
CNP, the Third Natriuretic Peptide: Its Biology and Significance to the Cardiovascular System



{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
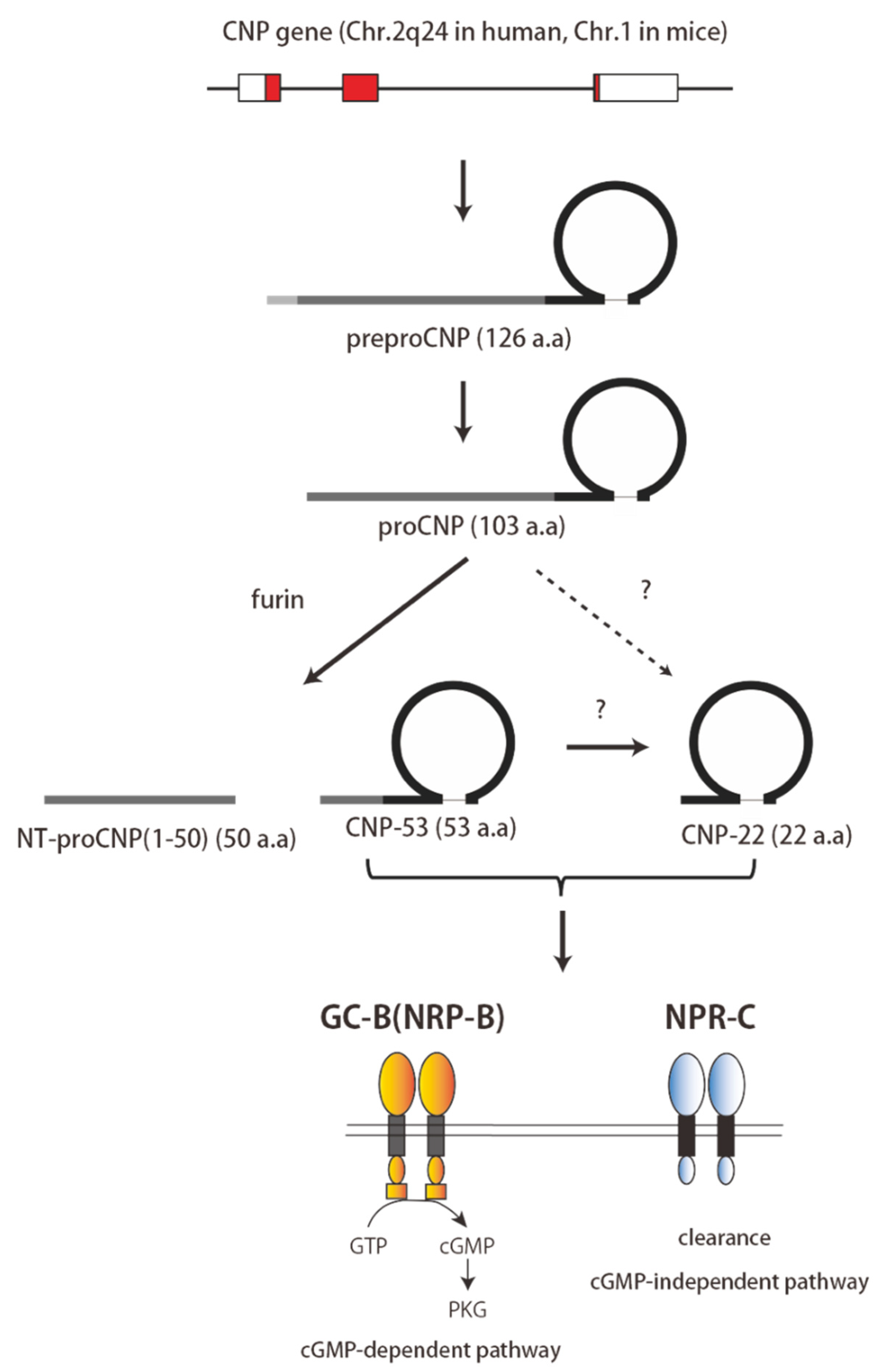
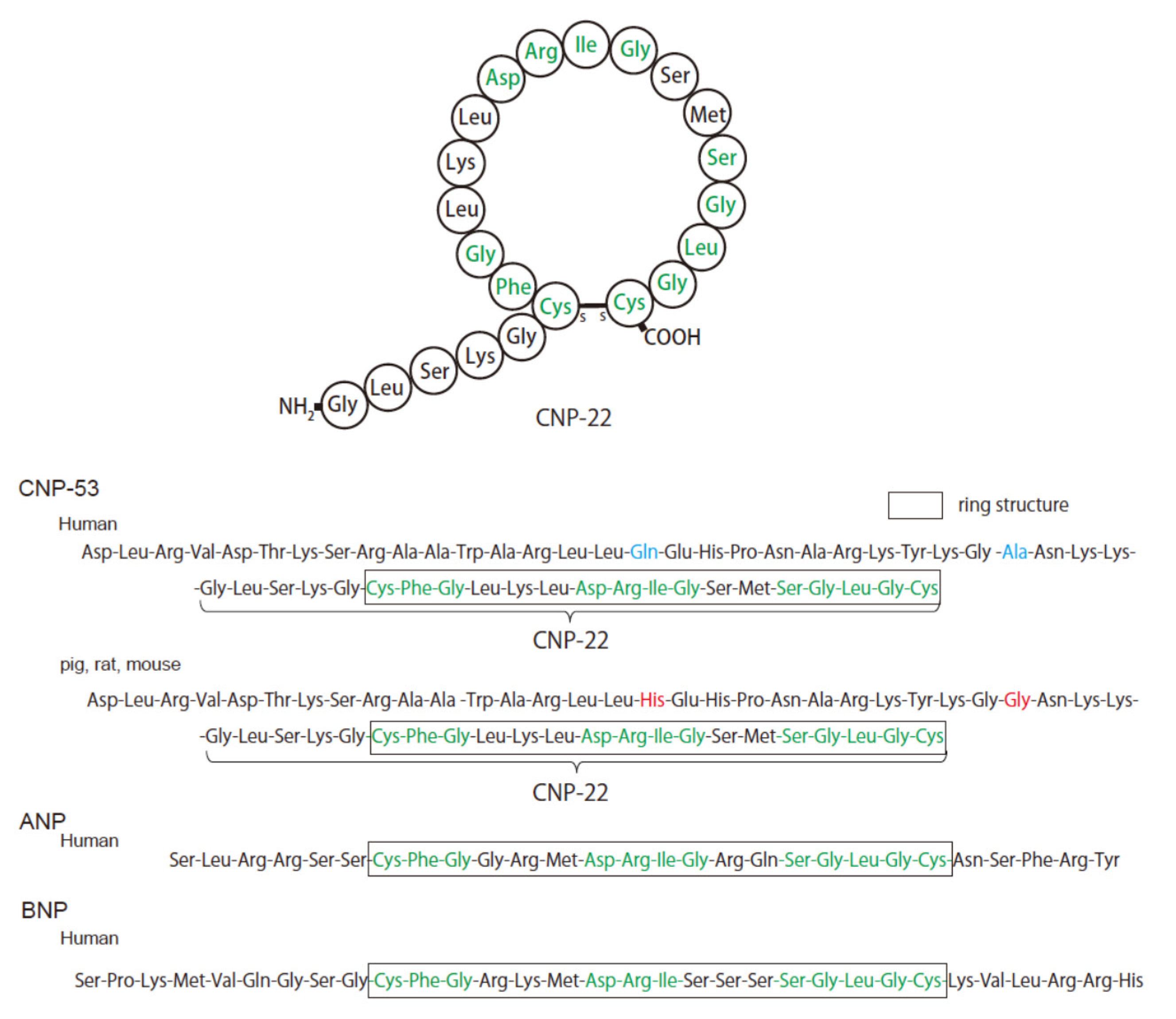
2. Structure and Physiology of CNP
3. Distribution of CNP and Its Receptor in the Heart
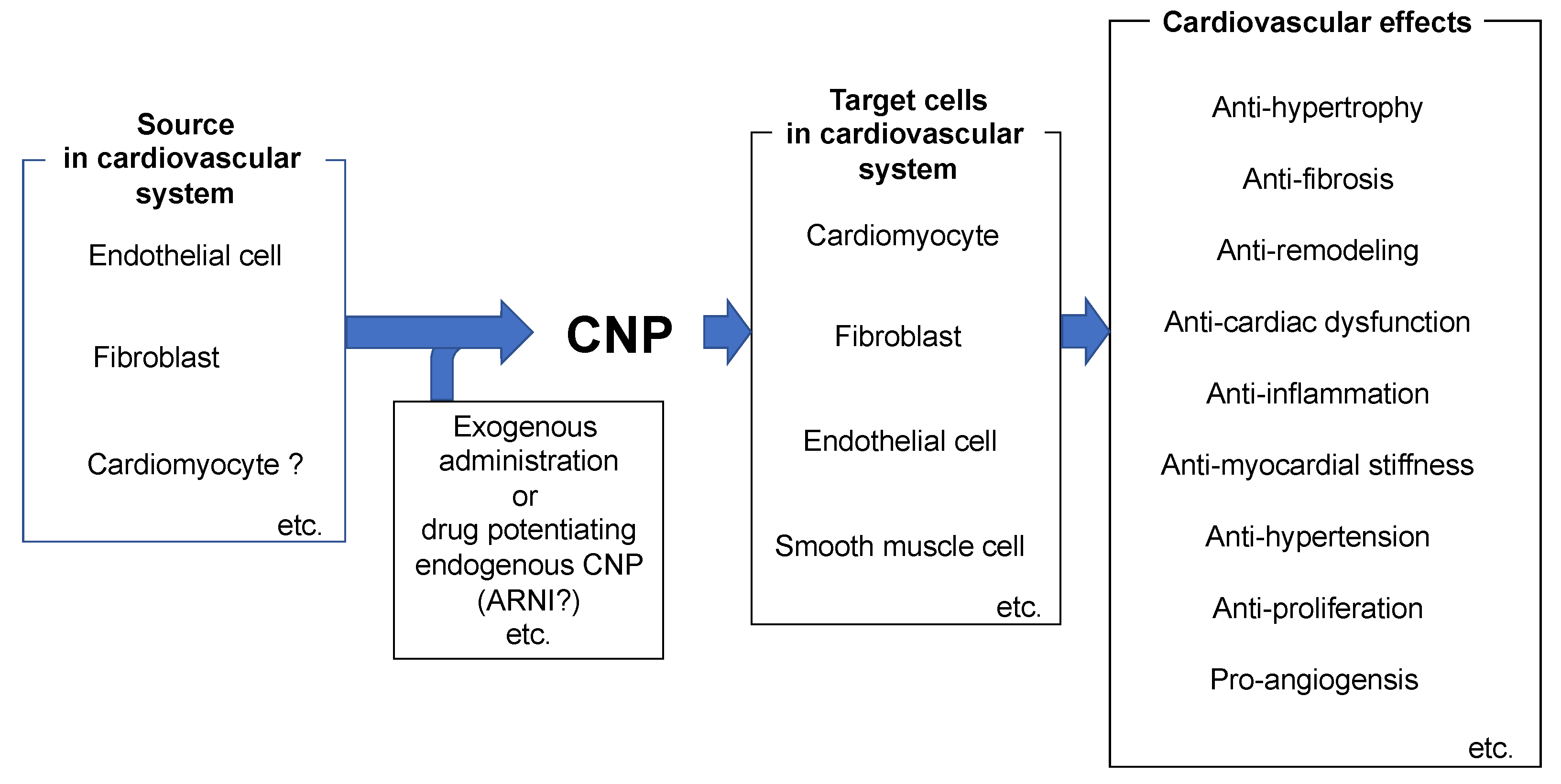
4. Role of CNP in the Pathophysiology of the Heart
5. Role of CNP in the Regulation of Vascular Tone and Blood Pressure
6. Role of CNP in Vascular Remodeling and Angiogenesis
7. Role of CNP in Fibrosis in Cardiovascular Disease
8. Increased Plasma CNP Levels in Heart Failure
9. The Source and Clinical Significance of CNP in Heart Failure
10. Role of CNP in Pathogenesis in Pulmonary Arterial Hypertension
11. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flynn, T.G.; de Bold, M.L.; de Bold, A.J. The amino acid sequence of an atrial peptide with potent diuretic and natriuretic properties. Biochem. Biophys. Res. Commun. 1983, 117, 859–865. [Google Scholar] [CrossRef]
- Kangawa, K.; Matsuo, H. Purification and complete amino acid sequence of alpha-human atrial natriuretic polypeptide (alpha-hANP). Biochem. Biophys. Res. Commun. 1984, 118, 131–139. [Google Scholar] [CrossRef]
- Sudoh, T.; Kangawa, K.; Minamino, N.; Matsuo, H. A new natriuretic peptide in porcine brain. Nature 1988, 332, 78–81. [Google Scholar] [CrossRef]
- Sudoh, T.; Minamino, N.; Kangawa, K.; Matsuo, H. C-type natriuretic peptide (CNP): A new member of natriuretic peptide family identified in porcine brain. Biochem. Biophys. Res. Commun. 1990, 168, 863–870. [Google Scholar] [CrossRef]
- Hata, N.; Seino, Y.; Tsutamoto, T.; Hiramitsu, S.; Kaneko, N.; Yoshikawa, T.; Yokoyama, H.; Tanaka, K.; Mizuno, K.; Nejima, J.; et al. Effects of carperitide on the long-term prognosis of patients with acute decompensated chronic heart failure: The PROTECT multicenter randomized controlled study. Circ. J. 2008, 72, 1787–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawase, Y.; Hata, R.; Tada, T.; Katoh, H.; Kadota, K. Effects of Carperitide on Degree of Pulmonary Congestion in Treatment of Acute Heart Failure. Circ. J. 2018, 82, 2079–2088. [Google Scholar] [CrossRef] [Green Version]
- Nogi, K.; Ueda, T.; Matsue, Y.; Nogi, M.; Ishihara, S.; Nakada, Y.; Kawakami, R.; Kagiyama, N.; Kitai, T.; Oishi, S.; et al. Effect of carperitide on the 1 year prognosis of patients with acute decompensated heart failure. ESC Heart Fail. 2022, 9, 1061–1070. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017, 136, e137–e161. [Google Scholar] [CrossRef]
- Tsutsui, H.; Ide, T.; Ito, H.; Kihara, Y.; Kinugawa, K.; Kinugawa, S.; Makaya, M.; Murohara, T.; Node, K.; Saito, Y.; et al. JCS/JHFS 2021 Guideline Focused Update on Diagnosis and Treatment of Acute and Chronic Heart Failure. J. Card. Fail. 2021, 27, 1404–1444. [Google Scholar] [CrossRef]
- Chusho, H.; Tamura, N.; Ogawa, Y.; Yasoda, A.; Suda, M.; Miyazawa, T.; Nakamura, K.; Nakao, K.; Kurihara, T.; Komatsu, Y.; et al. Dwarfism and early death in mice lacking C-type natriuretic peptide. Proc. Natl. Acad. Sci. USA 2001, 98, 4016–4021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savarirayan, R.; Irving, M.; Bacino, C.A.; Bostwick, B.; Charrow, J.; Cormier-Daire, V.; Le Quan Sang, K.H.; Dickson, P.; Harmatz, P.; Phillips, J.; et al. C-Type Natriuretic Peptide Analogue Therapy in Children with Achondroplasia. N. Engl. J. Med. 2019, 381, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wu, F.; Pan, J.; Morser, J.; Wu, Q. Furin-mediated processing of Pro-C-type natriuretic peptide. J. Biol. Chem. 2003, 278, 25847–25852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayek, S.; Nemer, M. Cardiac natriuretic peptides: From basic discovery to clinical practice. Cardiovasc. Ther. 2011, 29, 362–376. [Google Scholar] [CrossRef]
- Zakeri, R.; Sangaralingham, S.J.; Sandberg, S.M.; Heublein, D.M.; Scott, C.G.; Burnett, J.C., Jr. Urinary C-type natriuretic peptide: A new heart failure biomarker. JACC Heart Fail. 2013, 1, 170–177. [Google Scholar] [CrossRef]
- Matsuo, A.; Nagai-Okatani, C.; Nishigori, M.; Kangawa, K.; Minamino, N. Natriuretic peptides in human heart: Novel insight into their molecular forms, functions, and diagnostic use. Peptides 2019, 111, 3–17. [Google Scholar] [CrossRef]
- Moyes, A.J.; Hobbs, A.J. C-type Natriuretic Peptide: A Multifaceted Paracrine Regulator in the Heart and Vasculature. Int. J. Mol. Sci. 2019, 20, 2281. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, Y.; Chusho, H.; Tamura, N.; Yasoda, A.; Miyazawa, T.; Suda, M.; Miura, M.; Ogawa, Y.; Nakao, K. Significance of C-type natriuretic peptide (CNP) in endochondral ossification: Analysis of CNP knockout mice. J. Bone Miner. Metab. 2002, 20, 331–336. [Google Scholar] [CrossRef]
- Kojima, M.; Minamino, N.; Kangawa, K.; Matsuo, H. Cloning and sequence analysis of a cDNA encoding a precursor for rat C-type natriuretic peptide (CNP). FEBS Lett. 1990, 276, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Tawaragi, Y.; Fuchimura, K.; Tanaka, S.; Minamino, N.; Kangawa, K.; Matsuo, H. Gene and precursor structures of human C-type natriuretic peptide. Biochem. Biophys. Res. Commun. 1991, 175, 645–651. [Google Scholar] [CrossRef]
- Kuhn, M. Molecular Physiology of Membrane Guanylyl Cyclase Receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef] [PubMed]
- Murthy, K.S.; Teng, B.Q.; Zhou, H.; Jin, J.G.; Grider, J.R.; Makhlouf, G.M. G(i-1)/G(i-2)-dependent signaling by single-transmembrane natriuretic peptide clearance receptor. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G974–G980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, A.J.; Bourne, A.; Ingram, J. Hydrolysis of human and pig brain natriuretic peptides, urodilatin, C-type natriuretic peptide and some C-receptor ligands by endopeptidase-24.11. Biochem. J. 1993, 291, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Bayes-Genis, A.; Barallat, J.; Richards, A.M. A Test in Context: Neprilysin: Function, Inhibition, and Biomarker. J. Am. Coll. Cardiol. 2016, 68, 639–653. [Google Scholar] [CrossRef]
- Del Ry, S.; Cabiati, M.; Vozzi, F.; Battolla, B.; Caselli, C.; Forini, F.; Segnani, C.; Prescimone, T.; Giannessi, D.; Mattii, L. Expression of C-type natriuretic peptide and its receptor NPR-B in cardiomyocytes. Peptides 2011, 32, 1713–1718. [Google Scholar] [CrossRef]
- Horio, T.; Tokudome, T.; Maki, T.; Yoshihara, F.; Suga, S.; Nishikimi, T.; Kojima, M.; Kawano, Y.; Kangawa, K. Gene expression, secretion, and autocrine action of C-type natriuretic peptide in cultured adult rat cardiac fibroblasts. Endocrinology 2003, 144, 2279–2284. [Google Scholar] [CrossRef] [Green Version]
- Nishikimi, T.; Maeda, N.; Matsuoka, H. The role of natriuretic peptides in cardioprotection. Cardiovasc. Res. 2006, 69, 318–328. [Google Scholar] [CrossRef]
- Tokudome, T.; Horio, T.; Soeki, T.; Mori, K.; Kishimoto, I.; Suga, S.; Yoshihara, F.; Kawano, Y.; Kohno, M.; Kangawa, K. Inhibitory effect of C-type natriuretic peptide (CNP) on cultured cardiac myocyte hypertrophy: Interference between CNP and endothelin-1 signaling pathways. Endocrinology 2004, 145, 2131–2140. [Google Scholar] [CrossRef] [Green Version]
- Fujisaki, H.; Ito, H.; Hirata, Y.; Tanaka, M.; Hata, M.; Lin, M.; Adachi, S.; Akimoto, H.; Marumo, F.; Hiroe, M. Natriuretic peptides inhibit angiotensin II-induced proliferation of rat cardiac fibroblasts by blocking endothelin-1 gene expression. J. Clin. Investig. 1995, 96, 1059–1065. [Google Scholar] [CrossRef]
- Cao, L.; Gardner, D.G. Natriuretic peptides inhibit DNA synthesis in cardiac fibroblasts. Hypertension 1995, 25, 227–234. [Google Scholar] [CrossRef]
- Izumiya, Y.; Araki, S.; Usuku, H.; Rokutanda, T.; Hanatani, S.; Ogawa, H. Chronic C-Type Natriuretic Peptide Infusion Attenuates Angiotensin II-Induced Myocardial Superoxide Production and Cardiac Remodeling. Int. J. Vasc. Med. 2012, 2012, 246058. [Google Scholar] [CrossRef] [PubMed]
- Soeki, T.; Kishimoto, I.; Okumura, H.; Tokudome, T.; Horio, T.; Mori, K.; Kangawa, K. C-type natriuretic peptide, a novel antifibrotic and antihypertrophic agent, prevents cardiac remodeling after myocardial infarction. J. Am. Coll. Cardiol. 2005, 45, 608–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obata, H.; Yanagawa, B.; Tanaka, K.; Ohnishi, S.; Kataoka, M.; Miyahara, Y.; Ishibashi-Ueda, H.; Kodama, M.; Aizawa, Y.; Kangawa, K.; et al. CNP infusion attenuates cardiac dysfunction and inflammation in myocarditis. Biochem. Biophys. Res. Commun. 2007, 356, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; de Waard, M.C.; Sterner-Kock, A.; Stepan, H.; Schultheiss, H.P.; Duncker, D.J.; Walther, T. Cardiomyocyte-restricted over-expression of C-type natriuretic peptide prevents cardiac hypertrophy induced by myocardial infarction in mice. Eur. J. Heart Fail. 2007, 9, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Langenickel, T.H.; Buttgereit, J.; Pagel-Langenickel, I.; Lindner, M.; Monti, J.; Beuerlein, K.; Al-Saadi, N.; Plehm, R.; Popova, E.; Tank, J.; et al. Cardiac hypertrophy in transgenic rats expressing a dominant-negative mutant of the natriuretic peptide receptor B. Proc. Natl. Acad. Sci. USA 2006, 103, 4735–4740. [Google Scholar] [CrossRef] [Green Version]
- Michel, K.; Herwig, M.; Werner, F.; Spiranec Spes, K.; Abesser, M.; Schuh, K.; Dabral, S.; Mugge, A.; Baba, H.A.; Skryabin, B.V.; et al. C-type natriuretic peptide moderates titin-based cardiomyocyte stiffness. JCI Insight 2020, 5, e139910. [Google Scholar] [CrossRef]
- Tsuchihashi-Makaya, M.; Hamaguchi, S.; Kinugawa, S.; Yokota, T.; Goto, D.; Yokoshiki, H.; Kato, N.; Takeshita, A.; Tsutsui, H.; Investigators, J.-C. Characteristics and outcomes of hospitalized patients with heart failure and reduced vs preserved ejection fraction. Report from the Japanese Cardiac Registry of Heart Failure in Cardiology (JCARE-CARD). Circ. J. 2009, 73, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Koser, F.; Loescher, C.; Linke, W.A. Posttranslational modifications of titin from cardiac muscle: How, where, and what for? FEBS J. 2019, 286, 2240–2260. [Google Scholar] [CrossRef]
- Van Heerebeek, L.; Hamdani, N.; Falcao-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.; Bronzwaer, J.G.; van der Velden, J.; Stienen, G.J.; Laarman, G.J.; Somsen, A.; et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Dorey, T.W.; Mackasey, M.; Jansen, H.J.; McRae, M.D.; Bohne, L.J.; Liu, Y.; Belke, D.D.; Atkinson, L.; Rose, R.A. Natriuretic peptide receptor B maintains heart rate and sinoatrial node function via cyclic GMP-mediated signaling. Cardiovasc. Res. 2021, 118, 1917–1931. [Google Scholar] [CrossRef]
- Vaandrager, A.B.; Bot, A.G.; Ruth, P.; Pfeifer, A.; Hofmann, F.; De Jonge, H.R. Differential role of cyclic GMP-dependent protein kinase II in ion transport in murine small intestine and colon. Gastroenterology 2000, 118, 108–114. [Google Scholar] [CrossRef]
- Kuhn, M.; Voss, M.; Mitko, D.; Stypmann, J.; Schmid, C.; Kawaguchi, N.; Grabellus, F.; Baba, H.A. Left ventricular assist device support reverses altered cardiac expression and function of natriuretic peptides and receptors in end-stage heart failure. Cardiovasc. Res. 2004, 64, 308–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, S.D.; Nilsson, H.; Ahluwalia, A.; Hobbs, A.J. Release of C-type natriuretic peptide accounts for the biological activity of endothelium-derived hyperpolarizing factor. Proc. Natl. Acad. Sci. USA 2003, 100, 1426–1431. [Google Scholar] [CrossRef] [Green Version]
- Lopez, M.J.; Garbers, D.L.; Kuhn, M. The guanylyl cyclase-deficient mouse defines differential pathways of natriuretic peptide signaling. J. Biol. Chem. 1997, 272, 23064–23068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, K.; Kuwahara, K.; Nishikimi, T.; Nakagawa, Y.; Kinoshita, H.; Minami, T.; Kuwabara, Y.; Yamada, C.; Yamada, Y.; Tokudome, T.; et al. Endothelium-Derived C-Type Natriuretic Peptide Contributes to Blood Pressure Regulation by Maintaining Endothelial Integrity. Hypertension 2017, 69, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Moyes, A.J.; Khambata, R.S.; Villar, I.; Bubb, K.J.; Baliga, R.S.; Lumsden, N.G.; Xiao, F.; Gane, P.J.; Rebstock, A.S.; Worthington, R.J.; et al. Endothelial C-type natriuretic peptide maintains vascular homeostasis. J. Clin. Investig. 2014, 124, 4039–4051. [Google Scholar] [CrossRef]
- Spiranec, K.; Chen, W.; Werner, F.; Nikolaev, V.O.; Naruke, T.; Koch, F.; Werner, A.; Eder-Negrin, P.; Dieguez-Hurtado, R.; Adams, R.H.; et al. Endothelial C-Type Natriuretic Peptide Acts on Pericytes to Regulate Microcirculatory Flow and Blood Pressure. Circulation 2018, 138, 494–508. [Google Scholar] [CrossRef]
- Thonsgaard, S.; Prickett, T.C.R.; Hansen, L.H.; Wewer Albrechtsen, N.J.; Andersen, U.O.; Terzic, D.; Plomgaard, P.; Gustafsson, F.; Goetze, J.P.; Mark, P.D. Circulating Concentrations of C-Type Natriuretic Peptides Increase with Sacubitril/Valsartan Treatment in Healthy Young Men. Clin. Chem. 2022, 68, 713–720. [Google Scholar] [CrossRef]
- Ruilope, L.M.; Dukat, A.; Bohm, M.; Lacourciere, Y.; Gong, J.; Lefkowitz, M.P. Blood-pressure reduction with LCZ696, a novel dual-acting inhibitor of the angiotensin II receptor and neprilysin: A randomised, double-blind, placebo-controlled, active comparator study. Lancet 2010, 375, 1255–1266. [Google Scholar] [CrossRef]
- Yamahara, K.; Itoh, H.; Chun, T.H.; Ogawa, Y.; Yamashita, J.; Sawada, N.; Fukunaga, Y.; Sone, M.; Yurugi-Kobayashi, T.; Miyashita, K.; et al. Significance and therapeutic potential of the natriuretic peptides/cGMP/cGMP-dependent protein kinase pathway in vascular regeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 3404–3409. [Google Scholar] [CrossRef] [Green Version]
- Bubb, K.J.; Aubdool, A.A.; Moyes, A.J.; Lewis, S.; Drayton, J.P.; Tang, O.; Mehta, V.; Zachary, I.C.; Abraham, D.J.; Tsui, J.; et al. Endothelial C-Type Natriuretic Peptide Is a Critical Regulator of Angiogenesis and Vascular Remodeling. Circulation 2019, 139, 1612–1628. [Google Scholar] [CrossRef] [PubMed]
- Huntley, B.K.; Sandberg, S.M.; Noser, J.A.; Cataliotti, A.; Redfield, M.M.; Matsuda, Y.; Burnett, J.C., Jr. BNP-induced activation of cGMP in human cardiac fibroblasts: Interactions with fibronectin and natriuretic peptide receptors. J. Cell. Physiol. 2006, 209, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Sangaralingham, S.J.; Huntley, B.K.; Martin, F.L.; McKie, P.M.; Bellavia, D.; Ichiki, T.; Harders, G.E.; Chen, H.H.; Burnett, J.C., Jr. The aging heart, myocardial fibrosis, and its relationship to circulating C-type natriuretic Peptide. Hypertension 2011, 57, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Nagaya, N.; Itoh, T.; Fujii, T.; Iwase, T.; Hamada, K.; Kimura, H.; Kangawa, K. C-type natriuretic peptide attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L1172–L1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Allen, P.D.; Izumo, S. Expression of A-, B-, and C-type natriuretic peptide genes in failing and developing human ventricles. Correlation with expression of the Ca(2+)-ATPase gene. Circ. Res. 1992, 71, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.M.; Heublein, D.M.; Perrella, M.A.; Lerman, A.; Rodeheffer, R.J.; McGregor, C.G.; Edwards, W.D.; Schaff, H.V.; Burnett, J.C., Jr. Natriuretic peptide system in human heart failure. Circulation 1993, 88, 1004–1009. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.J.; Rehfeld, J.F.; Goetze, J.P. Mismeasure of C-type natriuretic peptide. Clin. Chem. 2008, 54, 225–227. [Google Scholar] [CrossRef]
- Cargill, R.I.; Barr, C.S.; Coutie, W.J.; Struthers, A.D.; Lipworth, B.J. C-type natriuretic peptide levels in cor pulmonale and in congestive heart failure. Thorax 1994, 49, 1247–1249. [Google Scholar] [CrossRef] [Green Version]
- Hama, N.; Itoh, H.; Shirakami, G.; Suga, S.; Komatsu, Y.; Yoshimasa, T.; Tanaka, I.; Mori, K.; Nakao, K. Detection of C-type natriuretic peptide in human circulation and marked increase of plasma CNP level in septic shock patients. Biochem. Biophys. Res. Commun. 1994, 198, 1177–1182. [Google Scholar] [CrossRef]
- Prickett, T.C.; Yandle, T.G.; Nicholls, M.G.; Espiner, E.A.; Richards, A.M. Identification of amino-terminal pro-C-type natriuretic peptide in human plasma. Biochem. Biophys. Res. Commun. 2001, 286, 513–517. [Google Scholar] [CrossRef]
- Wright, S.P.; Prickett, T.C.; Doughty, R.N.; Frampton, C.; Gamble, G.D.; Yandle, T.G.; Sharpe, N.; Richards, M. Amino-terminal pro-C-type natriuretic peptide in heart failure. Hypertension 2004, 43, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalra, P.R.; Clague, J.R.; Bolger, A.P.; Anker, S.D.; Poole-Wilson, P.A.; Struthers, A.D.; Coats, A.J. Myocardial production of C-type natriuretic peptide in chronic heart failure. Circulation 2003, 107, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.J.; Rehfeld, J.F.; Pedersen, F.; Kastrup, J.; Videbaek, R.; Goetze, J.P. Measurement of pro-C-type natriuretic peptide in plasma. Clin. Chem. 2005, 51, 2173–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Ry, S.; Maltinti, M.; Piacenti, M.; Passino, C.; Emdin, M.; Giannessi, D. Cardiac production of C-type natriuretic peptide in heart failure. J. Cardiovasc. Med. 2006, 7, 397–399. [Google Scholar] [CrossRef]
- Charles, C.J.; Prickett, T.C.; Espiner, E.A.; Rademaker, M.T.; Richards, A.M.; Yandle, T.G. Regional sampling and the effects of experimental heart failure in sheep: Differential responses in A, B and C-type natriuretic peptides. Peptides 2006, 27, 62–68. [Google Scholar] [CrossRef]
- Nishikimi, T.; Kitamura, K.; Saito, Y.; Shimada, K.; Ishimitsu, T.; Takamiya, M.; Kangawa, K.; Matsuo, H.; Eto, T.; Omae, T.; et al. Clinical studies on the sites of production and clearance of circulating adrenomedullin in human subjects. Hypertension 1994, 24, 600–604. [Google Scholar] [CrossRef] [Green Version]
- Nishikimi, T.; Saito, Y.; Kitamura, K.; Ishimitsu, T.; Eto, T.; Kangawa, K.; Matsuo, H.; Omae, T.; Matsuoka, H. Increased plasma levels of adrenomedullin in patients with heart failure. J. Am. Coll. Cardiol. 1995, 26, 1424–1431. [Google Scholar] [CrossRef] [Green Version]
- Suga, S.; Nakao, K.; Itoh, H.; Komatsu, Y.; Ogawa, Y.; Hama, N.; Imura, H. Endothelial production of C-type natriuretic peptide and its marked augmentation by transforming growth factor-beta. Possible existence of “vascular natriuretic peptide system”. J. Clin. Investig. 1992, 90, 1145–1149. [Google Scholar] [CrossRef] [Green Version]
- Isumi, Y.; Shoji, H.; Sugo, S.; Tochimoto, T.; Yoshioka, M.; Kangawa, K.; Matsuo, H.; Minamino, N. Regulation of adrenomedullin production in rat endothelial cells. Endocrinology 1998, 139, 838–846. [Google Scholar] [CrossRef]
- Dick, S.A.; Epelman, S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ. Res. 2016, 119, 159–176. [Google Scholar] [CrossRef] [Green Version]
- Del Ry, S.; Maltinti, M.; Cabiati, M.; Emdin, M.; Giannessi, D.; Morales, M.A. C-type natriuretic peptide and its relation to non-invasive indices of left ventricular function in patients with chronic heart failure. Peptides 2008, 29, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Del Ry, S.; Giannessi, D.; Maltinti, M.; Prontera, C.; Iervasi, A.; Colotti, C.; Emdin, M.; L’Abbate, A.; Neglia, D. Increased levels of C-type natriuretic peptide in patients with idiopathic left ventricular dysfunction. Peptides 2007, 28, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Del Ry, S.; Cabiati, M.; Stefano, T.; Catapano, G.; Caselli, C.; Prescimone, T.; Passino, C.; Emdin, M.; Giannessi, D. Comparison of NT-proCNP and CNP plasma levels in heart failure, diabetes and cirrhosis patients. Regul. Pept. 2011, 166, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Passino, C.; Del Ry, S.; Severino, S.; Gabutti, A.; Prontera, C.; Clerico, A.; Giannessi, D.; Emdin, M. C-type natriuretic peptide expression in patients with chronic heart failure: Effects of aerobic training. Eur. J. Cardiovasc. Prev. Rehabil. 2008, 15, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Lok, D.J.; Klip, I.T.; Voors, A.A.; Lok, S.I.; Bruggink-Andre de la Porte, P.W.; Hillege, H.L.; Jaarsma, T.; van Veldhuisen, D.J.; van der Meer, P. Prognostic value of N-terminal pro C-type natriuretic peptide in heart failure patients with preserved and reduced ejection fraction. Eur. J. Heart Fail. 2014, 16, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, N.E.; McCarthy, C.P.; Shrestha, S.; Gaggin, H.K.; Mukai, R.; Szymonifka, J.; Apple, F.S.; Burnett, J.C., Jr.; Iyer, S.; Januzzi, J.L., Jr. Effect of Neprilysin Inhibition on Various Natriuretic Peptide Assays. J. Am. Coll. Cardiol. 2019, 73, 1273–1284. [Google Scholar] [CrossRef]
- Moyes, A.J.; Chu, S.M.; Aubdool, A.A.; Dukinfield, M.S.; Margulies, K.B.; Bedi, K.C.; Hodivala-Dilke, K.; Baliga, R.S.; Hobbs, A.J. C-type natriuretic peptide co-ordinates cardiac structure and function. Eur. Heart J. 2020, 41, 1006–1020. [Google Scholar] [CrossRef] [Green Version]
- Hassoun, P.M. Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 385, 2361–2376. [Google Scholar] [CrossRef]
- Adnot, S.; Chabrier, P.E.; Brun-Buisson, C.; Viossat, I.; Braquet, P. Atrial natriuretic factor attenuates the pulmonary pressor response to hypoxia. J. Appl. Physiol. (1985) 1988, 65, 1975–1983. [Google Scholar] [CrossRef]
- Ou, L.C.; Yen, S.; Sardella, G.L.; Hill, N.S. Does atrial natriuretic factor protect against right ventricular overload? II. Tissue binding. J. Appl. Physiol. (1985) 1989, 67, 1612–1616. [Google Scholar] [CrossRef]
- Jin, H.; Yang, R.H.; Chen, Y.F.; Jackson, R.M.; Oparil, S. Atrial natriuretic peptide attenuates the development of pulmonary hypertension in rats adapted to chronic hypoxia. J. Clin. Investig. 1990, 85, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Raffestin, B.; Levame, M.; Eddahibi, S.; Viossat, I.; Braquet, P.; Chabrier, P.E.; Cantin, M.; Adnot, S. Pulmonary vasodilatory action of endogenous atrial natriuretic factor in rats with hypoxic pulmonary hypertension. Effects of monoclonal atrial natriuretic factor antibody. Circ. Res. 1992, 70, 184–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, N.S.; Klinger, J.R.; Warburton, R.R.; Pietras, L.; Wrenn, D.S. Brain natriuretic peptide: Possible role in the modulation of hypoxic pulmonary hypertension. Am. J. Physiol. 1994, 266, L308–L315. [Google Scholar] [CrossRef] [PubMed]
- Klinger, J.R.; Warburton, R.R.; Pietras, L.; Hill, N.S. Brain natriuretic peptide inhibits hypoxic pulmonary hypertension in rats. J. Appl. Physiol. (1985) 1998, 84, 1646–1652. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Long, L.; Morrell, N.W.; Wilkins, M.R. NPR-A-Deficient mice show increased susceptibility to hypoxia-induced pulmonary hypertension. Circulation 1999, 99, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Klinger, J.R.; Siddiq, F.M.; Swift, R.A.; Jackson, C.; Pietras, L.; Warburton, R.R.; Alia, C.; Hill, N.S. C-type natriuretic peptide expression and pulmonary vasodilation in hypoxia-adapted rats. Am. J. Physiol. 1998, 275, L645–L652. [Google Scholar] [CrossRef]
- Itoh, T.; Nagaya, N.; Murakami, S.; Fujii, T.; Iwase, T.; Ishibashi-Ueda, H.; Yutani, C.; Yamagishi, M.; Kimura, H.; Kangawa, K. C-type natriuretic peptide ameliorates monocrotaline-induced pulmonary hypertension in rats. Am. J. Respir. Crit. Care Med. 2004, 170, 1204–1211. [Google Scholar] [CrossRef]
- Casserly, B.; Mazer, J.M.; Vang, A.; Harrington, E.O.; Klinger, J.R.; Rounds, S.; Choudhary, G. C-type natriuretic peptide does not attenuate the development of pulmonary hypertension caused by hypoxia and VEGF receptor blockade. Life Sci. 2011, 89, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Nawa, N.; Ishida, H.; Katsuragi, S.; Baden, H.; Takahashi, K.; Higeno, R.; Torigoe, F.; Mihara, S.; Narita, J.; Miura, K.; et al. Constitutively active form of natriuretic peptide receptor 2 ameliorates experimental pulmonary arterial hypertension. Mol. Ther. Methods Clin. Dev. 2016, 3, 16044. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagawa, Y.; Nishikimi, T. CNP, the Third Natriuretic Peptide: Its Biology and Significance to the Cardiovascular System. Biology 2022, 11, 986. https://doi.org/10.3390/biology11070986
Nakagawa Y, Nishikimi T. CNP, the Third Natriuretic Peptide: Its Biology and Significance to the Cardiovascular System. Biology. 2022; 11(7):986. https://doi.org/10.3390/biology11070986
Chicago/Turabian StyleNakagawa, Yasuaki, and Toshio Nishikimi. 2022. "CNP, the Third Natriuretic Peptide: Its Biology and Significance to the Cardiovascular System" Biology 11, no. 7: 986. https://doi.org/10.3390/biology11070986
APA StyleNakagawa, Y., & Nishikimi, T. (2022). CNP, the Third Natriuretic Peptide: Its Biology and Significance to the Cardiovascular System. Biology, 11(7), 986. https://doi.org/10.3390/biology11070986