
AlphaFold2 Reveals Structural Patterns of Seasonal Haplotype Diversification in SARS-CoV-2 Spike Protein Variants



Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. AlphaFold2 Modeling
2.2. Structural Analysis
2.3. Validating the AlphaFold2 Models
3. Results
3.1. Full-Length S-Protein Analysis Using Backbone RMSD
3.2. Characterization of Types of Structural Deviation
3.3. Regional Analysis with TM Scores Using USalign
3.4. Significant Structural Changes Suggestive of Recruitments across VOCs and Haplotypes
3.5. Benchmarking AlphaFold2 Reference Structures against Experimental Cryo-EM Models
4. Discussion
4.1. Structural Changes Involve Regions of Protein Flexibility
4.2. Structural Changes Are Important for Disease Mitigation
4.3. Recruitments of Structural Changes That Improve Molecular Function Appear Pervasive
4.4. Haplotypes Altering NTD Structure Impact Seasonal Behavior
4.5. Patterns of Molecular Evolution Can Be Synergistic or Antagonistic
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ciotti, M.; Ciccozzi, M.; Terrinoni, A.; Jiang, W.-C.; Wang, C.-B. The COVID-19 pandemic. Crit. Rev. Clin. Lab. Sci. 2020, 57, 365–388. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Low, Z.Y.; Yip, A.J.W.; Sharma, A.; Lal, S.K. SARS coronavirus outbreaks past and present–A comparative analysis of SARS-CoV-2 and its predecessors. Virus Genes 2021, 57, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Hodgens, A.; Gupta, V. Severe Acute Respiratory Syndrome; StatPearls: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK558977/ (accessed on 31 December 2023).
- Ramadan, N.; Shaib, H. Middle East respiratory syndrome coronavirus (MERS-CoV): A review. Germs 2019, 9, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Inf. 2021, 54, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Müller, M.A.; Allen, P.; Soilleux, E. Evidence that TMPRSS2 activates the severe acute respira-tory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, 3000003. [Google Scholar] [CrossRef] [PubMed]
- Amicone, M.; Borges, V.; Alves, M.J.; Isidro, J.; Zé-Zé, L.; Duarte, S.; Vieira, L.; Guiomar, R.; Gomes, J.P.; Gordo, I. Mutation rate of SARS-CoV-2 and emergence of mutators dur-ing experimental evolution. Evol. Med. Public Health 2022, 10, 142–155. [Google Scholar] [CrossRef]
- MacLean, O.A.; Orton, R.J.; Singer, J.B.; Robertson, D.L. No evidence for distinct types in the evolution of SARS-CoV-2. Virus Evol. 2020, 6, 034. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; de Silva, T.I.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, T.; Ali, M.A.; Caetano-Anollés, K.; Caetano-Anollés, G. Seasonal effects decouple SARS-CoV-2 haplotypes worldwide. F1000Research 2023, 12, 267. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Peng, P.; Cao, X.; Wu, K.; Chen, J. Increased immune escape of the new SARS-CoV-2 variant of concern Omicron. Cell. Mol. Immunol. 2022, 19, 293–295. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T. Structure, function, and antigenicity of the SARS-CoV-2 ppike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, 3055. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Yang, Q.; Jian, X.; Syed, A.A.S.; Fahira, A.; Zheng, C. Structural comparison and drug screening of spike proteins of ten SARS-CoV-2 variants. Research 2022, 2022, 9781758. [Google Scholar] [CrossRef]
- Heo, L.; Feig, M. Modeling of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) proteins by machine learning and physics-based refinement. bioRxiv 2020. 2020.03.25.008904. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Thornton, J.M. PDBsum extras: SARS-CoV-2 and AlphaFold models. Protein Sci. 2022, 31, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Singanallur, N.B.; Vuren, P.J.; McAuley, A.J.; Bruce, M.P.; Kuiper, M.J. At least three doses of leading vaccines essential for neutralisation of SARS-CoV-2 Omicron variant. Front. Immunol. 2022, 13, 883612. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.-M.A.; Esswein, S.R. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Caetano-Anollés, K.; Hernandez, N.; Mughal, F.; Tomaszewski, T.; Caetano-Anollés, G. The seasonal behaviour of COVID-19 and its galectin-like culprit of the viral spike. In Methods in Microbiology; Pavia, C.S., Gurtler, V., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 27–81. [Google Scholar]
- Tang, T.; Jaimes, J.A.; Bidon, M.K.; Straus, M.R.; Daniel, S. Proteolytic activation of SARS-CoV-2 spike at the S1/S2 boundary: Potential role of proteases beyond furin. ACS Infect. Dis. 2021, 7, 264–272. [Google Scholar] [CrossRef]
- Tomaszewski, T.; DeVries, R.S.; Dong, M.; Bhatia, G.; Norsworthy, M.D.; Zheng, X.; Caetano-Anollés, G. New pathways of mutational change in SARS-CoV-2 proteomes involve regions of intrinsic disorder important for virus replication and release. Evol. Bioinform. 2020, 16, 1176934320965149. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data – from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Zhang, C.; Shine, M.; Pyle, A.M.; Zhang, Y. US-Align: Universal structure alignments of proteins, nucleic acids, and macromolecular complexes. Nat. Methods 2022, 19, 1109–1115. [Google Scholar] [CrossRef]
- Zemla, A. LGA: A Method for finding 3D Similarities in protein structures. Nucleic Acids Res. 2003, 31, 3370–3374. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins: Struct. Funct. Bioinf. 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Yang, Q.; Syed, A.A.S.; Fahira, A.; Shi, Y. Structural analysis of the SARS-CoV-2 Omicron variant proteins. Research 2021, 2021, 9769586. [Google Scholar] [CrossRef] [PubMed]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017, 27, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, T.; Gurtler, V.; Caetano-Anollés, K.; Caetano-Anollés, G. The emergence of SARS-CoV-2 Variants of concern in Australia by haplotype coalescence reveals a continental link to COVID-19 seasonality. In Methods in Microbiology; Pavia, C.S., Gurtler, V., Eds.; COVID-19: Biomedical Perspectives; Academic Press: Cambridge, MA, USA, 2022; Volume 50, pp. 233–268. [Google Scholar]
- Ito, J.; Suzuki, R.; Uriu, K.; Itakura, Y.; Zahradnik, J.; Kimura, K.T.; Deguchi, S.; Wang, L.; Lytras, S.; Tamura, T.; et al. Convergent evolution of SARS-CoV-2 Omicron subvariants leading to the emergence of BQ.1.1 variant. Nat. Commun. 2023, 14, 2671. [Google Scholar] [CrossRef] [PubMed]
- Neto, D.F.d.L.; Fonseca, V.; Jesus, R.; Dutra, L.H.; de Oliveria Portela, L.M.; Freitas, C.; Fillizola, E.; Soares, B.; de Abreu, A.L.; Twiari, S.; et al. Molecular dynamics simulations of the SARS-CoV-2 spike protein and variants of concern: Structural evidence for convergent adaptive evolution. J. Biomol. Struct. Dyn. 2023, 41, 5789–5801. [Google Scholar] [CrossRef]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.; Bhardwaj, T.; Shegane, M.; Gehi, B.R.; Kumar, P. Understanding COVID-19 via comparative analysis of dark proteomes of SARS-CoV-2, human SARS and bat SARS-like coronaviruses. Cell. Mol. Life Sci. 2021, 78, 1655–1688. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L. Cryo-EM structure of the 2019-nCoV spike in the pre-fusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Zhu, Y.; Feng, F.; Hu, G.; Wang, Y.; Yu, Y.; Zhu, Y.; Xu, W.; Cai, X.; Sun, Z.; Han, W.; et al. A genome-wide CRISPR screen identifies host factors that regulate SARS-CoV-2 entry. Nat. Commun. 2021, 12, 961. [Google Scholar] [CrossRef]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Hadizadeh, N.; Naderi, M.; Khezri, J.; Yazdani, M.; Shamsara, M.; Hashemi, E. Appraisal of SARS-CoV-2 mutations and their impact on vaccination efficacy: An overview. J. Diabetes Metab. Dis. 2022, 21, 1763–1783. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y. A neutralizing human antibody binds to the N-terminal domain of the spike protein of SARS-CoV-2. Science 2020, 369, 650–655. [Google Scholar] [CrossRef]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, G.; Guo, Y.; Zhou, T.; Gorman, J.; Lee, M. Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a sngle supersite. Cell Host Microbe 2021, 29, 819–833.e7. [Google Scholar] [CrossRef]
- Qing, E.; Kicmal, T.; Kumar, B.; Hawkins, G.M.; Timm, E.; Perlman, S.; Gallagher, T. Dynamics of SARS-CoV-2 spike proteins in cell entry: Control elements in the amino-terminal domains. mBio 2021, 12, e0159021. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Meng, B.; Datir, R.; Choi, J.; Bradley, J.R.; Smith, K.G.C.; Lee, J.H.; Gupta, R.K.; Baker, S.; Dougan, G.; Hess, C.; et al. SARS-CoV-2 spike N-terminal domain modulates TMPRSS2-dependent viral entry and fusogenicity. Cell Rep. 2022, 40, 111220. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.T.M.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Li, Y.; Ma, M.; Lei, Q.; Wang, F.; Hong, W.; Lai, D.; Hou, H.; Xu, Z.; Zhang, B.; Chen, H.; et al. Linear epitope landscape of the SARS-CoV-2 spike protein constructed from 1051 COVID-19 patients. Cell Rep. 2021, 34, 108915. [Google Scholar] [CrossRef] [PubMed]
- Poh, C.M.; Carissimo, G.; Wang, B.; Amrun, S.N.; Lee, C.Y.-P.; Chee, R.S.-L.; Fong, S.-W.; Yeo, N.K.-W.; Lee, W.-H.; Torres-Ruesta, A.; et al. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat. Commun. 2020, 11, 2806. [Google Scholar] [CrossRef] [PubMed]
- Tarigan, S.; Dharmayanti, N.L.P.I.; Sugiartanti, D.; Putri, R.; Andriani; Nuradji, H.; Robinson, M.; Wiendayanthi, N.; Djufri, F. Characterization of two linear epitopes SARS CoV-2 spike protein formulated in tandem repeat. PLoS ONE 2023, 18, e0280627. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y.; et al. Potent Binding of 2019 Novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 2020, 9, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M. Fusion peptides and the mechanism of viral fusion. Biochim. Biophys. Acta Biomembr. 2003, 1614, 116–121. [Google Scholar] [CrossRef]
- Zhu, J.; Xiao, G.; Xu, Y.; Yuan, F.; Zheng, C.; Liu, Y.; Yan, H.; Cole, D.K.; Bell, J.I.; Rao, Z.; et al. Following the rule: Formation of the 6-helix bundle of the fusion core from severe acute respiratory syndrome coronavirus spike protein and identification of potent peptide inhibitors. Biochem. Biophys. Res. Commun. 2004, 319, 283–288. [Google Scholar] [CrossRef]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.-T.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A.; et al. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef]
- Guruprasad, K. Mutations in Human SARS-CoV-2 spike proteins, potential drug binding and epitope sites for COVID-19 therapeutics development. Curr. Res. Struct. Biol. 2022, 4, 41–50. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Vu, M.N.; Alvarado, R.E.; Morris, D.R.; Lokugamage, K.G.; Zhou, Y.; Morgan, A.L.; Estes, L.K.; McLeland, A.M.; Schindewolf, C.; Plante, J.A.; et al. Loss-of-function mutation in Omicron variants reduces spike protein expression and attenuates SARS-CoV-2 infection. bioRxiv 2023. 2023.04.17.536926. [Google Scholar] [CrossRef]
- McCallum, M.; Marco, A.; Lempp, F.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347. [Google Scholar] [CrossRef]
- Suryadevara, N.; Shrihari, S.; Gilchuk, P.; VanBlargan, L.A.; Binshtein, E.; Zost, S.J.; Nargi, R.S.; Sutton, R.E.; Winkler, E.S.; Chen, E.C. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell 2021, 184, 2316–2331. [Google Scholar] [CrossRef]
- Burra, P.; Soto-Díaz, K.; Chalen, I.; Gonzalez-Ricon, R.J.; Istanto, D.; Caetano-Anollés, G. Temperature and latitude correlate with SARS-COV-2 epidemiological variables but not with genomic change worldwide. Evol. Bioinform. 2021, 17, 1176934321989695. [Google Scholar] [CrossRef]
- Hernandez, N.; Caetano-Anollés, G. Worldwide correlations support COVID-19 seasonal behavior and impact of global change. Evol. Bioinform. 2023, 19, 11769343231169377. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, R.W.; Lewer, D.; Beale, S.; Johnson, A.M.; Zambon, M.; Hayward, A.C.; Fragaszy, E.B. Flu watch group seasonality and immunity to laboratory-confirmed seasonal coronaviruses (HCoV-NL63, HCoV-OC43, and HCoV-229E): Results from the flu watch cohort study. Wellcome Open Res. 2020, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Nickbakhsh, S.; Ho, A.; Marques, D.F.P.; McMenamin, J.; Gunson, R.N.; Murcia, P.R. Epidemiology of seasonal coronaviruses: Establishing the context for the emergence of coronavirus disease 2019. J. Infect. Dis. 2020, 222, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Nair, H. Global seasonality of human seasonal coronaviruses: A clue for postpandemic circulating season of severe acute respiratory syndrome coronavirus 2? J. Infect. Dis. 2020, 222, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Price, R.H.M.; Graham, C.; Ramalingam, S. Association between Viral Seasonality and meteorological factors. Sci. Rep. 2019, 9, 929. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.M.; Winn, A.; Dahl, R.M.; Kniss, K.L.; Silk, B.J.; Killerby, M.E. Seasonality of common human coronaviruses, United States, 2014–20211. Emerg. Infect. Dis. 2022, 28, 1970–1976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiao, T.; Cai, Y.; Chen, B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021, 50, 173–182. [Google Scholar] [CrossRef]
- Cummings, R.D.; Liu, F.-T. Galectins. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; ISBN 978-0-87969-770-9. [Google Scholar]
- Wu, Y.; Zhou, Z.; Wang, J.; Luo, J.; Wang, L.; Zhang, Y. Temperature regulates the recognition activities of a galectin to pathogen and symbiont in the scleractinian coral Pocillopora damicornis. Dev. Comp. Immunol. 2019, 96, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Ben-Haim, Y.; Zicherman-Keren, M.; Rosenberg, E. Temperature-regulated bleaching and lysis of the coral Pocillopora damicomis by the novel pathogen Vibrio coralliilyticus. Appl. Environ. Microbiol. 2003, 69, 4236–4242. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.L.; Kumar, K. Investigation of the effect of temperature on the structure of SARS-CoV-2 spike protein by molecular dynamics simulations. Front. Mol. Biosci. 2020, 7, 583523. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
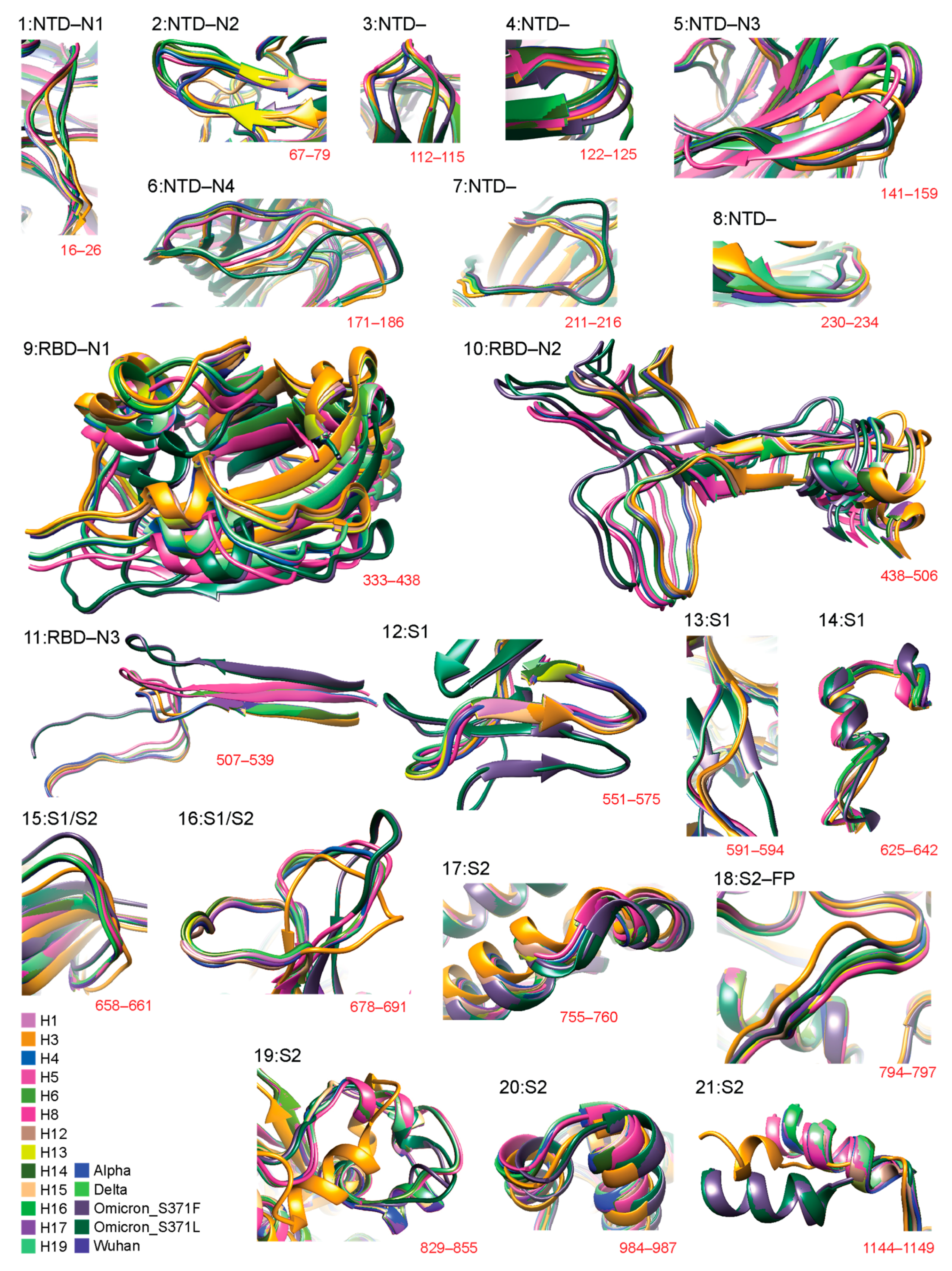{kind=link}
{kind=link}
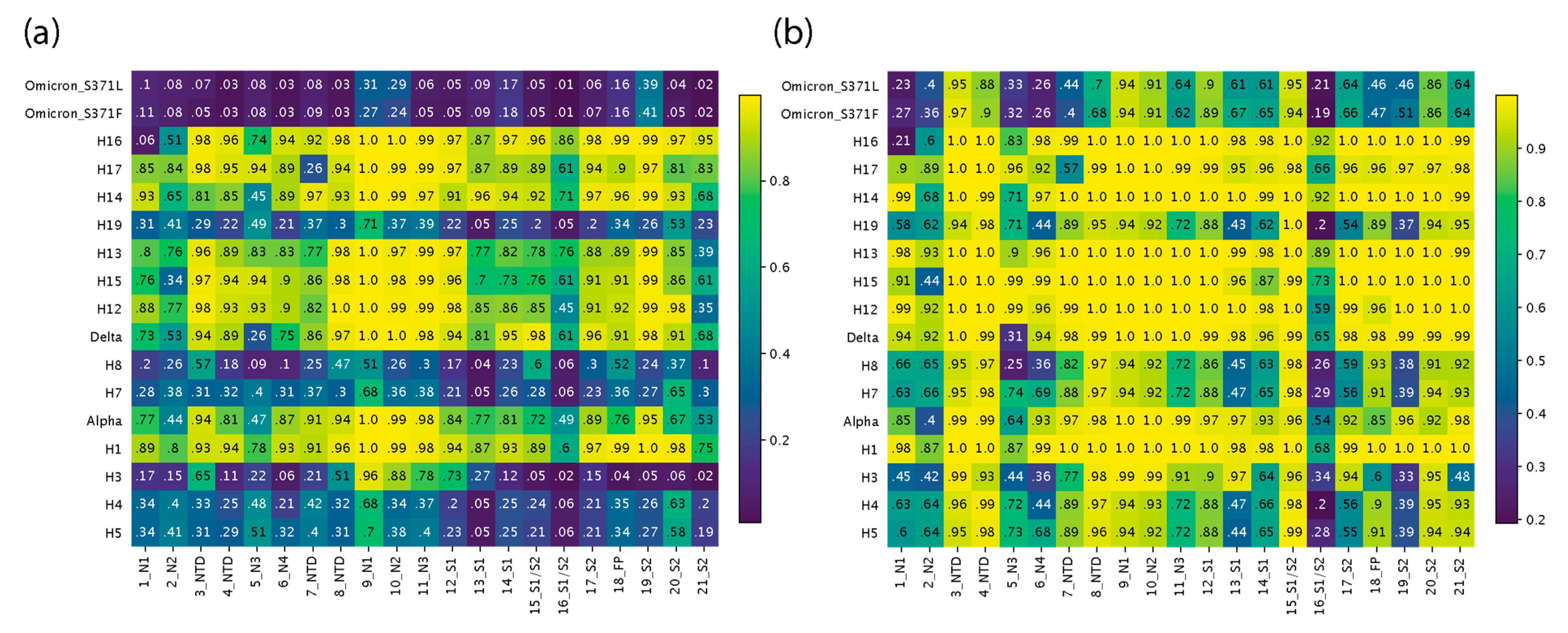{kind=link}
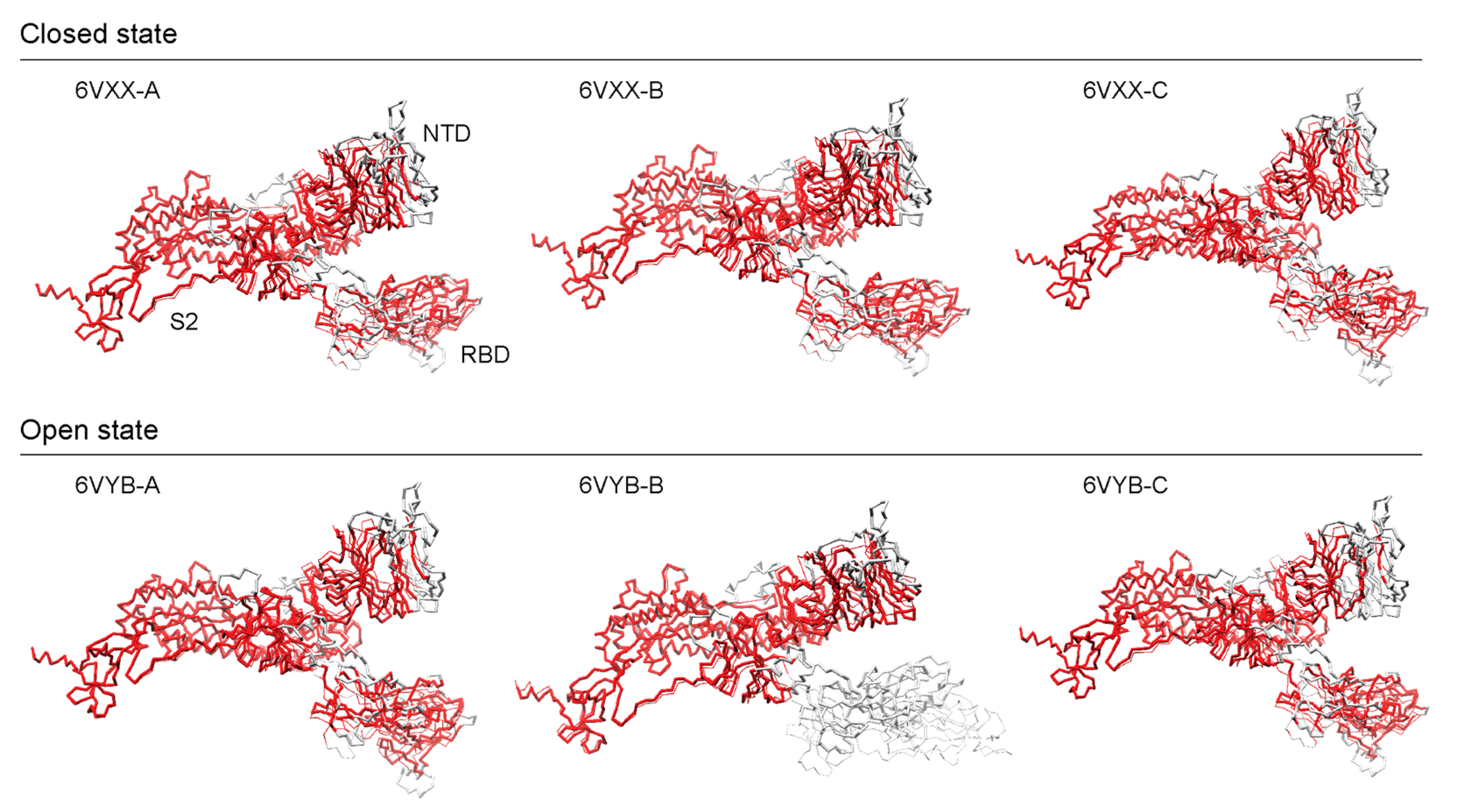{kind=link}
| V/H | 2 | 5 | 6 | 7 | 9 | 10 | 11 | 13 | 14 | 16 | 18 | 19 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Omicron | 1.75 | 6 | 3 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 2 | 3 |
| H16 | 0.5 | 0.25 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| H17 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 | 0 |
| H14 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| H19 | 0.5 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 |
| H13 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| H15 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 |
| H12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0.5 | 0 |
| Delta | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 | 0 |
| H8 | 0.5 | 3 | 1 | 0 | 1.5 | 1.5 | 1.5 | 1 | 1 | 1 | 0 | 1 |
| H7 | 0.5 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 |
| Alpha | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 | 0 |
| H1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 | 0 |
| H3 | 1.5 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 1 | 2 |
| H4 | 0.5 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 |
| H5 | 0.5 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 |
| State-Chain | LGDT-TS (AS2S) | TM Score/L (US-Align Server) | Superimposed RMSD < 5 Å (TM-Score Server) |
|---|---|---|---|
| Closed-A (6VXX-A) L = 972 | 74.434 | *L1: 0.82683, L2: 0.94899 | 2.10/884 |
| Closed-B (6VXX-B) L = 972 | 74.357 | 0.82682, 0.94899 | 2.10/884 |
| Closed-C (6VXX-C) L = 972 | 74.357 | 0.82682, 0.94899 | 2.10/884 |
| Open-A (6VYB-A) L = 966 | 77.019 | 0.82432, 0.95200 | 2.07/894 |
| Open-B (6VYB-B) L = 944 | 65.859 | 0.69501, 0.81421 | 1.43/711 |
| Open-C (6VYB-C) L = 960 | 71.146 | 0.80577, 0.93502 | 2.26/836 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.A.; Caetano-Anollés, G. AlphaFold2 Reveals Structural Patterns of Seasonal Haplotype Diversification in SARS-CoV-2 Spike Protein Variants. Biology 2024, 13, 134. https://doi.org/10.3390/biology13030134
Ali MA, Caetano-Anollés G. AlphaFold2 Reveals Structural Patterns of Seasonal Haplotype Diversification in SARS-CoV-2 Spike Protein Variants. Biology. 2024; 13(3):134. https://doi.org/10.3390/biology13030134
Chicago/Turabian StyleAli, Muhammad Asif, and Gustavo Caetano-Anollés. 2024. "AlphaFold2 Reveals Structural Patterns of Seasonal Haplotype Diversification in SARS-CoV-2 Spike Protein Variants" Biology 13, no. 3: 134. https://doi.org/10.3390/biology13030134
APA StyleAli, M. A., & Caetano-Anollés, G. (2024). AlphaFold2 Reveals Structural Patterns of Seasonal Haplotype Diversification in SARS-CoV-2 Spike Protein Variants. Biology, 13(3), 134. https://doi.org/10.3390/biology13030134