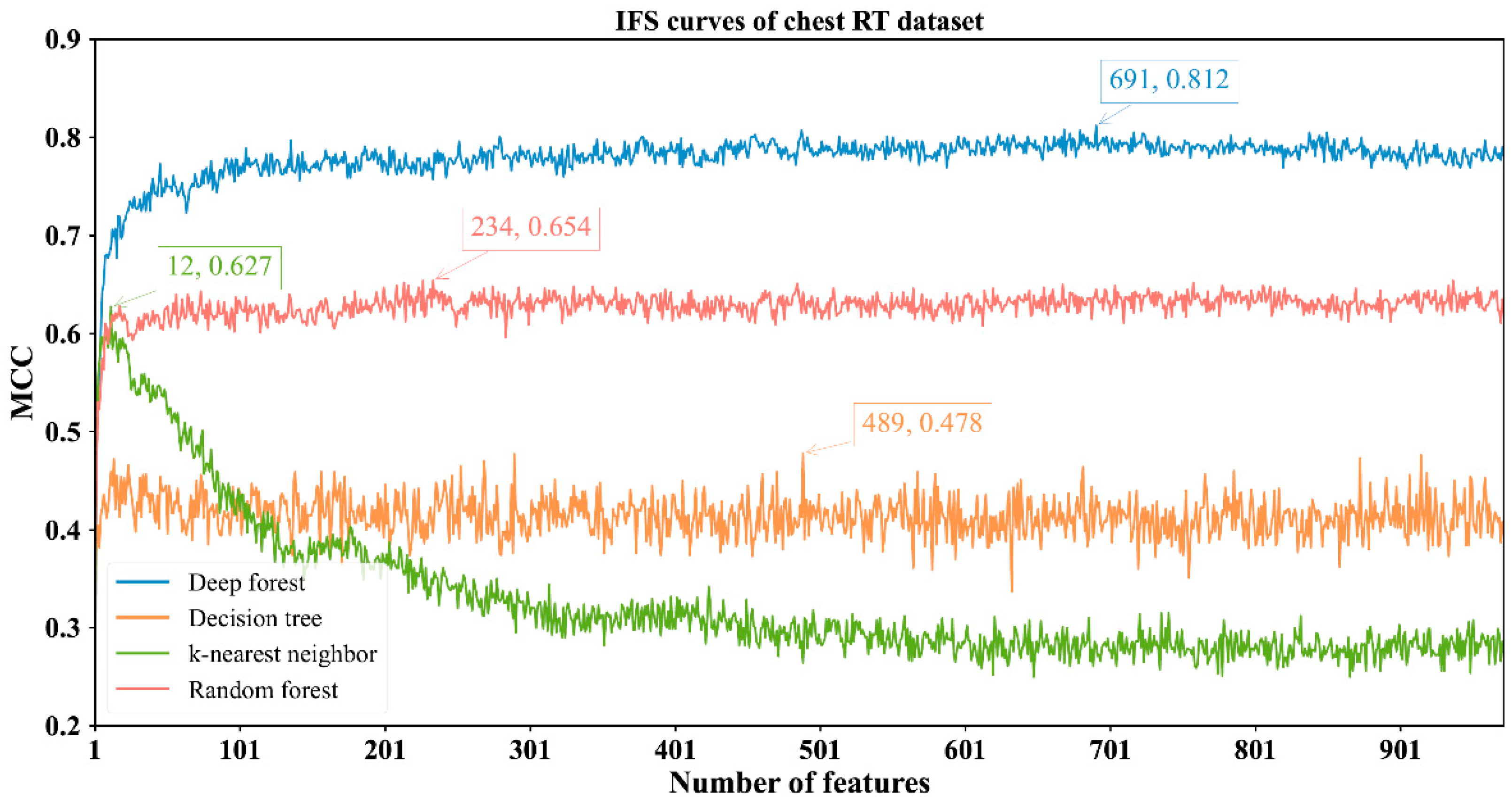
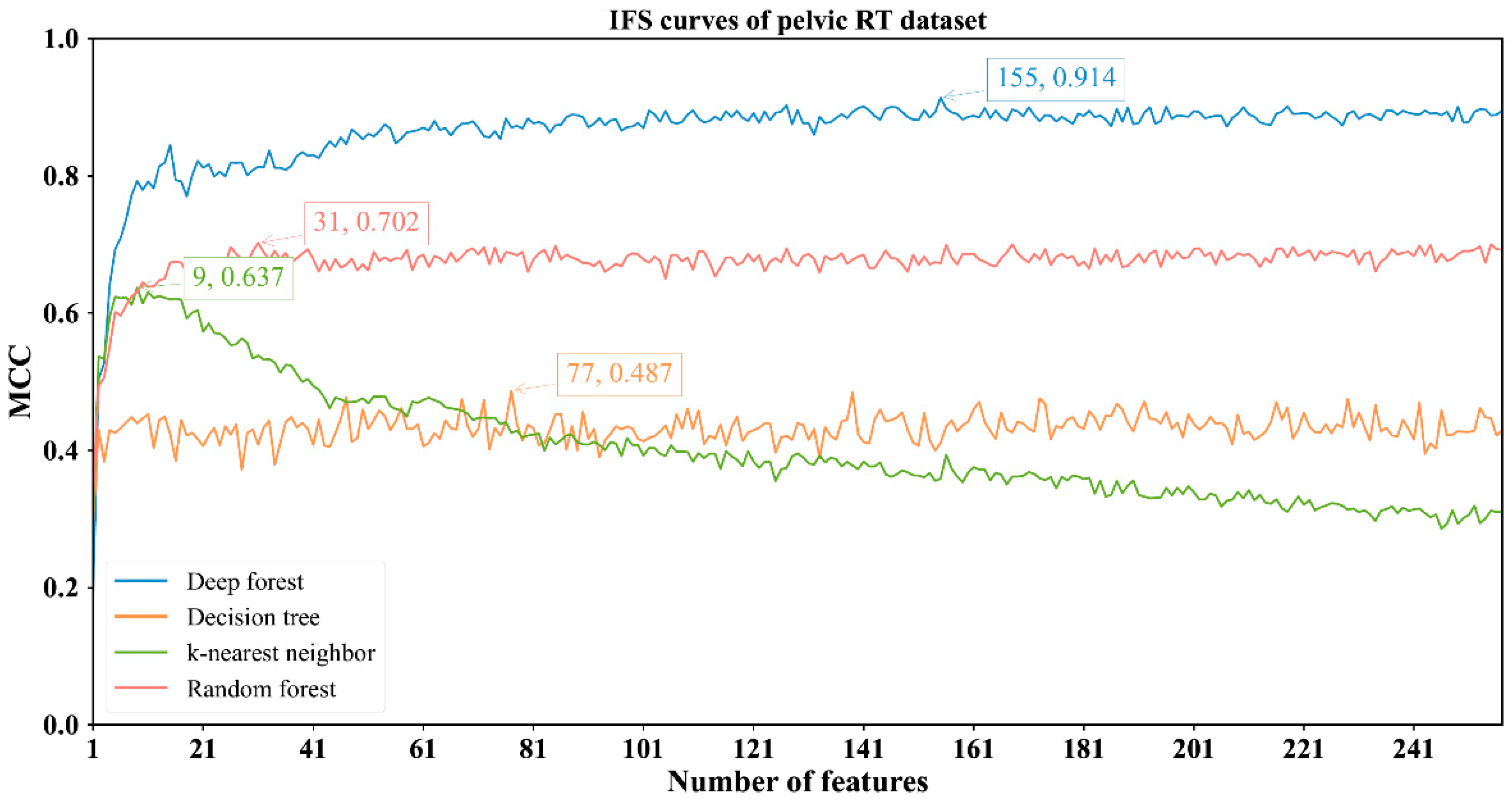
3.2. Results of IFS Method with Classification Algorithms
3.3. Classification Rules Extracted by the Optimal DT Classifiers
4. Discussion
4.1. Key Methylation Alteration Related to Abdominal RT
4.2. Key Methylation Alteration Related to Brain RT
4.3. Key Methylation Alteration Related to Chest RT
4.4. Key Methylation Alteration Related to Pelvic RT
4.5. Limitations of This Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jacks, T.; Jaffee, E.; Singer, D. Cancer Moonshot Blue Ribbon Panel Report 2016; National Cancer Institute: Tokyo, Japan, 2016.
- Kirsch, D.G.; Diehn, M.; Kesarwala, A.H.; Maity, A.; Morgan, M.A.; Schwarz, J.K.; Bristow, R.; Demaria, S.; Eke, I.; Griffin, R.J. The future of radiobiology. JNCI J. Natl. Cancer Inst. 2018, 110, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Abshire, D.; Lang, M.K. The Evolution of Radiation Therapy in Treating Cancer, Seminars in Oncology Nursing; Elsevier: Amsterdam, The Netherlands, 2018; pp. 151–157. [Google Scholar]
- Oeffinger, K.C.; Mertens, A.C.; Sklar, C.A.; Kawashima, T.; Hudson, M.M.; Meadows, A.T.; Friedman, D.L.; Marina, N.; Hobbie, W.; Kadan-Lottick, N.S. Chronic health conditions in adult survivors of childhood cancer. N. Engl. J. Med. 2006, 355, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Jung, S.W.; Huh, S.; Chung, Y.-S.; Cho, H.; Kang, H. Alteration of DNA methylation in gastric cancer with chemotherapy. J. Microbiol. Biotechnol. 2017, 27, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Relton, C.L.; Smith, G.D. Epigenetic epidemiology of common complex disease: Prospects for prediction, prevention, and treatment. PLoS Med. 2010, 7, e1000356. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Kalinich, J.F.; Catravas, G.N.; Snyder, S.L. The effect of γ radiation on DNA methylation. Radiat. Res. 1989, 117, 185–197. [Google Scholar] [CrossRef]
- Pogribny, I.; Raiche, J.; Slovack, M.; Kovalchuk, O. Dose-dependence, sex-and tissue-specificity, and persistence of radiation-induced genomic DNA methylation changes. Biochem. Biophys. Res. Commun. 2004, 320, 1253–1261. [Google Scholar] [CrossRef]
- Peng, Q.; Weng, K.; Li, S.; Xu, R.; Wang, Y.; Wu, Y. A perspective of epigenetic regulation in radiotherapy. Front. Cell Dev. Biol. 2021, 9, 261. [Google Scholar] [CrossRef]
- Song, N.; Hsu, C.-W.; Pan, H.; Zheng, Y.; Hou, L.; Sim, J.-a.; Li, Z.; Mulder, H.; Easton, J.; Walker, E. Persistent variations of blood DNA methylation associated with treatment exposures and risk for cardiometabolic outcomes in long-term survivors of childhood cancer in the st. Jude lifetime cohort. Genome Med. 2021, 13, 53. [Google Scholar] [CrossRef]
- Kursa, M.B.; Rudnicki, W.R. Feature selection with the boruta package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Setiono, R. Incremental feature selection. Appl. Intell. 1998, 9, 217–230. [Google Scholar] [CrossRef]
- Kohavi, R. A study of cross-validation and bootstrap for accuracy estimation and model selection. In Proceedings of the International Joint Conference on Artificial Intelligence, Montreal, QC, Canada, 20–25 August 1995; Lawrence Erlbaum Associates Ltd.: Mahwah, NJ, USA, 1995; pp. 1137–1145. [Google Scholar]
- Zhou, Z.-H.; Feng, J. Deep forest. Natl. Sci. Rev. 2018, 6, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.; Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Safavian, S.R.; Landgrebe, D. A survey of decision tree classifier methodology. IEEE Trans. Syst. Man Cybern. 1991, 21, 660–674. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-H.; Li, H.; Zeng, T.; Chen, L.; Li, Z.; Huang, T.; Cai, Y.-D. Identifying transcriptomic signatures and rules for sars-cov-2 infection. Front. Cell Dev. Biol. 2021, 8, 1763. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Li, Z.; Zeng, T.; Chen, L.; Li, H.; Huang, T.; Cai, Y.-D. Detecting the multiomics signatures of factor-specific inflammatory effects on airway smooth muscles. Front. Genet. 2021, 11, 599970. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Zeng, T.; Chen, L.; Huang, T.; Cai, Y.-D. Determining protein–protein functional associations by functional rules based on gene ontology and kegg pathway. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2021, 1869, 140621. [Google Scholar] [CrossRef]
- Pan, X.; Li, H.; Zeng, T.; Li, Z.; Chen, L.; Huang, T.; Cai, Y.-D. Identification of protein subcellular localization with network and functional embeddings. Front. Genet. 2021, 11, 626500. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, L. Identification of drug–disease associations by using multiple drug and disease networks. Curr. Bioinform. 2022, 17, 48–59. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, L.; Lu, J. A similarity-based method for prediction of drug side effects with heterogeneous information. Math. Biosci. 2018, 306, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Farooq, A.; Khan, M.U.G.; Shabbir, M.Z.; Rabbani, M.; Hussain, I. Identification of most relevant features for classification of francisella tularensis using machine learning. Curr. Bioinform. 2020, 15, 1197–1212. [Google Scholar] [CrossRef]
- Baranwal, M.; Magner, A.; Elvati, P.; Saldinger, J.; Violi, A.; Hero, A.O. A deep learning architecture for metabolic pathway prediction. Bioinformatics 2019, 36, 2547–2553. [Google Scholar] [CrossRef]
- Chen, L.; Li, Z.; Zhang, S.; Zhang, Y.-H.; Huang, T.; Cai, Y.-D. Predicting rna 5-methylcytosine sites by using essential sequence features and distributions. BioMed Res. Int. 2022, 2022, 4035462. [Google Scholar] [CrossRef]
- Liu, H.; Hu, B.; Chen, L.; Lu, L. Identifying protein subcellular location with embedding features learned from networks. Curr. Proteom. 2021, 18, 646–660. [Google Scholar] [CrossRef]
- Chen, W.; Chen, L.; Dai, Q. Impt-fdnpl: Identification of membrane protein types with functional domains and a natural language processing approach. Comput. Math. Methods Med. 2021, 2021, 7681497. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. Smote: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Matthews, B. Comparison of the predicted and observed secondary structure of t4 phage lysozyme. Biochim. Biophys. Acta (BBA)-Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Chen, L.; Wang, S.; Zhang, Y.-H.; Li, J.; Xing, Z.-H.; Yang, J.; Huang, T.; Cai, Y.-D. Identify key sequence features to improve crispr sgrna efficacy. IEEE Access 2017, 5, 26582–26590. [Google Scholar] [CrossRef]
- Jia, Y.; Zhao, R.; Chen, L. Similarity-based machine learning model for predicting the metabolic pathways of compounds. IEEE Access 2020, 8, 130687–130696. [Google Scholar] [CrossRef]
- Liang, H.; Chen, L.; Zhao, X.; Zhang, X. Prediction of drug side effects with a refined negative sample selection strategy. Comput. Math. Methods Med. 2020, 2020, 1573543. [Google Scholar] [CrossRef] [PubMed]
- Khor, C.C.; Vannberg, F.O.; Chapman, S.J.; Guo, H.; Wong, S.H.; Walley, A.J.; Vukcevic, D.; Rautanen, A.; Mills, T.C.; Chang, K.-C. Cish and susceptibility to infectious diseases. N. Engl. J. Med. 2010, 362, 2092–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, M.G.; Jacob, A.; O’Donnell, L.C.; Agler, A.; Druhan, L.J.; Coggeshall, K.M.; Avalos, B.R. Loss of ship and cis recruitment to the granulocyte colony-stimulating factor receptor contribute to hyperproliferative responses in severe congenital neutropenia/acute myelogenous leukemia. J. Immunol. 2004, 173, 5036–5045. [Google Scholar] [CrossRef]
- Joehanes, R.; Just, A.C.; Marioni, R.E.; Pilling, L.C.; Reynolds, L.M.; Mandaviya, P.R.; Guan, W.; Xu, T.; Elks, C.E.; Aslibekyan, S. Epigenetic signatures of cigarette smoking. Circ. Cardiovasc. Genet. 2016, 9, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.A.; King, B.; Lieberman, N.A.; Bobbili, P.J.; Rudloff, M.; Berndsen, C.E.; Wright, N.T.; Hecker, P.A.; Kontrogianni-Konstantopoulos, A. Novel obscurins mediate cardiomyocyte adhesion and size via the pi3k/akt/mtor signaling pathway. J. Mol. Cell. Cardiol. 2017, 111, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Mulder, R.H.; Neumann, A.; Cecil, C.A.; Walton, E.; Houtepen, L.C.; Simpkin, A.J.; Rijlaarsdam, J.; Heijmans, B.T.; Gaunt, T.R.; Felix, J.F. Epigenome-wide change and variation in DNA methylation in childhood: Trajectories from birth to late adolescence. Hum. Mol. Genet. 2021, 30, 119–134. [Google Scholar] [CrossRef]
- Liu, C.; Marioni, R.E.; Hedman, Å.K.; Pfeiffer, L.; Tsai, P.-C.; Reynolds, L.M.; Just, A.C.; Duan, Q.; Boer, C.G.; Tanaka, T. A DNA methylation biomarker of alcohol consumption. Mol. Psychiatry 2018, 23, 422–433. [Google Scholar] [CrossRef]
- Joubert, B.R.; Felix, J.F.; Yousefi, P.; Bakulski, K.M.; Just, A.C.; Breton, C.; Reese, S.E.; Markunas, C.A.; Richmond, R.C.; Xu, C.-J. DNA methylation in newborns and maternal smoking in pregnancy: Genome-wide consortium meta-analysis. Am. J. Hum. Genet. 2016, 98, 680–696. [Google Scholar] [CrossRef] [Green Version]
- Gracilla, D.E.; Korla, P.K.; Lai, M.T.; Chiang, A.J.; Liou, W.S.; Sheu, J.J.C. Overexpression of wild type or a q311e mutant mb21d2 promotes a pro-oncogenic phenotype in hnscc. Mol. Oncol. 2020, 14, 3065–3082. [Google Scholar] [CrossRef]
- Xu, C.-Z.; Shi, R.-J.; Chen, D.; Sun, Y.-Y.; Wu, Q.-W.; Wang, T.; Wang, P.-H. Potential biomarkers for paclitaxel sensitivity in hypopharynx cancer cell. Int. J. Clin. Exp. Pathol. 2013, 6, 2745. [Google Scholar] [PubMed]
- Pandi, G.; Nakka, V.P.; Dharap, A.; Roopra, A.; Vemuganti, R. Microrna mir-29c down-regulation leading to de-repression of its target DNA methyltransferase 3a promotes ischemic brain damage. PLoS ONE 2013, 8, e58039. [Google Scholar] [CrossRef] [PubMed]
- Bonder, M.J.; Kasela, S.; Kals, M.; Tamm, R.; Lokk, K.; Barragan, I.; Buurman, W.A.; Deelen, P.; Greve, J.-W.; Ivanov, M. Genetic and epigenetic regulation of gene expression in fetal and adult human livers. BMC Genom. 2014, 15, 860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, S.A.; Goodman, S.J.; MacIsaac, J.L.; Obradović, J.; Barr, R.G.; Boyce, W.T.; Kobor, M.S. Integration of DNA methylation patterns and genetic variation in human pediatric tissues help inform ewas design and interpretation. Epigenet. Chromatin 2019, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Dugué, P.A.; Wilson, R.; Lehne, B.; Jayasekara, H.; Wang, X.; Jung, C.H.; Joo, J.E.; Makalic, E.; Schmidt, D.F.; Baglietto, L. Alcohol consumption is associated with widespread changes in blood DNA methylation: Analysis of cross-sectional and longitudinal data. Addict. Biol. 2021, 26, e12855. [Google Scholar] [CrossRef]
- Gross, A.M.; Jaeger, P.A.; Kreisberg, J.F.; Licon, K.; Jepsen, K.L.; Khosroheidari, M.; Morsey, B.M.; Swindells, S.; Shen, H.; Ng, C.T. Methylome-wide analysis of chronic hiv infection reveals five-year increase in biological age and epigenetic targeting of hla. Mol. Cell 2016, 62, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Elias, D.; Vever, H.; Laenkholm, A.; Gjerstorff, M.; Yde, C.; Lykkesfeldt, A.; Ditzel, H. Gene expression profiling identifies fyn as an important molecule in tamoxifen resistance and a predictor of early recurrence in patients treated with endocrine therapy. Oncogene 2015, 34, 1919–1927. [Google Scholar] [CrossRef]
- Xie, Y.-G.; Yu, Y.; Hou, L.-K.; Wang, X.; Zhang, B.; Cao, X.-C. Fyn promotes breast cancer progression through epithelial-mesenchymal transition. Oncol. Rep. 2016, 36, 1000–1006. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Xu, L.; Chen, L.; Wang, Y.; Jiang, H.; Wang, Y.; Yan, Y.; Luo, S.; Zhai, Z. Fyn is required for arhgef16 to promote proliferation and migration in colon cancer cells. Cell Death Dis. 2020, 11, 652. [Google Scholar] [CrossRef]
- Nones, K.; Waddell, N.; Song, S.; Patch, A.M.; Miller, D.; Johns, A.; Wu, J.; Kassahn, K.S.; Wood, D.; Bailey, P. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of slit-robo, itga2 and met signaling. Int. J. Cancer 2014, 135, 1110–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ortiz, E.; Iglesias, E.; Soriano, A.; Buján-Rivas, S.; Español-Rego, M.; Castellanos-Moreira, R.; Tomé, A.; Yagüe, J.; Antón, J.; Hernández-Rodríguez, J. Disease phenotype and outcome depending on the age at disease onset in patients carrying the r92q low-penetrance variant in tnfrsf1a gene. Front. Immunol. 2017, 8, 299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Pan, Y.-B.; Ma, Y.-B.; Chu, S.-H. Integrated transcriptome analyses and experimental verifications of mesenchymal-associated tnfrsf1a as a diagnostic and prognostic biomarker in gliomas. Front. Oncol. 2020, 10, 250. [Google Scholar] [CrossRef] [PubMed]
- Brzozowska, A.; Powrózek, T.; Homa-Mlak, I.; Mlak, R.; Ciesielka, M.; Gołębiowski, P.; Małecka-Massalska, T. Polymorphism of promoter region of tnfrsf1a gene (−610 t > g) as a novel predictive factor for radiotherapy induced oral mucositis in hnc patients. Pathol. Oncol. Res. 2018, 24, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Eze, I.C.; Jeong, A.; Schaffner, E.; Rezwan, F.I.; Ghantous, A.; Foraster, M.; Vienneau, D.; Kronenberg, F.; Herceg, Z.; Vineis, P. Genome-wide DNA methylation in peripheral blood and long-term exposure to source-specific transportation noise and air pollution: The sapaldia study. Environ. Health Perspect. 2020, 128, 067003. [Google Scholar] [CrossRef]
- Templin, T.; Paul, S.; Amundson, S.A.; Young, E.F.; Barker, C.A.; Wolden, S.L.; Smilenov, L.B. Radiation-induced micro-rna expression changes in peripheral blood cells of radiotherapy patients. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 549–557. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Cha, H.J.; Lee, E.-M.; Jung, J.H.; Lee, S.-J.; Park, I.-C.; Jin, Y.-W.; An, S. Micrornas are significantly influenced by p53 and radiation in hct116 human colon carcinoma cells. Int. J. Oncol. 2009, 34, 1645–1652. [Google Scholar]
- Yang, J.; Gong, X.; Ouyang, L.; He, W.; Xiao, R.; Tan, L. Prex2 promotes the proliferation, invasion and migration of pancreatic cancer cells by modulating the pi3k signaling pathway. Oncol. Lett. 2016, 12, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Casado-Medrano, V.; Baker, M.J.; Lopez-Haber, C.; Cooke, M.; Wang, S.; Caloca, M.J.; Kazanietz, M.G. The role of rac in tumor susceptibility and disease progression: From biochemistry to the clinic. Biochem. Soc. Trans. 2018, 46, 1003–1012. [Google Scholar] [CrossRef]
- Liu, Y.; Aryee, M.J.; Padyukov, L.; Fallin, M.D.; Hesselberg, E.; Runarsson, A.; Reinius, L.; Acevedo, N.; Taub, M.; Ronninger, M. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol. 2013, 31, 142. [Google Scholar] [CrossRef]
- Gao, G.; Shi, X.; Yao, Z.; Shen, J.; Shen, L. Identification of lymph node metastasis-related micrornas in breast cancer using bioinformatics analysis. Medicine 2020, 99, e22105. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.-J.; Yang, Y.-L.; Wen, Z.-P.; Chen, P.; Chen, X.-P.; Gong, Z.-C. Identification of gene expression and DNA methylation of serpina5 and timp1 as novel prognostic markers in lower-grade gliomas. PeerJ 2020, 8, e9262. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Positive Sample | Negative Sample | Total |
|---|---|---|---|
| Abdominal RT | 412 | 1640 | 2052 |
| Brain RT | 629 | 1423 | 2052 |
| Chest RT | 577 | 1475 | 2052 |
| Pelvic RT | 352 | 1700 | 2052 |
| Dataset | Classifiers | Number of Features | Accuracy | Sensitivity | Specificity | MCC |
|---|---|---|---|---|---|---|
| Abdominal RT | DF | 744 | 0.966 | 0.910 | 0.980 | 0.895 |
| kNN | 10 | 0.846 | 0.971 | 0.814 | 0.662 | |
| RF | 753 | 0.903 | 0.913 | 0.900 | 0.739 | |
| DT | 761 | 0.791 | 0.825 | 0.783 | 0.515 | |
| Brain RT | DF | 128 | 0.869 | 0.736 | 0.928 | 0.686 |
| kNN | 8 | 0.749 | 0.863 | 0.699 | 0.519 | |
| RF | 115 | 0.811 | 0.765 | 0.832 | 0.577 | |
| DT | 150 | 0.690 | 0.693 | 0.688 | 0.355 | |
| Chest RT | DF | 691 | 0.925 | 0.828 | 0.963 | 0.812 |
| kNN | 12 | 0.804 | 0.945 | 0.749 | 0.627 | |
| RF | 234 | 0.851 | 0.823 | 0.862 | 0.654 | |
| DT | 489 | 0.762 | 0.747 | 0.768 | 0.478 | |
| Pelvic RT | DF | 155 | 0.976 | 0.923 | 0.986 | 0.914 |
| kNN | 9 | 0.841 | 0.977 | 0.813 | 0.637 | |
| RF | 31 | 0.896 | 0.906 | 0.894 | 0.702 | |
| DT | 77 | 0.798 | 0.795 | 0.798 | 0.487 |
| Dataset | Number of Rules | Number of Rules for Positive Class | Number of Rules for Negative Class |
|---|---|---|---|
| Abdominal RT | 151 | 87 | 64 |
| Brain RT | 239 | 132 | 107 |
| Chest RT | 166 | 93 | 73 |
| Pelvic RT | 183 | 99 | 84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Guo, W.; Ding, S.; Feng, K.; Lu, L.; Huang, T.; Cai, Y. Detecting Blood Methylation Signatures in Response to Childhood Cancer Radiotherapy via Machine Learning Methods. Biology 2022, 11, 607. https://doi.org/10.3390/biology11040607
Li Z, Guo W, Ding S, Feng K, Lu L, Huang T, Cai Y. Detecting Blood Methylation Signatures in Response to Childhood Cancer Radiotherapy via Machine Learning Methods. Biology. 2022; 11(4):607. https://doi.org/10.3390/biology11040607
Chicago/Turabian StyleLi, Zhandong, Wei Guo, Shijian Ding, Kaiyan Feng, Lin Lu, Tao Huang, and Yudong Cai. 2022. "Detecting Blood Methylation Signatures in Response to Childhood Cancer Radiotherapy via Machine Learning Methods" Biology 11, no. 4: 607. https://doi.org/10.3390/biology11040607
APA StyleLi, Z., Guo, W., Ding, S., Feng, K., Lu, L., Huang, T., & Cai, Y. (2022). Detecting Blood Methylation Signatures in Response to Childhood Cancer Radiotherapy via Machine Learning Methods. Biology, 11(4), 607. https://doi.org/10.3390/biology11040607