
Roles of Natriuretic Peptides and the Significance of Neprilysin in Cardiovascular Diseases


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Physiological Roles of Natriuretic Peptides: Lessons from Genetically Modified Mice
2.1. Effect of Natriuretic Peptides on Blood Pressure
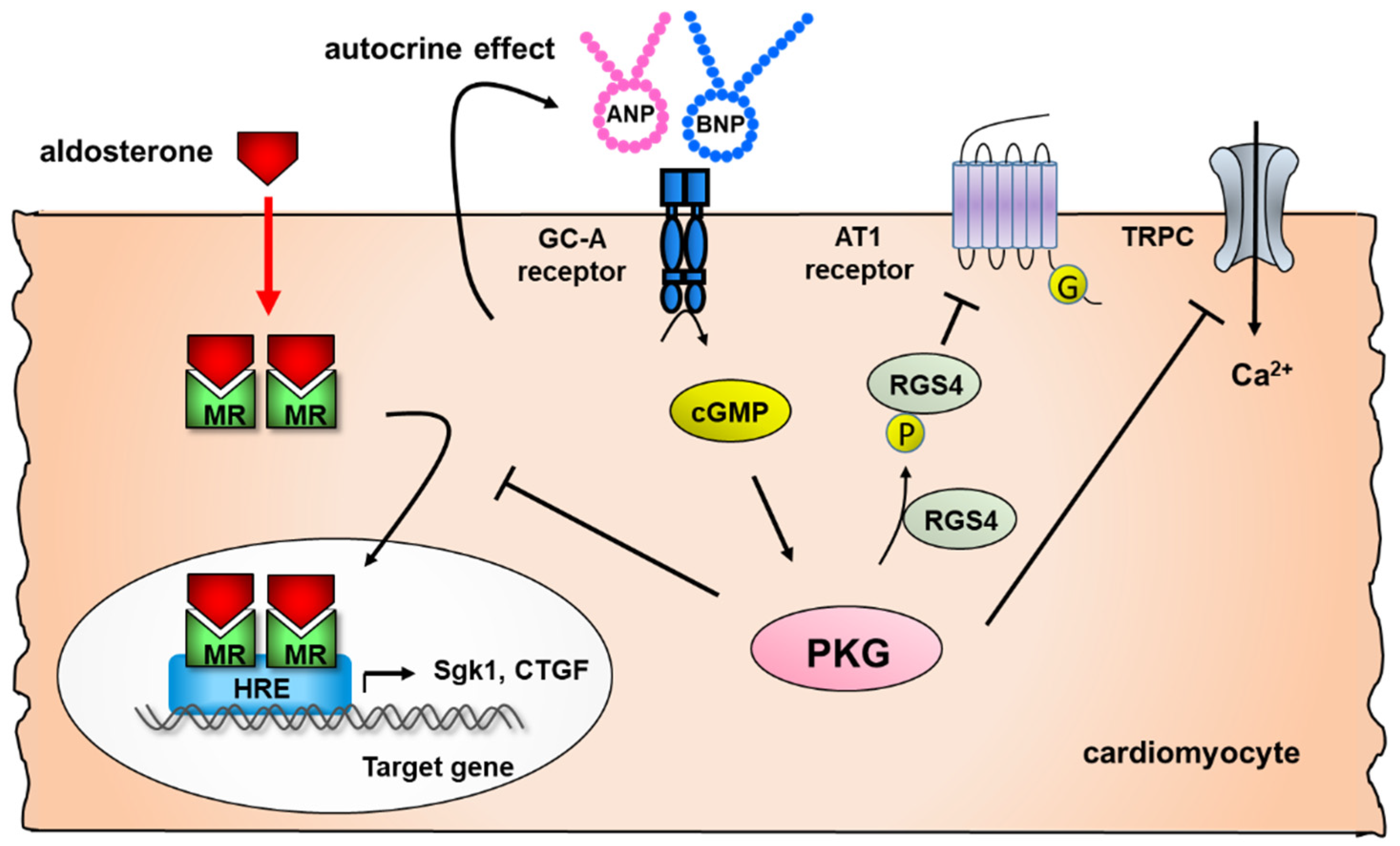
2.2. Effect of Natriuretic Peptides on Cardiac Remodeling
2.3. Effect of Natriuretic Peptides on Acute Myocardial Infarction (AMI)
3. Neprilysin Inhibitor and Revival of Natriuretic Peptides
3.1. Role of Neprilysin and Significance of Its Inhibition
3.2. Natriuretic Peptides as a Substrate of Neprilysin
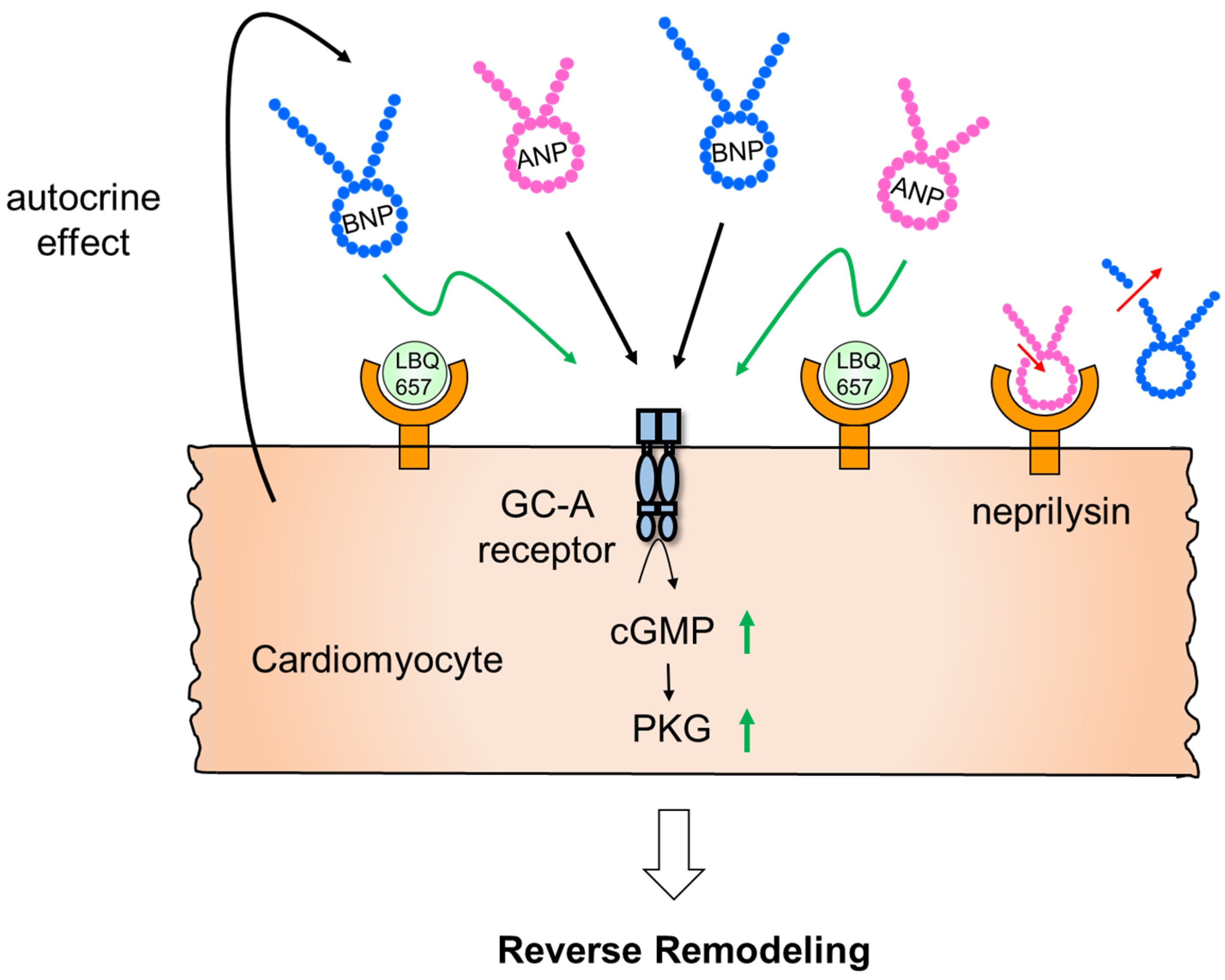
3.3. Effect of Neprilysin on Cardiac Remodeling
3.4. Effect of Neprilysin Inhibitor on AMI
3.5. Effect of Neprilysin Inhibitor on Hypertension
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kisch, B. Electron microscopy of the atrium of the heart. I. Guinea Pig. Exp. Med. Surg. 1956, 14, 99–112. [Google Scholar] [PubMed]
- de Bold, A.J.; Borenstein, H.B.; Veress, A.T.; Sonnenberg, H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 1981, 28, 89–94. [Google Scholar] [CrossRef]
- Flynn, T.G.; de Bold, M.L.; de Bold, A.J. The amino acid sequence of an atrial peptide with potent diuretic and natriuretic prop-erties. Biochem. Biophys. Res. Commun. 1983, 117, 859–865. [Google Scholar] [CrossRef]
- Kangawa, K.; Matsuo, H. Purification and complete amino acid sequence of alpha-human atrial natriuretic polypeptide (al-pha-hANP). Biochem. Biophys. Res. Commun. 1984, 118, 131–139. [Google Scholar] [CrossRef]
- Sudoh, T.; Kangawa, K.; Minamino, N.; Matsuo, H. A new natriuretic peptide in porcine brain. Nature 1988, 332, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Sudoh, T.; Minamino, N.; Kangawa, K.; Matsuo, H. C-type natriuretic peptide (CNP): A new member of natriuretic peptide fam-ily identified in porcine brain. Biochem. Biophys. Res. Commun 1990, 168, 863–870. [Google Scholar] [CrossRef]
- Sugawara, A.; Nakao, K.; Morii, N.; Sakamoto, M.; Suda, M.; Shimokura, M.; Kiso, Y.; Kihara, M.; Yamori, Y.; Nishimura, K.; et al. Al-pha-human atrial natriuretic polypeptide is released from the heart and circulates in the body. Biochem. Biophys. Res. Commun. 1985, 129, 439–446. [Google Scholar] [CrossRef]
- Ogawa, Y.; Nakao, K.; Mukoyama, M.; Hosoda, K.; Shirakami, G.; Arai, H.; Saito, Y.; Suga, S.; Jougasaki, M.; Imura, H. Natriuretic pep-tides as cardiac hormones in normotensive and spontaneously hypertensive rats. The ventricle is a major site of synthesis and secretion of brain natriuretic peptide. Circ. Res. 1991, 69, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Nakao, K.; Ogawa, Y.; Suga, S.-I.; Imura, H. Molecular biology and biochemistry of the natriuretic peptide system. J. Hypertens. 1996, 10, 74–82. [Google Scholar] [CrossRef]
- Mukoyama, M.; Nakao, K.; Hosoda, K.; Suga, S.; Saito, Y.; Ogawa, Y.; Shirakami, G.; Jougasaki, M.; Obata, K.; Yasue, H.; et al. Brain natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J. Clin. Investig. 1991, 87, 1402–1412. [Google Scholar] [CrossRef]
- Yasue, H.; Yoshimura, M.; Sumida, H.; Kikuta, K.; Kugiyama, K.; Jougasaki, M.; Ogawa, H.; Okumura, K.; Mukoyama, M.; Nakao, K. Localization and mechanism of secretion of B-type natriuretic peptide in comparison with those of A-type natriuretic pep-tide in normal subjects and patients with heart failure. Circulation 1994, 90, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Nakao, K.; Nishimura, K.; Sugawara, A.; Okumura, K.; Obata, K.; Sonoda, R.; Ban, T.; Yasue, H.; Imura, H. Clinical application of atrial natriuretic polypeptide in patients with congestive heart failure: Beneficial effects on left ventricular function. Circulation 1987, 76, 115–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M. Molecular Physiology of Membrane Guanylyl Cyclase Receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef] [PubMed]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac natriuretic peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef]
- Koller, K.J.; Lowe, D.G.; Bennett, G.L.; Minamino, N.; Kangawa, K.; Matsuo, H.; Goeddel, D.V. Selective activation of the B natriu-retic peptide receptor by C-type natriuretic peptide (CNP). Science 1991, 252, 120–123. [Google Scholar] [CrossRef]
- Campbell, D.J. Long-term neprilysin inhibition—implications for ARNIs. Nat. Rev. Cardiol. 2016, 14, 171–186. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; Barallat, J.; Richards, A.M. A Test in context: Neprilysin: Function, inhibition, and biomarker. J. Am. Coll. Cardiol. 2016, 68, 639–653. [Google Scholar] [CrossRef]
- Piedimonte, G.; Nadel, J.A.; Long, C.S.; Hoffman, J.I. Neutral endopeptidase in the heart. Neutral endopeptidase inhibition prevents isoproterenol-induced myocardial hypoperfusion in rats by reducing bradykinin degradation. Circ. Res. 1994, 75, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Noe, A.; Chandra, P.; Al-Fayoumi, S.; Ligueros-Saylan, M.; Sarangapani, R.; Maahs, S.; Ksander, G.; Rigel, D.F.; Jeng, A.Y.; et al. Pharmacokinetics and pharmacodynamics of LCZ696, a novel dual-acting angiotensin recep-tor-neprilysin inhibitor (ARNi). J. Clin. Pharmacol. 2010, 50, 401–414. [Google Scholar] [CrossRef]
- Packer, M.; McMurray, J.J.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin Receptor Neprilysin Inhibition Compared With Enalapril on the Risk of Clinical Progression in Surviving Patients With Heart Failure. Circulation 2015, 131, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Myhre, P.L.; Vaduganathan, M.; Claggett, B.; Packer, M.; Desai, A.S.; Rouleau, J.L.; Zile, M.R.; Swedberg, K.; Lefkowitz, M.; Shi, V.; et al. B-Type natriuretic peptide during treatment with sacubitril/valsartan: The PARADIGM-HFtrial. J. Am. Coll. Cardiol. 2019, 73, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Mcmurray, J.J.V.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure. PARADIGM-HF Investigators and Committees. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, D.G.; Aizenberg, D.; Gorbunov, V.; Hafeez, K.; Chen, C.-W.; Zhang, J. Efficacy and safety of sacubitril/valsartan in patients with essential hypertension uncontrolled by olmesartan: A randomized, double-blind, 8-week study. J. Clin. Hypertens. 2018, 20, 150–158. [Google Scholar] [CrossRef] [Green Version]
- John, S.W.; Krege, J.H.; Oliver, P.M.; Hagaman, J.R.; Hodgin, J.B.; Pang, S.C.; Flynn, T.G.; Smithies, O. Genetic decreases in atrial natriu-retic peptide and salt-sensitive hypertension. Science 1995, 267, 679–681. [Google Scholar] [CrossRef]
- Lopez, M.J.; Wong, S.K.-F.; Kishimoto, I.; Dubois, S.; Mach, V.; Friesen, J.; Garbers, D.L.; Beuve, A. Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature 1995, 378, 65–68. [Google Scholar] [CrossRef]
- Holtwick, R.; Gotthardt, M.; Skryabin, B.; Steinmetz, M.; Potthast, R.; Zetsche, B.; Hammer, R.E.; Herz, J.; Kuhn, M. Smooth musclese-lective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc. Natl. Acad. Sci. USA 2002, 99, 7142–7147. [Google Scholar] [CrossRef] [Green Version]
- Sabrane, K.; Kruse, M.N.; Fabritz, L.; Zetsche, B.; Mitko, D.; Skryabin, B.V.; Zwiener, M.; Baba, H.A.; Yanagisawa, M.; Kuhn, M. Vascular endothelium is critically involved in the hypotensive and hypovolemic actions of atrial natriuretic peptide. J. Clin. Investig. 2005, 115, 1666–1674. [Google Scholar] [CrossRef] [Green Version]
- Skryabin, B.V.; Holtwick, R.; Fabritz, L.; Kruse, M.N.; Veltrup, I.; Stypmann, J.; Kirchhof, P.; Sabrane, K.; Bubikat, A.; Voss, M.; et al. Hypervolemic hypertension in mice with systemic inactivation of the (floxed) guanylyl cyclase-A gene by alphaM-HCCre-mediated recombination. Genesis 2004, 39, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gaßner, B.; Börner, S.; Nikolaev, V.O.; Schlegel, N.; Waschke, J.; Steinbronn, N.; Strasser, R.; Kuhn, M.; Gassner, B. Atrial natriuretic peptide enhances microvascular albumin permeability by the caveolae-mediated transcellular pathway. Cardiovasc. Res. 2011, 93, 141–151. [Google Scholar] [CrossRef]
- Nakao, K.; Kuwahara, K.; Nishikimi, T.; Nakagawa, Y.; Kinoshita, H.; Minami, T.; Kuwabara, Y.; Yamada, C.; Yamada, Y.; Tokudome, T.; et al. Endotheli-um-Derived C-Type Natriuretic Peptide Contributes to Blood Pressure Regulation by Maintaining Endothelial Integrity. Hypertension 2017, 69, 286–296. [Google Scholar] [CrossRef]
- Piranec, K.; Chen, W.; Werner, F.; Nikolaev, V.O.; Naruke, T.; Koch, F.; Werner, A.; Eder-Negrin, P.; Diéguez-Hurtado, R.; Adams, R.H.; et al. Endothelial C-Type Natriuretic Peptide Acts on Pericytes to Regulate Microcirculatory Flow and Blood Pressure. Circulation 2018, 138, 494–508. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakao, K.; Arai, H.; Nishimura, K.; Okumura, K.; Obata, K.; Takemura, G.; Fujiwara, H.; Sugawara, A.; Yamada, T. Augmented expression of atrial natriuretic polypeptide gene in ventricle of human failing heart. J. Clin. Investig. 1989, 83, 298–305. [Google Scholar] [CrossRef]
- Li, Y.; Kishimoto, I.; Saito, Y.; Harada, M.; Kuwahara, K.; Izumi, T.; Takahashi, N.; Kawakami, R.; Tanimoto, K.; Nakagawa, Y.; et al. Guanylyl Cyclase-A Inhibits Angiotensin II Type 1A Receptor-Mediated Cardiac Remodeling, an Endogenous Protective Mechanism in the Heart. Circulation 2002, 106, 1722–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokudome, T.; Kishimoto, I.; Horio, T.; Arai, Y.; Schwenke, D.O.; Hino, J.; Okano, I.; Kawano, Y.; Kohno, M.; Miyazato, M.; et al. Regulator of G-Protein Signaling Subtype 4 Mediates Antihypertrophic Effect of Locally Secreted Natriuretic Peptides in the Heart. Circulation 2008, 117, 2329–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, H.; Kuwahara, K.; Nishida, M.; Jian, Z.; Rong, X.; Kiyonaka, S.; Kuwabara, Y.; Kurose, H.; Inoue, R.; Mori, Y.; et al. Inhibition of TRPC6 Channel Activity Contributes to the Antihypertrophic Effects of Natriuretic Peptides-Guanylyl Cyclase-A Signaling in the Heart. Circ. Res. 2010, 106, 1849–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtwick, R.; van Eickels, M.; Skryabin, B.V.; Baba, H.A.; Bubikat, A.; Begrow, F.; Schneider, M.D.; Garbers, D.L.; Kuhn, M. Pres-sure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J. Clin. Investig. 2003, 111, 1399–1407. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Oberwinkler, H.; Nikolaev, V.O.; Gaßner, B.; Umbenhauer, S.; Wagner, H.; Saito, Y.; Baba, H.A.; Frantz, S.; Kuhn, M. Atrial Natriuretic Peptide Locally Counteracts the Deleterious Effects of Cardiomyocyte Mineralocorticoid Receptor Activation. Circ. Heart Fail. 2014, 7, 814–821. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Somekawa, S.; Onoue, K.; Kumazawa, T.; Ueda, T.; Seno, A.; Nakada, Y.; Nakano, T.; Matsui, M.; Soeda, T.; et al. Salt accelerates aldosterone-induced cardiac remodeling in the absence of guan-ylyl cyclase-A signaling. Life Sci. 2016, 165, 9–15. [Google Scholar] [CrossRef]
- Michel, K.; Herwig, M.; Werner, F.; Spes, K.; Abeßer, M.; Schuh, K.; Dabral, S.; Mügge, A.; Baba, H.A.; Skryabin, B.V.; et al. C-type natriuretic peptide moderates titin-based cardiomyocyte stiffness. JCI Insight 2020, 5, e139910. [Google Scholar] [CrossRef]
- Nakanishi, M.; Saito, Y.; Kishimoto, I.; Harada, M.; Kuwahara, K.; Takahashi, N.; Kawakami, R.; Nakagawa, Y.; Tanimoto, K.; Yasuno, S.; et al. Role of Natriuretic Peptide Receptor Guanylyl Cyclase-A in Myocardial Infarction Evaluated Using Genetically Engineered Mice. Hypertension 2005, 46, 441–447. [Google Scholar] [CrossRef] [Green Version]
- Izumi, T.; Saito, Y.; Kishimoto, I.; Harada, M.; Kuwahara, K.; Hamanaka, I.; Takahashi, N.; Kawakami, R.; Li, Y.; Takemura, G.; et al. Blockade of the natriuretic peptide receptor guanylyl cyclase-A inhibits NF-kappaB activation and alleviates myocardial ischemia/reperfusion injury. J. Clin. Investig. 2001, 108, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, R.; Saito, Y.; Kishimoto, I.; Harada, M.; Kuwahara, K.; Takahashi, N.; Nakagawa, Y.; Nakanishi, M.; Tanimoto, K.; Usami, S.; et al. Overexpression of Brain Natriuretic Peptide Facilitates Neutrophil Infiltration and Cardiac Matrix Metalloproteinase-9 Expression After Acute Myocardial Infarction. Circulation 2004, 110, 3306–3312. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Spitzl, A.; Mathes, D.; Nikolaev, V.O.; Werner, F.; Weirather, J.; Špiranec, K.; Röck, K.; Fischer, J.W.; Kämmerer, U.; et al. Endothelial Actions of ANP Enhance Myocardial Inflammatory Infiltration in the Early Phase After Acute Infarction. Circ. Res. 2016, 119, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndrepepa, G.; Braun, S.; Mehilli, J.; von Beckerath, N.; Nekolla, S.; Vogt, W.; Schwaiger, M.; Schömig, A.; Kastrati, A. N-terminal pro-brain natriuretic peptide on admission in patients with acute myocardial infarction and correlation with scintigraphic infarct size, efficacy of reperfusion, and prognosis. Am. J. Cardiol. 2006, 97, 1151–1156. [Google Scholar] [CrossRef]
- Arakawa, N.; Nakamura, M.; Endo, H.; Sugawara, S.; Suzuki, T.; Hiramori, K. Brain natriuretic peptide and cardiac rupture after acute myocardial infarction. Intern. Med. 2001, 40, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Malito, E.; Hulse, R.E.; Tang, W.J. Amyloid beta-degrading cryptidases: Insulin degrading enzyme, presequence peptidase, and neprilysin. Cell Mol. Life Sci. 2008, 65, 2574–2585. [Google Scholar] [CrossRef] [Green Version]
- George, S.G.; Kenny, A.J. Studies on the enzymology of purified preparations of brush border from rabbit kidney. Biochem. J. 1973, 134, 43–57. [Google Scholar] [CrossRef] [Green Version]
- Kerr, M.A.; Kenny, A.J. The purification and specificity of a neutral endopeptidase from rabbit kidney brush border. Biochem. J. 1974, 137, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Erdös, E.G.; Skidgel, R.A. Neutral endopeptidase 24.11 (enkephalinase) and related regulators of peptide hormones. FASEB J. 1989, 3, 145–151. [Google Scholar] [CrossRef]
- Lu, B.; Figini, M.; Emanueli, C.; Geppetti, P.; Grady, E.F.; Gerard, N.P.; Ansell, J.; Payan, D.G.; Gerard, C.; Bunnett, N. The control of mi-crovascular permeability and blood pressure by neutral endopeptidase. Nat. Med. 1997, 3, 904–907. [Google Scholar] [CrossRef]
- Packer, M.; Califf, R.M.; Konstam, M.A.; Krum, H.; McMurray, J.J.; Rouleau, J.L.; Swedberg, K. Comparison of omapatrilat and enal-april in patients with chronic heart failure: The Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE). Circulation 2002, 106, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R. Natriuretic peptide metabolism, clearance and degradation. FEBS J. 2011, 278, 1808–1817. [Google Scholar] [CrossRef] [Green Version]
- Pankow, K.; Schwiebs, A.; Becker, M.; Siems, W.E.; Krause, G.; Walther, T. Structural substrate conditions required for neutral en-dopeptidase-mediated natriuretic Peptide degradation. J. Mol. Biol. 2009, 393, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Yamaguchi, Y.; Horii, M.; Kawata, H.; Yamamoto, H.; Uemura, S.; Saito, Y. ANP is cleared much faster than BNP in patients with congestive heart failure. Eur. J. Clin. Pharmacol. 2007, 63, 699–702. [Google Scholar] [CrossRef]
- Hunt, P.J.; Richards, A.M.; Espiner, E.A.; Nicholls, M.G.; Yandle, T.G. Bioactivity and metabolism of C-type natriuretic peptide in normal man. J. Clin. Endocrinol. Metab. 1994, 78, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.J.; Espiner, E.A.; Richards, A.M.; Yandle, T.; Frampton, C. Renal, endocrine, and hemodynamic effects of human brain natriuretic peptide in normal man. J. Clin. Endocrinol. Metab. 1993, 76, 91–96. [Google Scholar] [CrossRef]
- Murphy, S.P.; Prescott, M.F.; Camacho, A.; Iyer, S.R.; Maisel, A.S.; Felker, G.M.; Butler, J.; Piña, I.L.; Ibrahim, N.E.; Abbas, C.; et al. Atrial Natriuretic Peptide and Treatment With Sacubitril/Valsartan in Heart Failure With Reduced Ejection Fraction. J. Am. Coll. Cardiol. HF 2021, 9, 127–136. [Google Scholar] [CrossRef]
- Nishikimi, T.; Okamoto, H.; Nakamura, M.; Ogawa, N.; Horii, K.; Nagata, K.; Nakagawa, Y.; Kinoshita, H.; Yamada, C.; Nakao, K.; et al. Direct immunochemiluminescent as-say for proBNP and total BNP in human plasma proBNP and total BNP levels in normal and heart failure. PLoS ONE 2013, 8, e53233. [Google Scholar] [CrossRef]
- Januzzi, J.L., Jr.; Prescott, M.F.; Butler, J.; Felker, G.M.; Maisel, A.S.; McCague, K.; Camacho, A.; Piña, I.L.; Rocha, R.A.; Shah, A.M.; et al. Association of Change in N-Terminal Pro-B-Type Natriuretic Peptide Fol-lowing Initiation of Sacubitril-Valsartan Treatment With Cardiac Structure and Function in Patients With Heart Failure With Reduced Ejection Fraction. JAMA 2019, 322, 1085–1095. [Google Scholar] [CrossRef]
- Martín-Garcia, A.; López-Fernández, T.; Mitroi, C.; Chaparro-Muñoz, M.; Moliner, P.; Martin-Garcia, A.C.; Martinez-Monzonis, A.; Castro, A.; Lopez-Sendon, J.L.; Sanchez, P.L. Effectiveness of sacubitril–valsartan in cancer patients with heart failure. ESC Heart Fail. 2020, 7, 763–767. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Kumazawa, T.; Onoue, K.; Nakada, Y.; Nakano, T.; Ishihara, S.; Minamino, N.; Hosoda, H.; Iwata, N.; Ueda, T.; et al. Local Action of Neprilysin Exacerbates Pressure Over-load Induced Cardiac Remodeling. Hypertension 2021, 77, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.I.; Burrell, M.; Olsson, L.-L.; Fowler, S.B.; Digby, S.; Sandercock, A.; Snijder, A.; Tebbe, J.; Haupts, U.; Grudzinska, J.; et al. Engineering Neprilysin Activity and Specificity to Create a Novel Therapeutic for Alzheimer’s Disease. PLoS ONE 2014, 9, e104001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, A.J.; Bourne, A.; Ingram, J. Hydrolysis of human and pig brain natriuretic peptides, urodilatin, C-type natriuretic pep-tide and some C-receptor ligands by endopeptidase-24. Biochem. J. 1993, 291, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Kitakaze, M.; Asakura, M.; Kim, J.; Shintani, Y.; Asanuma, H.; Hamasaki, T.; Seguchi, O.; Myoishi, M.; Minamino, T.; Ohara, T.; et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): Two randomised trials. Lancet 2007, 370, 1483–1493. [Google Scholar] [CrossRef]
- von Lueder, T.G.; Wang, B.H.; Kompa, A.R.; Huang, L.; Webb, R.; Jordaan, P.; Atar, D.; Krum, H. Angiotensin receptor neprilysin in-hibitor LCZ696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ. Heart Fail. 2015, 8, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Ishii, M.; Kaikita, K.; Sato, K.; Sueta, D.; Fujisue, K.; Arima, Y.; Oimatsu, Y.; Mitsuse, T.; Onoue, Y.; Araki, S.; et al. Cardioprotective Effects of LCZ696 (Sacubi-tril/Valsartan) After Experimental Acute Myocardial Infarction. JACC Basic Transl. Sci. 2017, 2, 655–668. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Claggett, B.; Lewis, E.F.; Granger, C.B.; Køber, L.; Maggioni, A.P.; Mann, D.L.; McMurray, J.J.V.; Rouleau, J.L.; Solomon, S.D.; et al. Investigators and Committees. Angiotensin Receptor-Neprilysin Inhibition in Acute Myocardial In-farction. N. Engl. J. Med. 2021, 385, 1845–1855. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Claggett, B.; Lewis, E.F.; Granger, C.B.; Køber, L.; Maggioni, A.P.; Mann, D.L.; McMurray, J.J.V.; Rouleau, J.L.; Solomon, S.D.; et al. Impact of Sacu-bitril/Valsartan Versus Ramipril on Total Heart Failure Events in the PARADISE-MI Trial. Circulation 2022, 145, 87–89. [Google Scholar] [CrossRef]
- Wang, T.-D.; Tan, R.S.; Lee, H.-Y.; Ihm, S.-H.; Rhee, M.-Y.; Tomlinson, B.; Pal, P.; Yang, F.; Hirschhorn, E.; Prescott, M.F.; et al. Effects of Sacubitril/Valsartan (LCZ696) on Natriuresis, Diuresis, Blood Pressures, and NT-proBNP in Salt-Sensitive Hypertension. Hypertension 2017, 69, 32–41. [Google Scholar] [CrossRef]
- Ruilope, L.M.; Dukat, A.; Böhm, M.; Lacourcière, Y.; Gong, J.; Lefkowitz, M.P. Blood-pressure reduction with LCZ696, a novel du-al-acting inhibitor of the angiotensin II receptor and neprilysin: A randomised, double-blind, placebo-controlled, active comparator study. Lancet 2010, 375, 1255–1266. [Google Scholar] [CrossRef]
- Rakugi, H.; Kario, K.; Yamaguchi, M.; Sasajima, T.; Gotou, H.; Zhang, J. Efficacy of sacubitril/valsartan versus olmesartan in Japa-nese patients with essential hypertension: A randomized, double-blind, multicenter study. Hypertens. Res. 2022, 45, 824–833. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagawa, H.; Saito, Y. Roles of Natriuretic Peptides and the Significance of Neprilysin in Cardiovascular Diseases. Biology 2022, 11, 1017. https://doi.org/10.3390/biology11071017
Nakagawa H, Saito Y. Roles of Natriuretic Peptides and the Significance of Neprilysin in Cardiovascular Diseases. Biology. 2022; 11(7):1017. https://doi.org/10.3390/biology11071017
Chicago/Turabian StyleNakagawa, Hitoshi, and Yoshihiko Saito. 2022. "Roles of Natriuretic Peptides and the Significance of Neprilysin in Cardiovascular Diseases" Biology 11, no. 7: 1017. https://doi.org/10.3390/biology11071017
APA StyleNakagawa, H., & Saito, Y. (2022). Roles of Natriuretic Peptides and the Significance of Neprilysin in Cardiovascular Diseases. Biology, 11(7), 1017. https://doi.org/10.3390/biology11071017