Identification of Driver Genes and miRNAs in Ovarian Cancer through an Integrated In-Silico Approach


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Acquisition of miRNA Expression Data and Ethical Compliance
2.2. Differentially Expressed MicroRNAs (DEMs) Screening
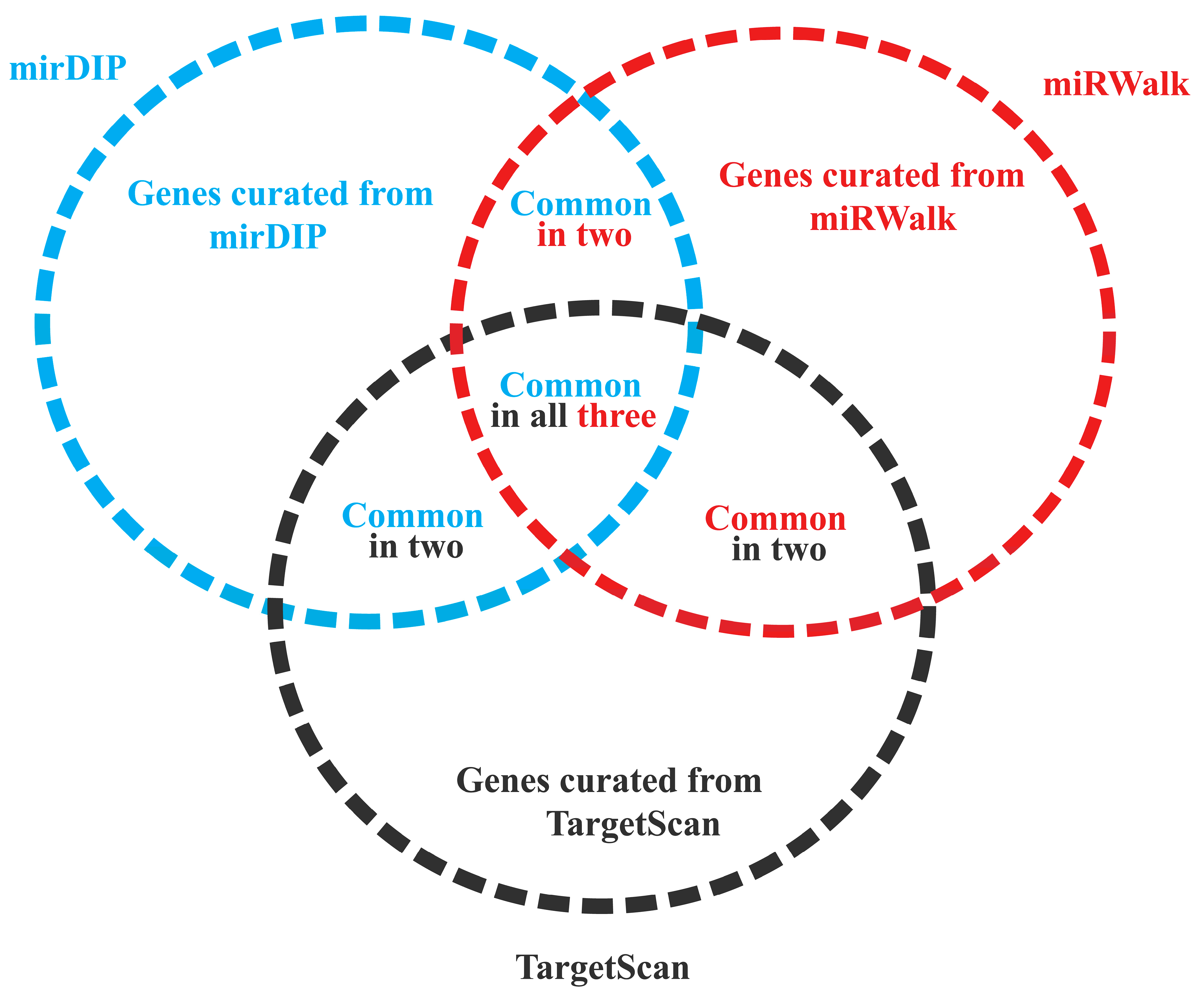
2.3. Identification of Target Genes of DEMs
2.4. DEM Target Gene Network Construction and Hub Gene Identification
2.5. Gene Ontology and Pathway Analysis
2.6. Transcription Factor and Feed Forward Loop Analysis
3. Results
3.1. Identification of DEMs Associated with Ovarian Cancer
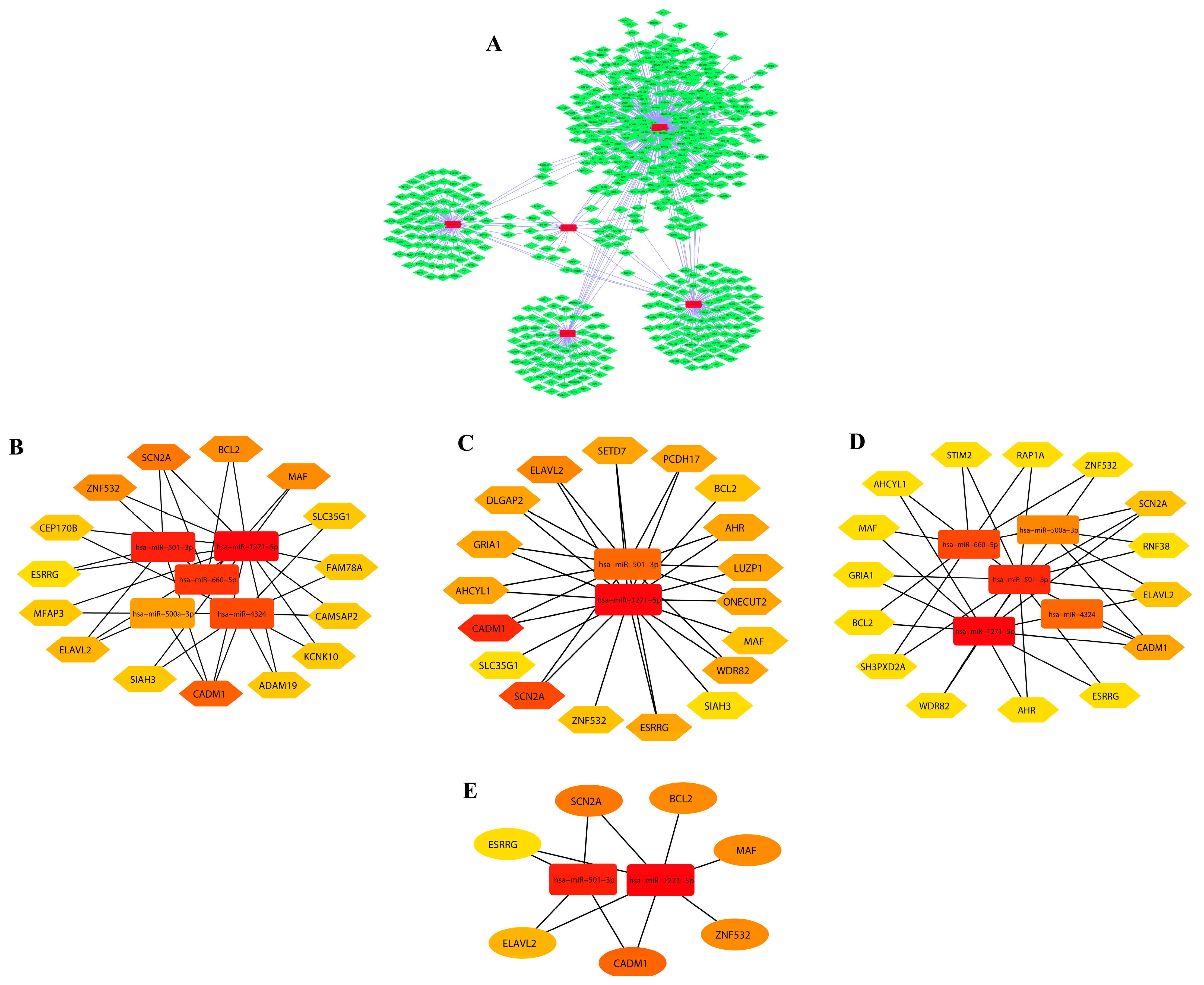
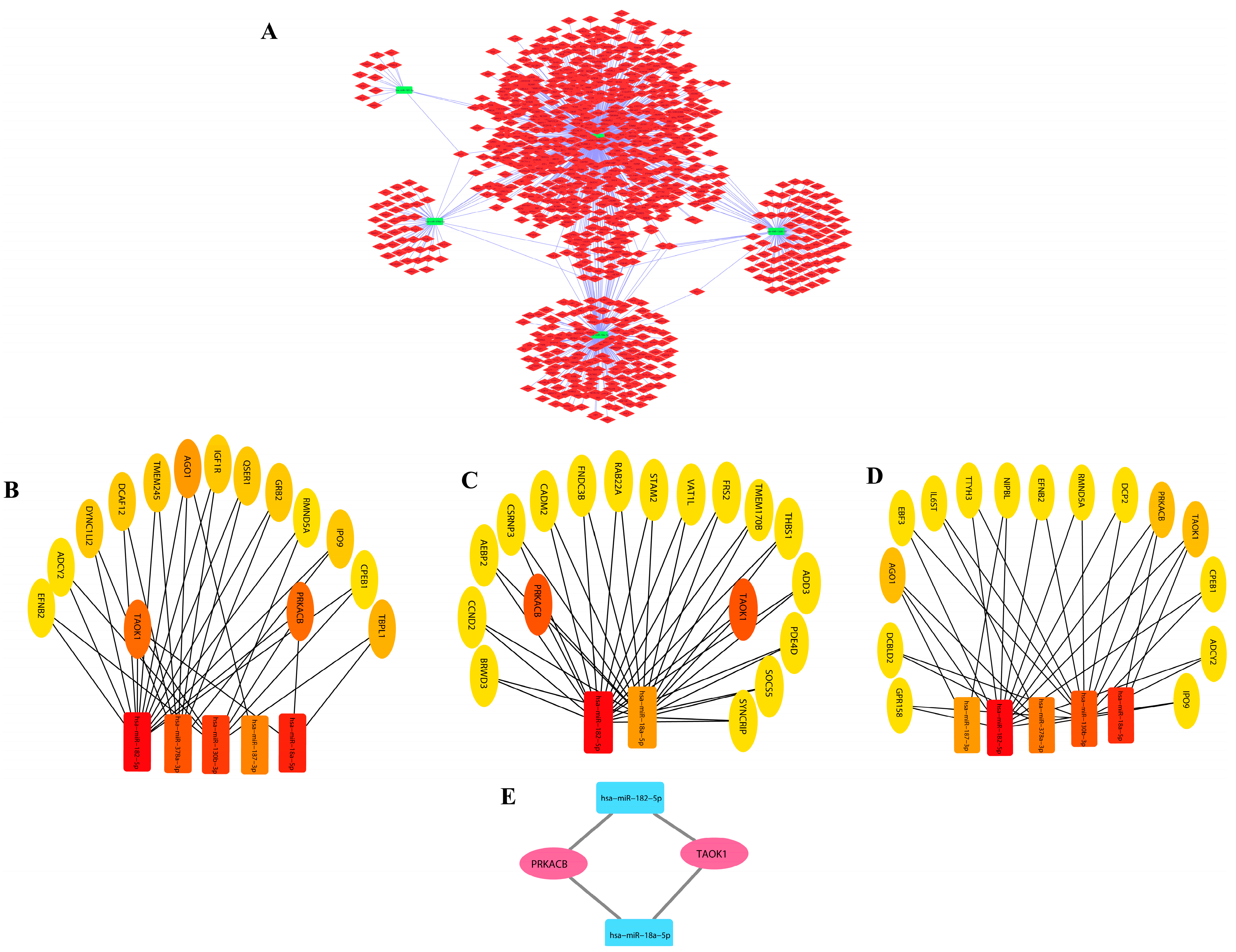
3.2. DEM–miRNA Network Construction and Extraction of Disease-Associated Genes
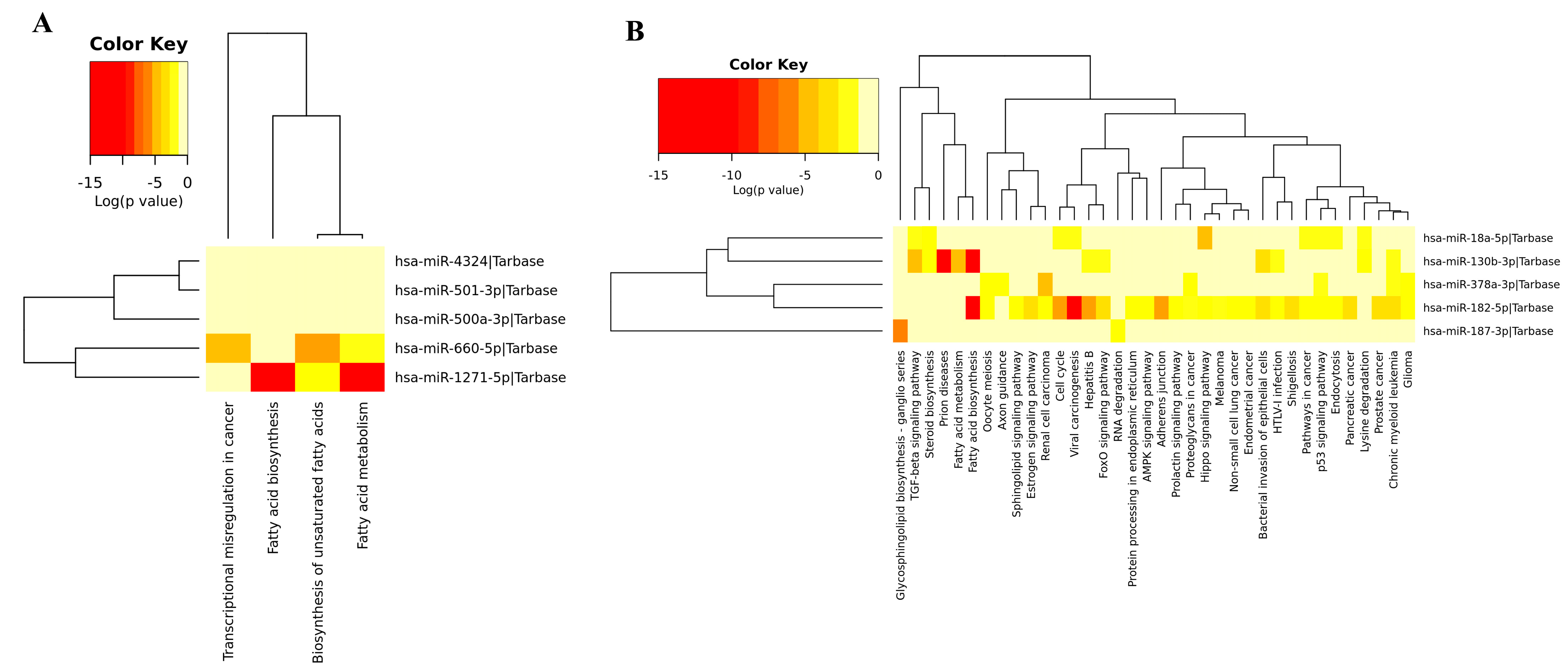
3.3. Module Detection and Pathway Enrichment Analysis
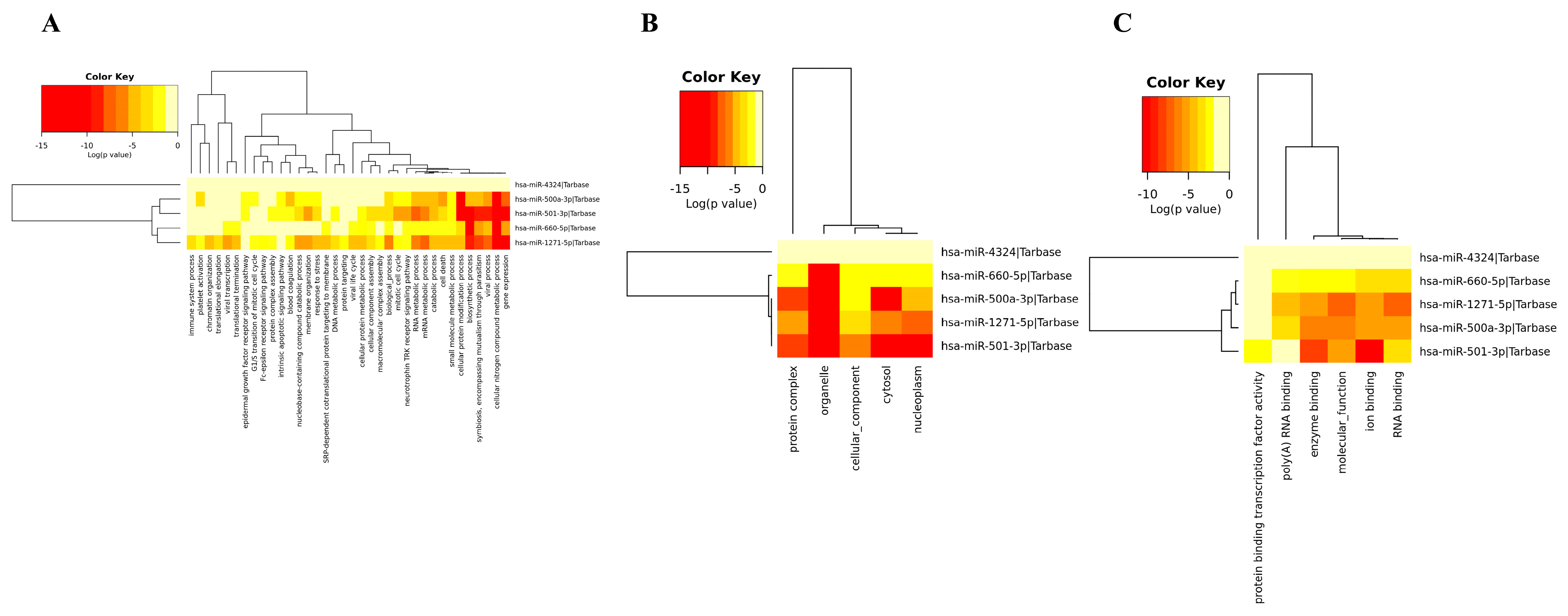
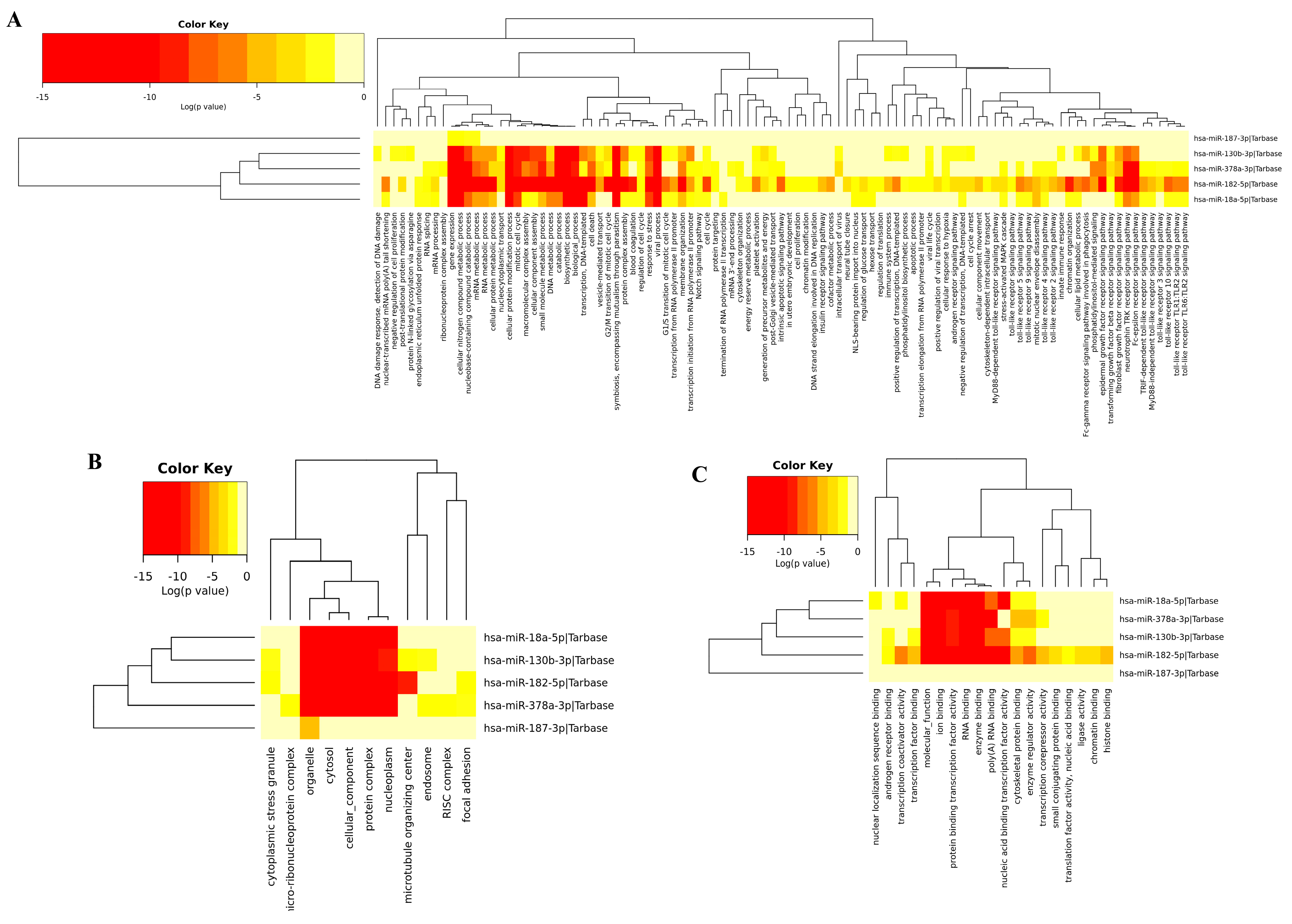
3.4. Gene Ontology of DEM
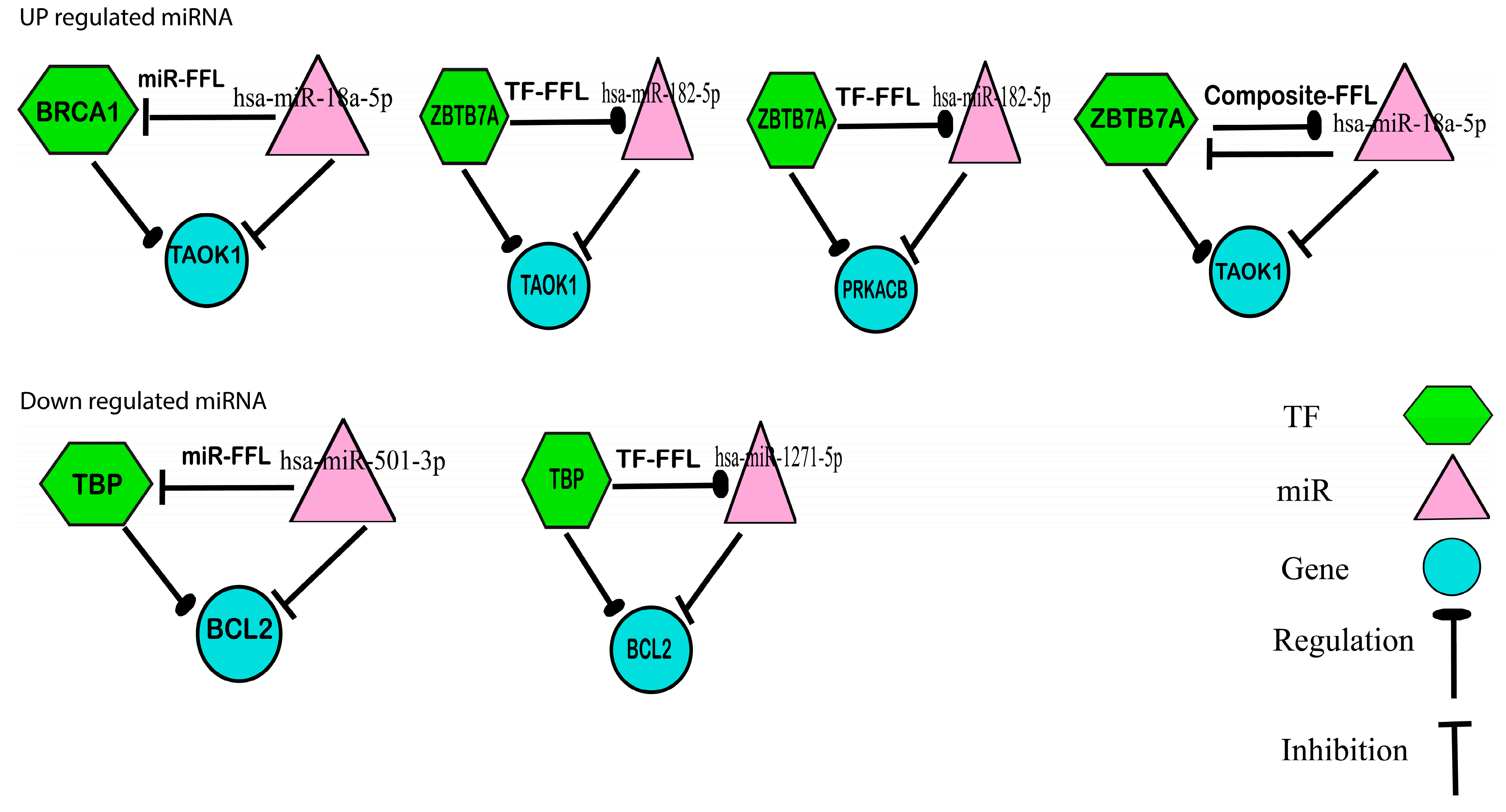
3.5. Transcription Factor Finding and Feed Forward Loop Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OC | Ovarian Cancer |
| GEO | Gene Expression Omnibus |
| miRNA | Micro RNA |
| DEMs | Differentially Expressed miRNAs |
| DEGs | Differentially Expressed Genes |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| TFs | Transcription Factor |
| TRRUST | Transcriptional Regulatory Relationships Unraveled by Sentence based Text mining |
| FFL Tool | Feed Forward loop |
References
- Ghafouri-Fard, S.; Shoorei, H.; Taheri, M. miRNA profile in ovarian cancer. Exp. Mol. Pathol. 2020, 113, 104381. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Wang, R.; Wang, H.; Ding, Q.; Zhou, X.; Wang, J.; Zhang, K.; Long, Y.; Lu, S.; Hong, T.; et al. HOXD-AS1 promotes the epithelial to mesenchymal transition of ovarian cancer cells by regulating miR-186-5p and PIK3R3. J. Exp. Clin. Cancer Res. 2019, 38, 110. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.; Jemal, A. Cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beg, A.; Parveen, R. Gene Expression Signature: An Influential Access to Drug Discovery in Ovarian Cancer. In Computational Intelligence in Oncology; Springer: Singapore, 2022; pp. 271–284. [Google Scholar]
- Dochez, V.; Caillon, H.; Vaucel, E.; Dimet, J.; Winer, N.; Ducarme, G. Biomarkers and algorithms for diagnosis of ovarian cancer: CA125, HE4, RMI and ROMA, a review. J. Ovarian Res. 2019, 12, 28. [Google Scholar] [CrossRef] [Green Version]
- Cannistra, S.A. Cancer of the ovary. N. Engl. J. Med. 2004, 351, 2519–2529. [Google Scholar] [CrossRef]
- Beg, A.; Parveen, R. Review of Bioinformatics Tools and Techniques to Accelerate Ovarian Cancer Research. Int. J. Bioinform. Intell. Comput. 2022, 1, 1–9. [Google Scholar]
- Beg, A.; Khan, F.I.; Lobb, K.A.; Islam, A.; Ahmad, F.; Hassan, M.I. High throughput screening, docking, and molecular dynamics studies to identify potential inhibitors of human calcium/calmodulin-dependent protein kinase IV. J. Biomol. Struct. Dyn. 2019, 37, 2179–2192. [Google Scholar] [CrossRef]
- Beg, A.; Parveen, R. Role of bioinformatics in cancer research and drug development. In Translational Bioinformatics in Healthcare and Medicine; Elsevier: Amsterdam, The Netherlands, 2021; pp. 141–148. [Google Scholar]
- Lan, H.; Lu, H.; Wang, H.; Jin, H. MicroRNAs as potential biomarkers in cancer: Opportunities and challenges. BioMed Res. Int. 2015, 2015, 125094. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-Y.; Li, C.-J.; Lin, L.-T.; Tsui, K.-H. Multi-omics analysis identifying key biomarkers in ovarian cancer. Cancer Control 2020, 27, 1073274820976671. [Google Scholar] [CrossRef]
- Beg, A.; Parveen, R.; Fouad, H.; Yahia, M.E.; Hassanein, A.S. Role of different non-coding RNAs as ovarian cancer biomarkers. J. Ovarian Res. 2022, 15, 1–11. [Google Scholar] [CrossRef]
- Ahmad, S.; Ahmed, M.M.; Hasan, P.M.Z.; Sharma, A.; Bilgrami, A.L.; Manda, K.; Ishrat, R.; Ali Syed, M. Identification and validation of potential miRNAs, as biomarkers for sepsis and associated lung injury: A network-based approach. Genes 2020, 11, 1327. [Google Scholar] [CrossRef] [PubMed]
- Kinose, Y.; Sawada, K.; Nakamura, K.; Kimura, T. The role of microRNAs in ovarian cancer. BioMed Res. Int. 2014, 2014, 249393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafat, M.; Moraghebi, M.; Afsa, M.; Malekzadeh, K. The outstanding role of miR-132-3p in carcinogenesis of solid tumors. Hum. Cell 2021, 34, 1051–1065. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, P.D.; Margiotti, K.; Fabiani, M.; Barros-Filho, M.C.; Sparacino, D.; Cima, A.; Longo, S.A.; Cupellaro, M.; Mesoraca, A.; Giorlandino, C. Multi-analytical test based on serum miRNAs and proteins quantification for ovarian cancer early detection. PLoS ONE 2021, 16, e0255804. [Google Scholar] [CrossRef]
- Lopacinska-Jørgensen, J.; Oliveira, D.V.N.P.; Novotny, C.W.; Høgdall, C.K.; Høgdall, E.V. Integrated microRNA and mRNA signatures associated with overall survival in epithelial ovarian cancer. PLoS ONE 2021, 16, e0255142. [Google Scholar] [CrossRef]
- Prahm, K.P.; Høgdall, C.; Karlsen, M.A.; Christensen, I.J.; Novotny, G.W.; Høgdall, E. Identification and validation of potential prognostic and predictive miRNAs of epithelial ovarian cancer. PloS ONE 2018, 13, e0207319. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-J.; Lin, L.-T.; Chu, P.-Y.; Chiang, A.-J.; Tsai, H.-W.; Chiu, Y.-H.; Huang, M.-S.; Wen, Z.-H.; Tsui, K.-H. Identification of novel biomarkers and candidate drug in ovarian cancer. J. Pers. Med. 2021, 11, 316. [Google Scholar] [CrossRef]
- Gentleman, R.; Carey, V.; Huber, W.; Irizarry, R.; Dudoit, S. Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: Berlin/Heidelberg, Germany, 2005; Volume 1. [Google Scholar]
- Sethupathy, P.; Megraw, M.; Hatzigeorgiou, A.G. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat. Methods 2006, 3, 881–886. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.-H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3. 0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef]
- Tokar, T.; Pastrello, C.; Rossos, A.E.M.; Abovsky, A.M.; Hauschild, A.C.; Tsay, M.; Lu, R.; Jurisica, I. mirDIP 4.1—Integrative database of human microRNA target predictions. Nucleic Acids Res. 2018, 46, D360–D370. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Cho, J.-W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.-Y.; Xia, M.; Miao, Y.-R.; Luo, M.; Zhang, Q.; Guo, A.-Y. FFLtool: A web server for transcription factor and miRNA feed forward loop analysis in human. Bioinformatics 2020, 36, 2605–2607. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Song, P.; Zhao, Y.; An, N.; Xia, Y.; Qi, Y.; Zhao, H.; Ge, J. miR-600 promotes ovarian cancer cells stemness, proliferation and metastasis via targeting KLF9. J. Ovarian Res. 2022, 15, 52. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Wang, K.X.; Xu, H.; Hong, Y. Integrative miRNA analysis identifies hsa-miR-3154, hsa-miR-7-3, and hsa-miR-600 as potential prognostic biomarker for cervical cancer. J. Cell. Biochem. 2018, 119, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Yong, W.; Xuan, Z.; Wang, B.; Zhang, D.; Zhang, C.; Sun, Y. Hsa_circ_0071589 promotes carcinogenesis via the miR-600/EZH2 axis in colorectal cancer. Biomed. Pharmacother. 2018, 102, 1188–1194. [Google Scholar] [CrossRef]
- Li, C.; Gao, X.; Zhao, Y.; Chen, X. High Expression of circ_0001821 Promoted Colorectal Cancer Progression Through miR-600/ISOC1 Axis. Biochem. Genet. 2022, 1–18. [Google Scholar] [CrossRef]
- Pishbin, F.; Ziamajidi, N.; Abbasalipourkabir, R.; Najafi, R.; Farhadian, M. Correlation of miR-600 with WT1 expression and its potential clinical significance in breast cancer. Pers. Med. 2021, 18, 31–42. [Google Scholar] [CrossRef]
- Chi, Y.; Luo, Q.; Song, Y.; Yang, F.; Wang, Y.; Jin, M.; Zhang, D. Circular RNA circPIP5K1A promotes non-small cell lung cancer proliferation and metastasis through miR-600/HIF-1α regulation. J. Cell. Biochem. 2019, 120, 19019–19030. [Google Scholar] [CrossRef]
- Arnold, J.; Engelmann, J.C.; Schneider, N.; Bosserhoff, A.K.; Kuphal, S. miR-488-5p and its role in melanoma. Exp. Mol. Pathol. 2020, 112, 104348. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.-T.; Yu, S.-Y.; Li, S.-C.; Lam, H.-C.; Chang, H.-T.; Chen, W.-S.; Yeh, C.-Y.; Hung, S.-F.; Liu, T.-C.; Wu, T.; et al. MicroRNA-324 in human cancer: miR-324-5p and miR-324-3p have distinct biological functions in human cancer. Anticancer Res. 2016, 36, 5189–5196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Wu, F.; Yang, H.; Deng, X.; Yuan, Y. MiR-9-5p promotes cell growth and metastasis in non-small cell lung cancer through the repression of TGFBR2. Biomed. Pharmacother. 2017, 96, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; He, Q.; Liang, J.; Li, W.; Su, Q.; Chen, Z.; Wan, Q.; Zhou, X. miR-31-5p is a potential circulating biomarker and therapeutic target for oral cancer. Mol. Ther.-Nucleic Acids 2019, 16, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Zhang, L.; Sun, W.; Guan, W.; Lin, Q.; Ren, W.; Zhang, J.; Xu, G. Cytidine monophosphate kinase is inhibited by the TGF-β signalling pathway through the upregulation of miR-130b-3p in human epithelial ovarian cancer. Cell. Signal. 2017, 35, 197–207. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.; Jiang, J.; Qiao, Z. The lncRNA SNHG15/miR-18a-5p axis promotes cell proliferation in ovarian cancer through activating Akt/mTOR signaling pathway. J. Cell. Biochem. 2020, 121, 4699–4710. [Google Scholar] [CrossRef]
- Sharma, B.; Randhawa, V.; Vaiphei, K.; Gupta, V.; Dahiya, D.; Agnihotri, N. Expression of miR-18a-5p, miR-144-3p, and miR-663b in colorectal cancer and their association with cholesterol homeostasis. J. Steroid Biochem. Mol. Biol. 2021, 208, 105822. [Google Scholar] [CrossRef]
- Ichihara, A.; Wang, Z.; Masatoshi, J.; Izuno, Y.; Shimozono, N.; Yamane, K.; Akihiko, Y. Upregulation of miR-18a-5p contributes to epidermal necrolysis in severe drug eruptions. J. Allergy Clin. Immunol. 2014, 133, 1065–1074. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, M.-J.; Ren, A.-M.; Wu, H.-F.; Han, W.-M.; Tan, R.-Y.; Tu, R.-Q. A ten-microRNA signature identified from a genome-wide microRNA expression profiling in human epithelial ovarian cancer. PloS One 2014, 9, e96472. [Google Scholar] [CrossRef]
- Yoshida, K.; Yokoi, A.; Kagawa, T.; Oda, S.; Hattori, S.; Tamauchi, S.; Ikeda, Y.; Nobuhisa Yoshikawa, N.; Nishino, K. Unique miRNA profiling of squamous cell carcinoma arising from ovarian mature teratoma: Comprehensive miRNA sequence analysis of its molecular background. Carcinogenesis 2019, 40, 1435–1444. [Google Scholar] [CrossRef]
- Soonthornchai, W.; Tangtanatakul, P.; Meesilpavikkai, K.; Dalm, V.; Kueanjinda, P.; Wongpiyabovorn, J. MicroRNA-378a-3p is overexpressed in psoriasis and modulates cell cycle arrest in keratinocytes via targeting BMP2 gene. Sci. Rep. 2021, 11, 14186. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Jin, X.; Wang, H.; Zhou, Q.; Li, G.; Lu, X. Expression, clinical significance, and prospective pathway signaling of miR-501-3p in ovarian cancer based on database and informatics analysis. Int. J. Gen. Med. 2021, 14, 5193. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, Y.; Chen, C.; Xue, Y.; Zheng, S.; Cai, L. Hsa-let-7c-5p, hsa-miR-130b-3p, and hsa-miR-142-3p as Novel miRNA Biomarkers for Melanoma Progression. Genet. Res. 2022, 2022, 5671562. [Google Scholar] [CrossRef] [PubMed]
- Bayat, Z.; Farhadi, Z.; Taherkhani, A. Identification of potential biomarkers associated with poor prognosis in oral squamous cell carcinoma through integrated bioinformatics analysis: A pilot study. Gene Rep. 2021, 24, 101243. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, K.; Liu, X.; Zhao, X.; Zhao, T.; Lu, G. Exosomal mir-625-3p derived from hypoxic lung cancer cells facilitates metastasis by targeting SCAI. Mol. Biol. Rep. 2022, 49, 9275–9281. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhao, S.; Shi, Z.; Cao, L.; Liu, J.; Pan, T.; Zhou, D.; Zhang, Y. Chemotherapy-elicited exosomal miR-378a-3p and miR-378d promote breast cancer stemness and chemoresistance via the activation of EZH2/STAT3 signaling. J. Exp. Clin. Cancer Res. 2021, 40, 120. [Google Scholar] [CrossRef]
- Sun, P.; Wei, L.; Sehouli, J.; Denkert, C.; Zhao, D.; Gao, M.; Sun, X.; Litchtenegger, W. Role of estrogen receptor-related receptors alpha, beta and gamma in ovarian cancer cells. Zhonghua Fu Chan Ke Za Zhi 2005, 40, 544–548. [Google Scholar]
- Schüler-Toprak, S.; Weber, F.; Skrzkypczak, M.; Ortmankn, O.; Treeck, O. Expression of estrogen-related receptors in ovarian cancer and impact on survival. J. Cancer Res. Clin. Oncol. 2021, 147, 2555–2567. [Google Scholar] [CrossRef]
- Wang, C.; Chung, B.; Yan, H.; Wang, H.; Lee, S.; Pitt, G. Structural analyses of Ca2+/CaM interaction with NaV channel C-termini reveal mechanisms of calcium-dependent regulation. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Antonsson, B.; Conti, F.; Ciavatta, A.; Montessuit, S.; Lewis, S.; Martinou, I.; Bernasconi, L.; Bernard, A.; Mermod, J.-J.; Mazzei, G.; et al. Inhibition of Bax channel-forming activity by Bcl-2. Science 1997, 277, 370–372. [Google Scholar] [CrossRef]
- Pouponnot, C.; Hmitou, I.; Rocques, N.; Lecoin, L.; Druillennec, S.; Felder-Schmittbuhl, M.; Eychène, A. Cell context reveals a dual role for Maf in oncogenesis. Oncogene 2006, 25, 1299–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.H.; Luo, Y.; Wei, R.; Yin, J.; Qin, Z.; Lu, L.; Ma, W. CircZNF532 knockdown protects retinal pigment epithelial cells against high glucose-induced apoptosis and pyroptosis by regulating the miR-20b-5p/STAT3 axis. J. Diabetes Investig. 2022, 13, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Shingai, T.; Ikeda, W.; Kakunaga, S.; Morimoto, K.; Takekuni, K.; Itoh, S.; Satoh, K.; Takeuchi, M.; Morito, I.; Takai, M. Implications of nectin-like molecule-2/IGSF4/RA175/SgIGSF/TSLC1/SynCAM1 in cell-cell adhesion and transmembrane protein localization in epithelial cells. J. Biol. Chem. 2003, 278, 35421–35427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boles, K.S.; Barchet, W.; Diacovo, T.; Cella, M.; Cella, M. The tumor suppressor TSLC1/NECL-2 triggers NK-cell and CD8+ T-cell responses through the cell-surface receptor CRTAM. Blood 2005, 106, 779–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatanaka, T.; Higashino, F.; Tei, K.; Yasuda, M. The neural ELAVL protein HuB enhances endogenous proto-oncogene activation. Biochem. Biophys. Res. Commun. 2019, 517, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Yang, L.; Stallcup, M.R. Hormone-independent transcriptional activation and coactivator binding by novel orphan nuclear receptor ERR3. J. Biol. Chem. 1999, 274, 22618–22626. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.-J.; Mattioli, M.; Morse, H.; Dalla-Favera, R. c-MYC activates protein kinase A (PKA) by direct transcriptional activation of the PKA catalytic subunit beta (PKA-Cβ) gene. Oncogene 2002, 21, 7872–7882. [Google Scholar] [CrossRef] [Green Version]
- Mitsopoulos, C.; Zihni, C.; Garg, R.; Ridley, A.; Morris, J.D.H. The prostate-derived sterile 20-like kinase (PSK) regulates microtubule organization and stability. J. Biol. Chem. 2003, 278, 18085–18091. [Google Scholar] [CrossRef] [Green Version]
- Timm, T.; Li, X.; Biernat, J.; Jiao, J.; Mandelkow, E.; Vandekerckhove, J.; Mandelkow, E. MARKK, a Ste20-like kinase, activates the polarity-inducing kinase MARK/PAR-1. EMBO J. 2003, 22, 5090–5101. [Google Scholar] [CrossRef] [Green Version]
- Johne, C.; Matenia, D.; Li, X.; Timm, T.; Balusamy, K.; Mandelkow, E. Spred1 and TESK1—Two new interaction partners of the kinase MARKK/TAO1 that link the microtubule and actin cytoskeleton. Mol. Biol. Cell 2008, 19, 1391–1403. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | miRNA ID | Log FC | p-Value |
|---|---|---|---|
| Up-regulated | |||
| 20501181 | hsa-miR-130b-3p | 2.00379 | 0.000894 |
| 20500132 | hsa-miR-18a-5p | 2.172073 | 0.006009 |
| 20500450 | hsa-miR-182-5p | 3.910191 | 0.006054 |
| 20500455 | hsa-miR-187-3p | 2.440276 | 0.008152 |
| 20501243 | hsa-miR-378a-3p | 2.4392 | 0.017887 |
| Down-regulated | |||
| 20503878 | hsa-miR-501-3p | −2.589788641 | 2.21 × 10−6 |
| 20517704 | hsa-miR-4324 | −2.773375005 | 3.00 × 10−5 |
| 20503876 | hsa-miR-500a-3p | −2.197486478 | 8.40 × 10−5 |
| 20504569 | hsa-miR-1271-5p | −2.517825869 | 0.00020669 |
| 20504431 | hsa-miR-660-5p | −2.392424335 | 0.00025731 |
| Genes | Chromosomal Location | Aliases | Expression | Gene Structure | Functions | Reference | |
|---|---|---|---|---|---|---|---|
| Downregulated Genes | SCN2A (sodium voltage-gated channel alpha subunit 2) | 2q24.3 | EA9; HBA; NAC2; BFIC3; BFIS3; Nav1.2; SCN2A1; | Biased expression in kidney and brain | 120kb in size and have 29 exons | Enables protein binding and voltage gated sodium channel activity | [52] |
| BCL2 (Apoptosis regulator) | 20q11.21 18q21.33 | Bcl-2; PPP1R50 | Broad expression is found in thyroid, spleen, and 20 different tissues such as ovary, prostate, colon, skin endometrium placenta, lungs, etc. | 6exons | Enables channel activity, protease binding, protein binding, etc. | [53] | |
| MAF (MAF bZIPtranscription factor) | 16q23.2 | CCA4; AYGRP; c-MAF; CTRCT21 | Ubiquitous expression in kidney, endometrium, and 24 other tissues | 7 exons | Role in RNA polymerase II-specific oncogenesis activator activity | [54] | |
| ZNF532 (zinc finger protein 532) | 18q21.32 | - | Ubiquitous expression in endometrium, prostate, and 22 other tissues | 21 exons | Enables DNA binding and metal ion binding, and along with ncRNAs it is involved in apoptosis | [55] | |
| CADM1 | 11q23.3 | BL2; ST17; IGSF4; NECL2; RA175; TSLC1; | Broad expression in lung, thyroid, and 23 other tissues | 13 exons | Enables signaling receptor binding, PDZ domain binding, and protein binding | [56,57] | |
| ELAVL2 | 9p21.3 | HUB; HELN1; HEL-N1 | Biased expression in testis and brain | 20 exons | Enables RNA binding, protein binding, and activation of protooncogenes | [58] | |
| ESRRG | 1q41 | ERR3; ERRg; NR3B3; ERRgamma; ERR-gamma | Biased expression in kidney, stomach, and 12 other tissues | 28exons | Enables nuclear receptor activity and protein binding | [59] | |
| Upregulated Genes | PRKACB (protein kinase cAMP-activated catalytic subunit beta) | 1p31.1 | CAFD2; PKACB; PKA C-beta | Broad expression in brain, colon, and 22 other tissues | 18 exons | Enables ATP binding and caMP dependent protein kinase activity | [60] |
| TAOK1 TAO kinase 1 | 17q11.2 | PSK2; TAO1; KFC-B; MARKK; PSK-2; MAP3K16 | Ubiquitous expression in brain, thyroid, and 25 other tissues | 20 exons | Enables ATP binding, kinase activity, and protein binding | [61,62,63]) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beg, A.; Parveen, R.; Fouad, H.; Yahia, M.E.; Hassanein, A.S. Identification of Driver Genes and miRNAs in Ovarian Cancer through an Integrated In-Silico Approach. Biology 2023, 12, 192. https://doi.org/10.3390/biology12020192
Beg A, Parveen R, Fouad H, Yahia ME, Hassanein AS. Identification of Driver Genes and miRNAs in Ovarian Cancer through an Integrated In-Silico Approach. Biology. 2023; 12(2):192. https://doi.org/10.3390/biology12020192
Chicago/Turabian StyleBeg, Anam, Rafat Parveen, Hassan Fouad, M. E. Yahia, and Azza S. Hassanein. 2023. "Identification of Driver Genes and miRNAs in Ovarian Cancer through an Integrated In-Silico Approach" Biology 12, no. 2: 192. https://doi.org/10.3390/biology12020192
APA StyleBeg, A., Parveen, R., Fouad, H., Yahia, M. E., & Hassanein, A. S. (2023). Identification of Driver Genes and miRNAs in Ovarian Cancer through an Integrated In-Silico Approach. Biology, 12(2), 192. https://doi.org/10.3390/biology12020192