
Pharmacological Inhibition of Inositol Hexakisphosphate Kinase 1 Protects Mice against Obesity-Induced Bone Loss

 ,
,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell and Cell-Based Assays
2.2. Mice and Experimental Design
2.3. Chemicals
2.4. Micro-Computed Tomography
2.5. Fluorescence-Activated Cell Sorting
2.6. Spermatogenesis
2.7. Statistical Analysis
3. Results
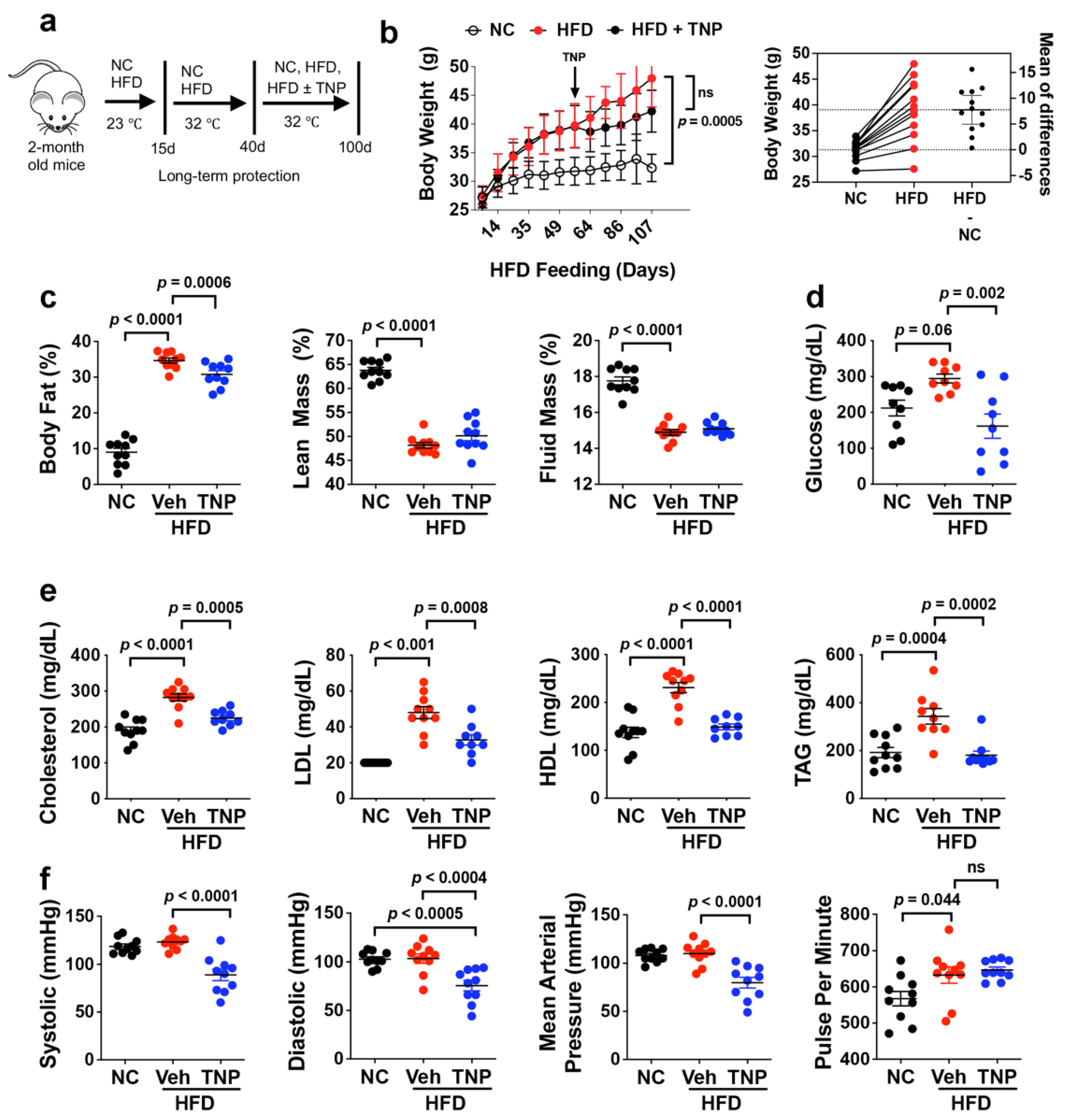
3.1. TNP Administration Reverses HFD-Induced Weight Gain and Hyperglycemia
3.2. TNP Administration Prevents HFD-Induced Bone Loss and MAT Accumulation
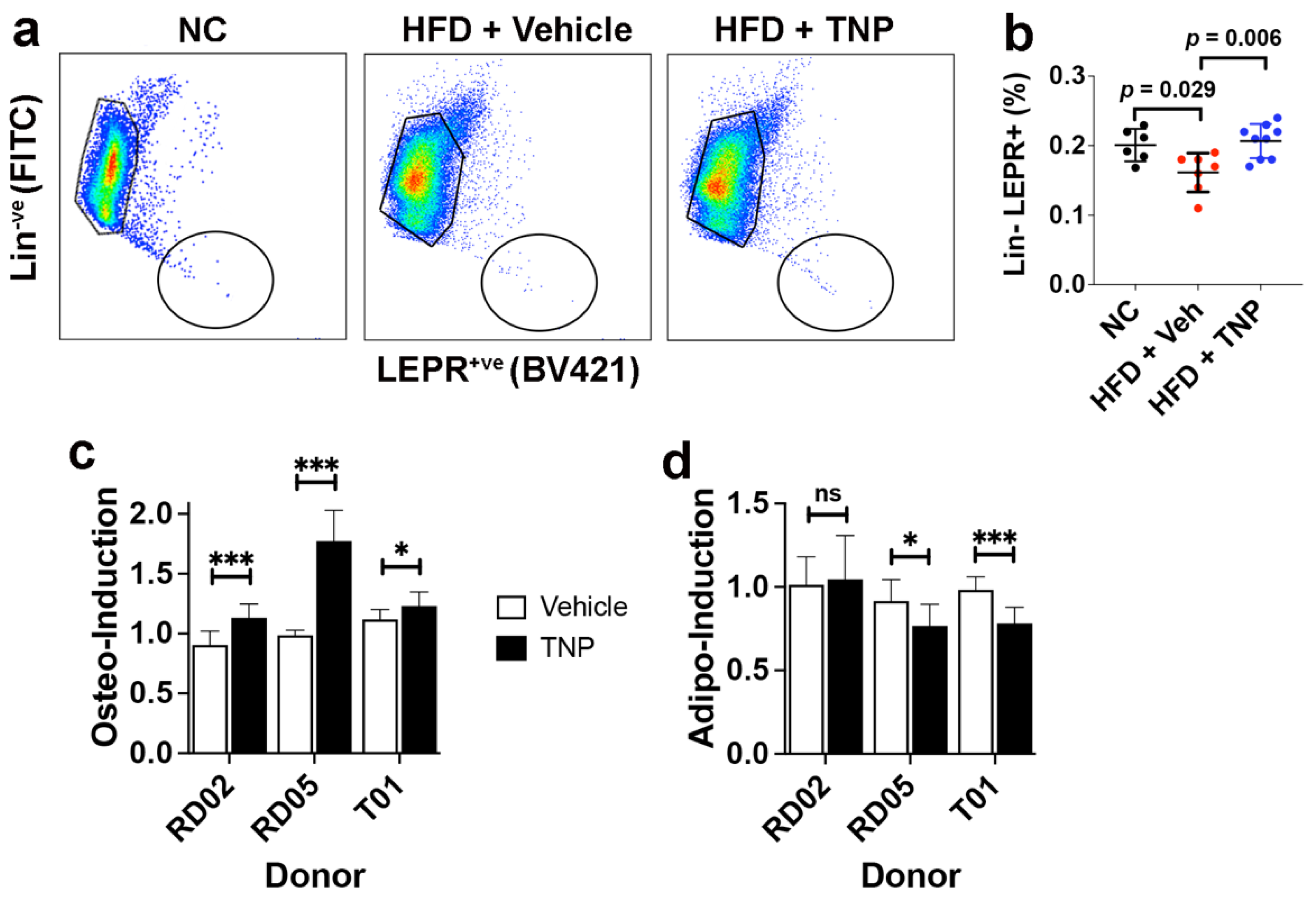
3.3. TNP Administration Preserves the SSPC Pool in the Bone Marrow of HFD-Fed Mice
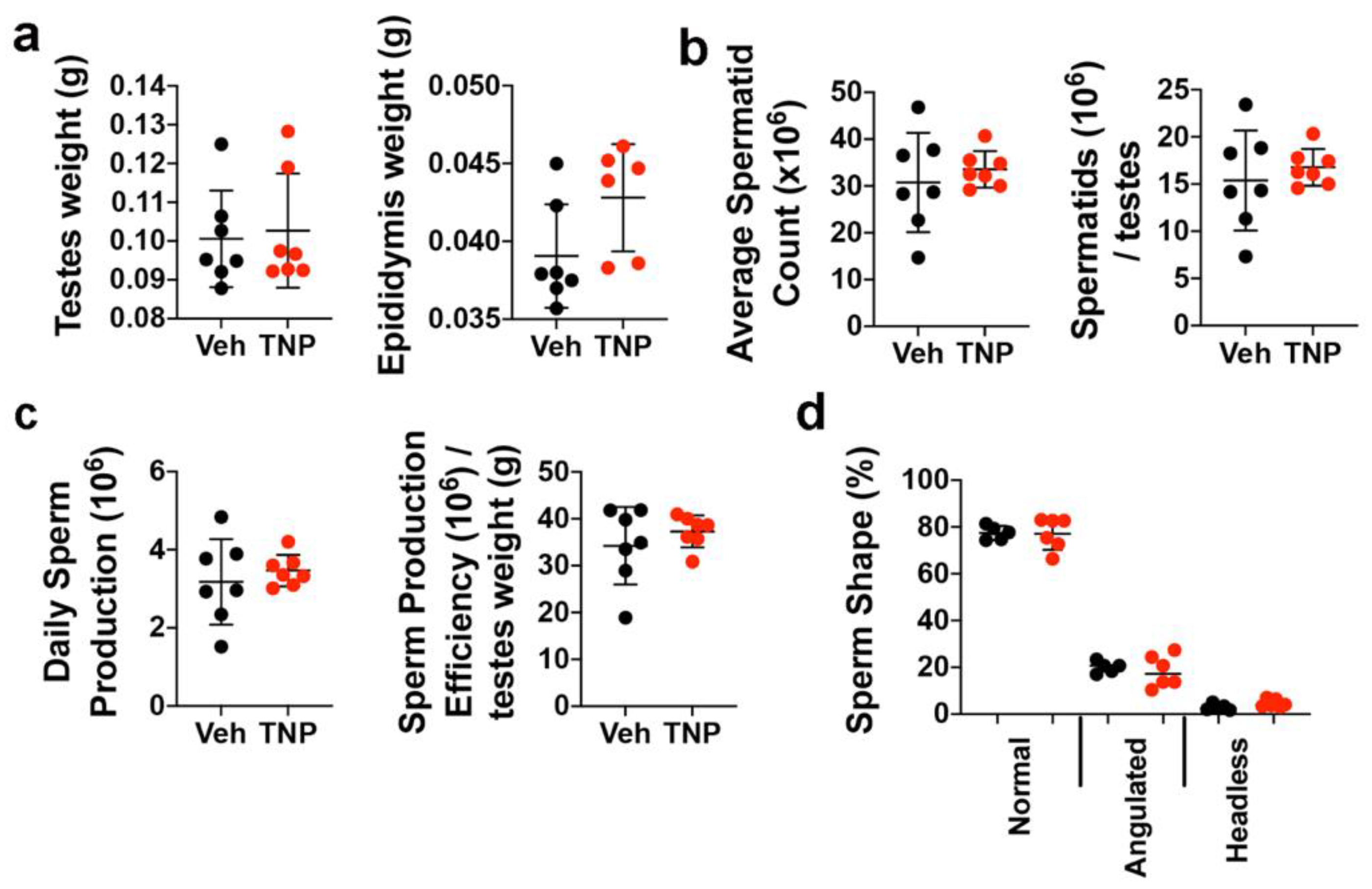
3.4. TNP Has No Long-Term Negative Impacts on Spermatogenesis in Adult Male Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiao, H.; Xiao, E.; Graves, D.T. Diabetes and Its Effect on Bone and Fracture Healing. Curr. Osteoporos. Rep. 2015, 13, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.C.; Shin, D.H.; Lee, S.Y.; Im, J.A.; Lee, D.C. Relation between Obesity and Bone Mineral Density and Vertebral Fractures in Korean Postmenopausal Women. Yonsei Med. J. 2010, 51, 857–863. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Sugimoto, T. Advanced Glycation End Products, Diabetes, and Bone Strength. Curr. Osteoporos. Rep. 2016, 14, 320–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.J.; Sun, L.; Gao, H. Diet-Induced Obesity Alters Bone Remodeling Leading to Decreased Femoral Trabecular Bone Mass in Mice. Ann. N. Y. Acad. Sci. 2010, 1192, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Scheller, E.L.; Khoury, B.; Moller, K.L.; Wee, N.K.; Khandaker, S.; Kozloff, K.M.; Abrishami, S.H.; Zamarron, B.F.; Singer, K. Changes in Skeletal Integrity and Marrow Adiposity During High-Fat Diet and after Weight Loss. Front. Endocrinol. 2016, 7, 102. [Google Scholar] [CrossRef]
- Brown, M.L.; Yukata, K.; Farnsworth, C.W.; Chen, D.-G.; Awad, H.; Hilton, M.J.; O’ Keefe, R.J.; Xing, L.; Mooney, R.A.; Zuscik, M.J. Delayed Fracture Healing and Increased Callus Adiposity in a C57bl/6j Murine Model of Obesity-Associated Type 2 Diabetes Mellitus. PLoS ONE 2014, 9, e99656. [Google Scholar] [CrossRef] [Green Version]
- Ambrosi, T.H.; Scialdone, A.; Graja, A.; Gohlke, S.; Jank, A.-M.; Bocian, C.; Woelk, L.; Fan, H.; Logan, D.W.; Schürmann, A.; et al. Adipocyte Accumulation in the Bone Marrow During Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell 2017, 20, 771–784.e776. [Google Scholar] [CrossRef] [Green Version]
- Botolin, S.; Mccabe, L.R. Bone Loss and Increased Bone Adiposity in Spontaneous and Pharmacologically Induced Diabetic Mice. Endocrinology 2007, 148, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Meunier, P.; Aaron, J.; Edouard, C.; Vignon, G. Osteoporosis and the Replacement of Cell Populations of the Marrow by Adipose Tissue. A Quantitative Study of 84 Iliac Bone Biopsies. Clin. Orthop. Relat. Res. 1971, 80, 147–154. [Google Scholar] [CrossRef]
- Devlin, M.J.; Rosen, C.J. The Bone-Fat Interface: Basic and Clinical Implications of Marrow Adiposity. Lancet Diabetes Endocrinol. 2015, 3, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Rosen, C.J.; Bouxsein, M.L. Mechanisms of Disease: Is Osteoporosis the Obesity of Bone? Nat. Clin. Pract. Rheumatol. 2006, 2, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Janeckova, R. The Role of Leptin in Human Physiology and Pathophysiology. Physiol. Res. 2001, 50, 443–459. [Google Scholar] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum Immunoreactive-Leptin Concentrations in Normal-Weight and Obese Humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Hamrick, M.W. Leptin, Bone Mass, and the Thrifty Phenotype. J. Bone Miner. Res. 2004, 19, 1607–1611. [Google Scholar] [CrossRef]
- Reid, I.R.; Baldock, P.A.; Cornish, J. Effects of Leptin on the Skeleton. Endocr. Rev. 2018, 39, 938–959. [Google Scholar] [CrossRef]
- Elefteriou, F.; Takeda, S.; Ebihara, K.; Magre, J.; Patano, N.; Kim, C.A.; Ogawa, Y.; Liu, X.; Ware, S.M.; Craigen, W.J.; et al. Serum Leptin Level Is a Regulator of Bone Mass. Proc. Natl. Acad. Sci. USA 2004, 101, 3258–3263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.O.; Yue, R.; Murphy, M.M.; Peyer, J.G.; Morrison, S.J. Leptin-Receptor-Expressing Mesenchymal Stromal Cells Represent the Main Source of Bone Formed by Adult Bone Marrow. Cell Stem Cell 2014, 15, 154–168. [Google Scholar] [CrossRef] [Green Version]
- Yue, R.; Zhou, B.O.; Shimada, I.S.; Zhao, Z.; Morrison, S.J. Leptin Receptor Promotes Adipogenesis and Reduces Osteogenesis by Regulating Mesenchymal Stromal Cells in Adult Bone Marrow. Cell Stem Cell 2016, 18, 782–796. [Google Scholar] [CrossRef] [Green Version]
- Grey, A.; Beckley, V.; Doyle, A.; Fenwick, S.; Horne, A.; Gamble, G.; Bolland, M. Pioglitazone Increases Bone Marrow Fat in Type 2 Diabetes: Results from a Randomized Controlled Trial. Eur. J. Endocrinol. 2012, 166, 1087–1091. [Google Scholar] [CrossRef] [Green Version]
- Grey, A.; Bolland, M.; Gamble, G.; Wattie, D.; Horne, A.; Davidson, J.; Reid, I.R. The Peroxisome Proliferator-Activated Receptor-Gamma Agonist Rosiglitazone Decreases Bone Formation and Bone Mineral Density in Healthy Postmenopausal Women: A Randomized, Controlled Trial. J. Clin. Endocrinol. Metab. 2007, 92, 1305–1310. [Google Scholar] [CrossRef]
- Liu, L.; Aronson, J.; Huang, S.; Lu, Y.; Czernik, P.; Rahman, S.; Kolli, V.; Suva, L.J.; Lecka-Czernik, B. Rosiglitazone Inhibits Bone Regeneration and Causes Significant Accumulation of Fat at Sites of New Bone Formation. Calcif. Tissue Int. 2012, 91, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loke, Y.K.; Singh, S.; Furberg, C.D. Long-Term Use of Thiazolidinediones and Fractures in Type 2 Diabetes: A Meta-Analysis. CMAJ 2009, 180, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Sorocéanu, M.A.; Miao, D.; Bai, X.-Y.; Su, H.; Goltzman, D.; Karaplis, A.C. Rosiglitazone Impacts Negatively on Bone by Promoting Osteoblast/Osteocyte Apoptosis. J. Endocrinol. 2004, 183, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, D.; O’Brien, C.A.; Stewart, S.A.; Manolagas, S.C.; Weinstein, R.S. Glucocorticoids Act Directly on Osteoclasts to Increase Their Life Span and Reduce Bone Density. Endocrinology 2006, 147, 5592–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chotiyarnwong, P.; Mccloskey, E.V. Pathogenesis of Glucocorticoid-Induced Osteoporosis and Options for Treatment. Nat. Rev. Endocrinol. 2020, 16, 437–447. [Google Scholar] [CrossRef]
- Thrailkill, K.M.; Bunn, R.C.; Uppuganti, S.; Ray, P.; Popescu, I.; Kalaitzoglou, E.; Fowlkes, J.L.; Nyman, J.S. Canagliflozin, an Sglt2 Inhibitor, Corrects Glycemic Dysregulation in Tallyho Model of T2d but Only Partially Prevents Bone Deficits. Bone 2020, 141, 115625. [Google Scholar] [CrossRef]
- Chakraborty, A. The Inositol Pyrophosphate Pathway in Health and Diseases. Biol. Rev. Camb. Philos. Soc. 2018, 93, 1203–1227. [Google Scholar] [CrossRef]
- Wilson, M.S.; Livermore, T.M.; Saiardi, A. Inositol Pyrophosphates: Between Signalling and Metabolism. Biochem. J. 2013, 452, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Wundenberg, T.; Grabinski, N.; Lin, H.; Mayr, G.W. Discovery of Insp6-Kinases as Insp6-Dephosphorylating Enzymes Provides a New Mechanism of Cytosolic Insp6 Degradation Driven by the Cellular Atp/Adp Ratio. Biochem. J. 2014, 462, 173–184. [Google Scholar] [CrossRef]
- Rounge, T.B.; Page, C.M.; Lepistö, M.; Ellonen, P.; Andreassen, B.K.; Weiderpass, E. Genome-Wide DNA Methylation in Saliva and Body Size of Adolescent Girls. Epigenomics 2016, 8, 1495–1505. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Koldobskiy, M.A.; Bello, N.; Maxwell, M.; Potter, J.J.; Juluri, K.R.; Maag, D.; Kim, S.; Huang, A.S.; Dailey, M.J.; et al. Inositol Pyrophosphates Inhibit Akt Signaling, Thereby Regulating Insulin Sensitivity and Weight Gain. Cell 2010, 143, 897–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Ghoshal, S.; Rodrigues, A.; Gao, S.; Asterian, A.; Kamenecka, T.M.; Barrow, J.C.; Chakraborty, A. Adipocyte-Specific Deletion of Ip6k1 Reduces Diet-Induced Obesity by Enhancing Ampk-Mediated Thermogenesis. J. Clin. Invest. 2016, 126, 4273–4288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoshal, S.; Zhu, Q.; Asteian, A.; Lin, H.; Xu, H.; Ernst, G.; Barrow, J.C.; Xu, B.; Cameron, M.D.; Kamenecka, T.M.; et al. Tnp [N2-(M-Trifluorobenzyl), N6-(P-Nitrobenzyl)Purine] Ameliorates Diet Induced Obesity and Insulin Resistance Via Inhibition of the Ip6k1 Pathway. Mol. Metab. 2016, 5, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Ghoshal, S.; Tyagi, R.; Chakraborty, A. Global Ip6k1 Deletion Enhances Temperature Modulated Energy Expenditure Which Reduces Carbohydrate and Fat Induced Weight Gain. Mol. Metab. 2017, 6, 73–85. [Google Scholar] [CrossRef]
- Boregowda, S.V.; Krishnappa, V.; Haga, C.L.; Ortiz, L.A.; Phinney, D.G. A Clinical Indications Prediction Scale Based on Twist1 for Human Mesenchymal Stem Cells. EBioMedicine 2016, 4, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Nanjappa, M.K.; Hess, R.A.; Medrano, T.I.; Locker, S.H.; Levin, E.R.; Cooke, P.S. Membrane-Localized Estrogen Receptor 1 Is Required for Normal Male Reproductive Development and Function in Mice. Endocrinology 2016, 157, 2909–2919. [Google Scholar] [CrossRef] [Green Version]
- Padmanabhan, U.; Dollins, D.E.; Fridy, P.C.; York, J.D.; Downes, C.P. Characterization of a Selective Inhibitor of Inositol Hexakisphosphate Kinases: Use in Defining Biological Roles and Metabolic Relationships of Inositol Pyrophosphates. J. Biol. Chem. 2009, 284, 10571–10582. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, T.; Rüegsegger, P. Quantification of Bone Microarchitecture with the Structure Model Index. Comput. Methods Biomech. Biomed. Eng. 1997, 1, 15–23. [Google Scholar] [CrossRef]
- Patsch, J.M.; Burghardt, A.J.; Yap, S.P.; Baum, T.; Schwartz, A.V.; Joseph, G.B.; Link, T.M. Increased Cortical Porosity in Type 2 Diabetic Postmenopausal Women with Fragility Fractures. J. Bone Miner. Res. 2013, 28, 313–324. [Google Scholar] [CrossRef]
- Farr, J.N.; Khosla, S. Determinants of Bone Strength and Quality in Diabetes Mellitus in Humans. Bone 2016, 82, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Boregowda, S.V.; Ghoshal, S.; Booker, C.N.; Krishnappa, V.; Chakraborty, A.; Phinney, D.G. Ip6k1 Reduces Mesenchymal Stem/Stromal Cell Fitness and Potentiates High Fat Diet-Induced Skeletal Involution. Stem Cells 2017, 35, 1973–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandari, R.; Juluri, K.R.; Resnick, A.C.; Snyder, S.H. Gene Deletion of Inositol Hexakisphosphate Kinase 1 Reveals Inositol Pyrophosphate Regulation of Insulin Secretion, Growth, and Spermiogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 2349–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Liu, Y.; Xue, K.; Gu, G.; Fan, W.; Xu, Y.; Ding, Z. Diet-Induced Obesity in Male C57bl/6 Mice Decreases Fertility as a Consequence of Disrupted Blood-Testis Barrier. PLoS ONE 2015, 10, e0120775. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Yan, W.-J.; Yin, T.-L.; Zhang, Y.; Li, J.; Yang, J. Diet-Induced Obesity Impairs Spermatogenesis: A Potential Role for Autophagy. Sci. Rep. 2017, 7, 43475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecka-Czernik, B.; Stechschulte, L.A.; Czernik, P.J.; Dowling, A.R. High Bone Mass in Adult Mice with Diet-Induced Obesity Results from a Combination of Initial Increase in Bone Mass Followed by Attenuation in Bone Formation; Implications for High Bone Mass and Decreased Bone Quality in Obesity. Mol. Cell. Endocrinol. 2015, 410, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Styner, M.; Pagnotti, G.M.; McGrath, C.; Wu, X.; Sen, B.; Uzer, G.; Xie, Z.; Zong, X.; Styner, M.A.; Rubin, C.T.; et al. Exercise Decreases Marrow Adipose Tissue through S-Oxidation in Obese Running Mice. J. Bone Miner. Res. 2017, 32, 1692–1702. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-T.; Choi, G.; Bae, Y.-S.; Burdett, M.; Moon, H.-S.; Lee, J.W.; Gray, N.S.; Schultz, P.G.; Meijer, L.; Chung, S.-K.; et al. Purine-Based Inhibitors of Inositol-1,4,5-Trisphosphate-3-Kinase. ChemBioChem 2002, 3, 897–901. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boregowda, S.V.; Nanjappa, M.K.; Booker, C.N.; Strivelli, J.; Supper, V.M.; Cooke, P.S.; Phinney, D.G. Pharmacological Inhibition of Inositol Hexakisphosphate Kinase 1 Protects Mice against Obesity-Induced Bone Loss. Biology 2022, 11, 1257. https://doi.org/10.3390/biology11091257
Boregowda SV, Nanjappa MK, Booker CN, Strivelli J, Supper VM, Cooke PS, Phinney DG. Pharmacological Inhibition of Inositol Hexakisphosphate Kinase 1 Protects Mice against Obesity-Induced Bone Loss. Biology. 2022; 11(9):1257. https://doi.org/10.3390/biology11091257
Chicago/Turabian StyleBoregowda, Siddaraju V., Manjunatha K. Nanjappa, Cori N. Booker, Jacqueline Strivelli, Valentina M. Supper, Paul S. Cooke, and Donald G. Phinney. 2022. "Pharmacological Inhibition of Inositol Hexakisphosphate Kinase 1 Protects Mice against Obesity-Induced Bone Loss" Biology 11, no. 9: 1257. https://doi.org/10.3390/biology11091257
APA StyleBoregowda, S. V., Nanjappa, M. K., Booker, C. N., Strivelli, J., Supper, V. M., Cooke, P. S., & Phinney, D. G. (2022). Pharmacological Inhibition of Inositol Hexakisphosphate Kinase 1 Protects Mice against Obesity-Induced Bone Loss. Biology, 11(9), 1257. https://doi.org/10.3390/biology11091257