
ATF4 Signaling in HIV-1 Infection: Viral Subversion of a Stress Response Transcription Factor
 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
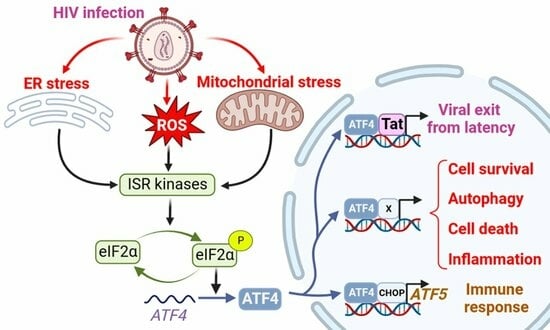
1. Introduction
2. HIV-1 Infection Regulates ATF4
2.1. ATF4 Is Up-Regulated during HIV-1 and SIV Infections
2.2. How Can HIV-1 Regulate ATF4?
2.2.1. HIV-1-Induced ISR/ATF4 Signaling
2.2.2. Mitochondrial Stress Response, ATF4 and HIV-1
2.2.3. The Viral Vpu Protein Stabilizes the ATF4 Protein
2.2.4. HIV-1 Antiretroviral Drugs Induce ATF4 Signaling
3. ATF4 Role during HIV-1 Replication
3.1. ATF4 Positively Regulates HIV-1 Cycle
3.2. How ATF4 Favorizes HIV-1 Replication
3.2.1. ATF4 Binds to the HIV-1 LTR and Promotes Viral Gene Transcription
3.2.2. ATF4, HIV-1 and Apoptosis
3.2.3. ATF4, HIV-1 and Autophagy
3.2.4. Immune Response and ATF4 Activation during HIV-1 Infection
4. ATF5 the Paralog of ATF4
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ameri, K.; Harris, A.L. Activating Transcription Factor 4. Int. J. Biochem. Cell Biol. 2008, 40, 14–21. [Google Scholar] [CrossRef]
- Kasai, S.; Yamazaki, H.; Tanji, K.; Engler, M.J.; Matsumiya, T.; Itoh, K. Role of the ISR-ATF4 Pathway and Its Cross Talk with Nrf2 in Mitochondrial Quality Control. J. Clin. Biochem. Nutr. 2019, 64, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The Integrated Stress Response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Angelastro, J.M. Targeting ATF5 in Cancer. Trends Cancer 2017, 3, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.A.; Lee, H.Y.; Angelastro, J.M. The Transcription Factor ATF5: Role in Neurodevelopment and Neural Tumors. J. Neurochem. 2009, 108, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; Benedetti, S.; Gentili, V.; Grigolato, J.; Di Luca, D. Short Communication: Activating Transcription Factor 4 (ATF4) Promotes HIV Type 1 Activation. AIDS Res. Hum. Retroviruses 2012, 28, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-D.; Yu, K.-L.; Park, S.-H.; Jung, Y.-M.; Kim, M.-J.; You, J.-C. Understanding of the Functional Role(s) of the Activating Transcription Factor 4(ATF4) in HIV Regulation and Production. BMB Rep. 2018, 51, 388–393. [Google Scholar] [CrossRef]
- Jiang, G.; Santos Rocha, C.; Hirao, L.A.; Mendes, E.A.; Tang, Y.; Thompson, G.R.; Wong, J.K.; Dandekar, S. HIV Exploits Antiviral Host Innate GCN2-ATF4 Signaling for Establishing Viral Replication Early in Infection. mBio 2017, 8, e01518-16. [Google Scholar] [CrossRef]
- Vallejo-Gracia, A.; Chen, I.P.; Perrone, R.; Besnard, E.; Boehm, D.; Battivelli, E.; Tezil, T.; Krey, K.; Raymond, K.A.; Hull, P.A.; et al. FOXO1 Promotes HIV Latency by Suppressing ER Stress in T Cells. Nat. Microbiol. 2020, 5, 1144–1157. [Google Scholar] [CrossRef]
- Li, D.; Wong, L.M.; Tang, Y.; Allard, B.; James, K.S.; Thompson, G.R.; Dandekar, S.; Browne, E.P.; Li, Q.; Simon, J.M.; et al. Depletion of HIV Reservoir by Activation of ISR Signaling in Resting CD4+T Cells. iScience 2023, 26, 105743. [Google Scholar] [CrossRef] [PubMed]
- Campestrini, J.; Silveira, D.B.; Pinto, A.R. HIV-1 Tat-Induced Bystander Apoptosis in Jurkat Cells Involves Unfolded Protein Responses. Cell Biochem. Funct. 2018, 36, 377–386. [Google Scholar] [CrossRef]
- Granberg, F.; Svensson, C.; Pettersson, U.; Zhao, H. Adenovirus-Induced Alterations in Host Cell Gene Expression Prior to the Onset of Viral Gene Expression. Virology 2006, 353, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Chai, Y.; Song, J.; Liu, T.; Chen, P.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Yang, H. Reprogramming the Unfolded Protein Response for Replication by Porcine Reproductive and Respiratory Syndrome Virus. PLoS Pathog. 2019, 15, e1008169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Moon, A.; Childs, K.; Goodbourn, S.; Dixon, L.K. The African Swine Fever Virus DP71L Protein Recruits the Protein Phosphatase 1 Catalytic Subunit to Dephosphorylate eIF2alpha and Inhibits CHOP Induction but Is Dispensable for These Activities during Virus Infection. J. Virol. 2010, 84, 10681–10689. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.L.; Lipkin, W.I. Endoplasmic Reticulum Stress and Neurodegeneration in Rats Neonatally Infected with Borna Disease Virus. J. Virol. 2006, 80, 8613–8626. [Google Scholar] [CrossRef] [PubMed]
- Tuñón, M.J.; San-Miguel, B.; Crespo, I.; Laliena, A.; Vallejo, D.; Álvarez, M.; Prieto, J.; González-Gallego, J. Melatonin Treatment Reduces Endoplasmic Reticulum Stress and Modulates the Unfolded Protein Response in Rabbits with Lethal Fulminant Hepatitis of Viral Origin. J. Pineal Res. 2013, 55, 221–228. [Google Scholar] [CrossRef]
- Zhou, Y.; Qi, B.; Gu, Y.; Xu, F.; Du, H.; Li, X.; Fang, W. Porcine Circovirus 2 Deploys PERK Pathway and GRP78 for Its Enhanced Replication in PK-15 Cells. Viruses 2016, 8, 56. [Google Scholar] [CrossRef]
- Zhou, Y.-S.; Gu, Y.-X.; Qi, B.-Z.; Zhang, Y.-K.; Li, X.-L.; Fang, W.-H. Porcine Circovirus Type 2 Capsid Protein Induces Unfolded Protein Response with Subsequent Activation of Apoptosis. J. Zhejiang Univ. Sci. B 2017, 18, 316–323. [Google Scholar] [CrossRef]
- Lv, J.; Jiang, Y.; Feng, Q.; Fan, Z.; Sun, Y.; Xu, P.; Hou, Y.; Zhang, X.; Fan, Y.; Xu, X.; et al. Porcine Circovirus Type 2 ORF5 Protein Induces Autophagy to Promote Viral Replication via the PERK-eIF2α-ATF4 and mTOR-ERK1/2-AMPK Signaling Pathways in PK-15 Cells. Front. Microbiol. 2020, 11, 320. [Google Scholar] [CrossRef]
- Liao, Y.; Fung, T.S.; Huang, M.; Fang, S.G.; Zhong, Y.; Liu, D.X. Upregulation of CHOP/GADD153 during Coronavirus Infectious Bronchitis Virus Infection Modulates Apoptosis by Restricting Activation of the Extracellular Signal-Regulated Kinase Pathway. J. Virol. 2013, 87, 8124–8134. [Google Scholar] [CrossRef]
- Chen, W.; Huang, C.; Shi, Y.; Li, N.; Wang, E.; Hu, R.; Li, G.; Yang, F.; Zhuang, Y.; Liu, P.; et al. Investigation of the Crosstalk between GRP78/PERK/ATF-4 Signaling Pathway and Renal Apoptosis Induced by Nephropathogenic Infectious Bronchitis Virus Infection. J. Virol. 2022, 96, e0142921. [Google Scholar] [CrossRef]
- Fang, P.; Tian, L.; Zhang, H.; Xia, S.; Ding, T.; Zhu, X.; Zhang, J.; Ren, J.; Fang, L.; Xiao, S. Induction and Modulation of the Unfolded Protein Response during Porcine Deltacoronavirus Infection. Vet. Microbiol. 2022, 271, 109494. [Google Scholar] [CrossRef]
- Chen, Y.-M.; Gabler, N.K.; Burrough, E.R. Porcine Epidemic Diarrhea Virus Infection Induces Endoplasmic Reticulum Stress and Unfolded Protein Response in Jejunal Epithelial Cells of Weaned Pigs. Vet. Pathol. 2022, 59, 82–90. [Google Scholar] [CrossRef]
- Christ, W.; Klingström, J.; Tynell, J. SARS-CoV-2 Variant-Specific Differences in Inhibiting the Effects of the PKR-Activated Integrated Stress Response. Virus Res. 2024, 339, 199271. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, K.-Y.; Wang, S.-H.; Liu, Y.; Zhao, Y.-Q.; Yang, L.; Yang, G.-H.; Wang, X.-J.; Zhu, Y.-H.; Yin, J.-H.; et al. Effects of Spatial Expression of Activating Transcription Factor 4 on the Pathogenicity of Two Phenotypes of Bovine Viral Diarrhea Virus by Regulating the Endoplasmic Reticulum-Mediated Autophagy Process. Microbiol. Spectr. 2023, 11, e0422522. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.E.; Wang, C.; Chan, K.W.K.; Vasudevan, S.G.; Jans, D.A. Novel Dengue Virus Inhibitor 4-HPR Activates ATF4 Independent of Protein Kinase R-like Endoplasmic Reticulum Kinase and Elevates Levels of eIF2α Phosphorylation in Virus Infected Cells. Antivir. Res. 2016, 130, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Diosa-Toro, M.; Troost, B.; van de Pol, D.; Heberle, A.M.; Urcuqui-Inchima, S.; Thedieck, K.; Smit, J.M. Tomatidine, a Novel Antiviral Compound towards Dengue Virus. Antivir. Res. 2019, 161, 90–99. [Google Scholar] [CrossRef]
- Ciccaglione, A.R.; Marcantonio, C.; Tritarelli, E.; Equestre, M.; Vendittelli, F.; Costantino, A.; Geraci, A.; Rapicetta, M. Activation of the ER Stress Gene Gadd153 by Hepatitis C Virus Sensitizes Cells to Oxidant Injury. Virus Res. 2007, 126, 128–138. [Google Scholar] [CrossRef]
- Merquiol, E.; Uzi, D.; Mueller, T.; Goldenberg, D.; Nahmias, Y.; Xavier, R.J.; Tirosh, B.; Shibolet, O. HCV Causes Chronic Endoplasmic Reticulum Stress Leading to Adaptation and Interference with the Unfolded Protein Response. PLoS ONE 2011, 6, e24660. [Google Scholar] [CrossRef]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C Virus Core Protein Activates Autophagy through EIF2AK3 and ATF6 UPR Pathway-Mediated MAP1LC3B and ATG12 Expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef]
- Li, Y.-C.; Zhang, M.-Q.; Zhang, J.-P. Opposite Effects of Two Human ATG10 Isoforms on Replication of a HCV Sub-Genomic Replicon Are Mediated via Regulating Autophagy Flux in Zebrafish. Front. Cell Infect. Microbiol. 2018, 8, 109. [Google Scholar] [CrossRef]
- Ríos-Ocampo, W.A.; Navas, M.-C.; Buist-Homan, M.; Faber, K.N.; Daemen, T.; Moshage, H. Hepatitis C Virus Proteins Core and NS5A Are Highly Sensitive to Oxidative Stress-Induced Degradation after eIF2α/ATF4 Pathway Activation. Viruses 2020, 12, 425. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Xu, A.; Wu, X.; Zhang, Y.; Guo, Y.; Guo, F.; Pan, Z.; Kong, L. Japanese Encephalitis Virus Induces Apoptosis by the IRE1/JNK Pathway of ER Stress Response in BHK-21 Cells. Arch. Virol. 2016, 161, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.; Gonzalez, G.; Martinelli, A.; Wastika, C.E.; Ito, K.; Orba, Y.; Sasaki, M.; Hall, W.W.; Sawa, H. Upregulated Expression of the Antioxidant Sestrin 2 Identified by Transcriptomic Analysis of Japanese Encephalitis Virus-Infected SH-SY5Y Neuroblastoma Cells. Virus Genes 2019, 55, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xin, X.; Wang, T.; Wan, J.; Ou, Y.; Yang, Z.; Yu, Q.; Zhu, L.; Guo, Y.; Wu, Y.; et al. Japanese Encephalitis Virus Induces Apoptosis and Encephalitis by Activating the PERK Pathway. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yang, J.; Han, K.; Liu, Q.; Wang, H.; Liu, Y.; Huang, X.; Zhang, L.; Li, Y. The Unfolded Protein Response Induced by Tembusu Virus Infection. BMC Vet. Res. 2019, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Basu, M.; Courtney, S.C.; Brinton, M.A. Arsenite-Induced Stress Granule Formation Is Inhibited by Elevated Levels of Reduced Glutathione in West Nile Virus-Infected Cells. PLoS Pathog. 2017, 13, e1006240. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Zhang, W.; Sun, J.; Fu, Z.; Ke, X.; Zheng, C.; Zhang, Y.; Li, P.; Liu, Y.; Hu, Q.; et al. ZIKV Infection Activates the IRE1-XBP1 and ATF6 Pathways of Unfolded Protein Response in Neural Cells. J. Neuroinflamm. 2018, 15, 275. [Google Scholar] [CrossRef] [PubMed]
- Mufrrih, M.; Chen, B.; Chan, S.-W. Zika Virus Induces an Atypical Tripartite Unfolded Protein Response with Sustained Sensor and Transient Effector Activation and a Blunted BiP Response. mSphere 2021, 6, e0036121. [Google Scholar] [CrossRef]
- Cho, H.K.; Cheong, K.J.; Kim, H.Y.; Cheong, J. Endoplasmic Reticulum Stress Induced by Hepatitis B Virus X Protein Enhances Cyclo-Oxygenase 2 Expression via Activating Transcription Factor 4. Biochem. J. 2011, 435, 431–439. [Google Scholar] [CrossRef]
- Li, J.; He, J.; Fu, Y.; Hu, X.; Sun, L.-Q.; Huang, Y.; Fan, X. Hepatitis B Virus X Protein Inhibits Apoptosis by Modulating Endoplasmic Reticulum Stress Response. Oncotarget 2017, 8, 96027–96034. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-M.; Kim, D.H.; Jang, J.; Choe, W.H.; Kim, B.-J. rt269L-Type Hepatitis B Virus (HBV) in Genotype C Infection Leads to Improved Mitochondrial Dynamics via the PERK-eIF2α-ATF4 Axis in an HBx Protein-Dependent Manner. Cell Mol. Biol. Lett. 2023, 28, 26. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Sugden, B. The LMP1 Oncogene of EBV Activates PERK and the Unfolded Protein Response to Drive Its Own Synthesis. Blood 2008, 111, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Isler, J.A.; Skalet, A.H.; Alwine, J.C. Human Cytomegalovirus Infection Activates and Regulates the Unfolded Protein Response. J. Virol. 2005, 79, 6890–6899. [Google Scholar] [CrossRef] [PubMed]
- Xuan, B.; Qian, Z.; Torigoi, E.; Yu, D. Human Cytomegalovirus Protein pUL38 Induces ATF4 Expression, Inhibits Persistent JNK Phosphorylation, and Suppresses Endoplasmic Reticulum Stress-Induced Cell Death. J. Virol. 2009, 83, 3463–3474. [Google Scholar] [CrossRef] [PubMed]
- Siddiquey, M.N.A.; Zhang, H.; Nguyen, C.C.; Domma, A.J.; Kamil, J.P. The Human Cytomegalovirus Endoplasmic Reticulum-Resident Glycoprotein UL148 Activates the Unfolded Protein Response. J. Virol. 2018, 92, e00896-18. [Google Scholar] [CrossRef]
- Sharon, E.; Frenkel, N. Human Herpesvirus 6A Exhibits Restrictive Propagation with Limited Activation of the Protein Kinase R-eIF2α Stress Pathway. J. Virol. 2017, 91, e02120-16. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; Benedetti, S.; Grigolato, J.; Caruso, A.; Di Luca, D. Activating Transcription Factor 4 (ATF4) Is Upregulated by Human Herpesvirus 8 Infection, Increases Virus Replication and Promotes Proangiogenic Properties. Arch. Virol. 2012, 157, 63–74. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, F.; Chen, Q.; Sarid, R.; Li, X.; Kuang, E. Upregulation of ATF4-LAMP3 Axis by ORF45 Facilitates Lytic Replication of Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2022, 96, e0145622. [Google Scholar] [CrossRef]
- Burnett, H.F.; Audas, T.E.; Liang, G.; Lu, R.R. Herpes Simplex Virus-1 Disarms the Unfolded Protein Response in the Early Stages of Infection. Cell Stress. Chaperones 2012, 17, 473–483. [Google Scholar] [CrossRef]
- Qian, Z.; Xuan, B.; Chapa, T.J.; Gualberto, N.; Yu, D. Murine Cytomegalovirus Targets Transcription Factor ATF4 to Exploit the Unfolded-Protein Response. J. Virol. 2012, 86, 6712–6723. [Google Scholar] [CrossRef]
- Zhou, X.-C.; Dong, S.-H.; Liu, Z.-S.; Liu, S.; Zhang, C.-C.; Liang, X.-Z. Regulation of Gammaherpesvirus Lytic Replication by Endoplasmic Reticulum Stress-Induced Transcription Factors ATF4 and CHOP. J. Biol. Chem. 2018, 293, 2801–2814. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhu, J.; Zhou, X.; Wang, H.; Li, X.; Zhao, A. Induction of the Unfolded Protein Response (UPR) during Pseudorabies Virus Infection. Vet. Microbiol. 2019, 239, 108485. [Google Scholar] [CrossRef]
- Chen, L.; Ni, M.; Ahmed, W.; Xu, Y.; Bao, X.; Zhuang, T.; Feng, L.; Guo, M. Pseudorabies Virus Infection Induces Endoplasmic Reticulum Stress and Unfolded Protein Response in Suspension-Cultured BHK-21 Cells. J. Gen. Virol. 2022, 103, 001818. [Google Scholar] [CrossRef]
- Liao, Y.; Gu, F.; Mao, X.; Niu, Q.; Wang, H.; Sun, Y.; Song, C.; Qiu, X.; Tan, L.; Ding, C. Regulation of de Novo Translation of Host Cells by Manipulation of PERK/PKR and GADD34-PP1 Activity during Newcastle Disease Virus Infection. J. Gen. Virol. 2016, 97, 867–879. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, R.; Li, Y.; Sun, Y.; Song, C.; Zhan, Y.; Tan, L.; Liao, Y.; Meng, C.; Qiu, X.; et al. Newcastle Disease Virus Induces G0/G1 Cell Cycle Arrest in Asynchronously Growing Cells. Virology 2018, 520, 67–74. [Google Scholar] [CrossRef]
- Liang, Q.; Deng, H.; Sun, C.-W.; Townes, T.M.; Zhu, F. Negative Regulation of IRF7 Activation by Activating Transcription Factor 4 Suggests a Cross-Regulation between the IFN Responses and the Cellular Integrated Stress Responses. J. Immunol. 2011, 186, 1001–1010. [Google Scholar] [CrossRef]
- Cao, L.; Xue, M.; Chen, J.; Shi, H.; Zhang, X.; Shi, D.; Liu, J.; Huang, L.; Wei, Y.; Liu, C.; et al. Porcine Parvovirus Replication Is Suppressed by Activation of the PERK Signaling Pathway and Endoplasmic Reticulum Stress-Mediated Apoptosis. Virology 2020, 539, 1–10. [Google Scholar] [CrossRef]
- Sun, P.; Zhang, S.; Qin, X.; Chang, X.; Cui, X.; Li, H.; Zhang, S.; Gao, H.; Wang, P.; Zhang, Z.; et al. Foot-and-Mouth Disease Virus Capsid Protein VP2 Activates the Cellular EIF2S1-ATF4 Pathway and Induces Autophagy via HSPB1. Autophagy 2018, 14, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Colli, M.L.; Paula, F.M.; Marselli, L.; Marchetti, P.; Roivainen, M.; Eizirik, D.L.; Op de Beeck, A. Coxsackievirus B Tailors the Unfolded Protein Response to Favour Viral Amplification in Pancreatic β Cells. J. Innate Immun. 2019, 11, 375–390. [Google Scholar] [CrossRef]
- Dunlap, K.M.; Bartee, M.Y.; Bartee, E. Myxoma Virus Attenuates Expression of Activating Transcription Factor 4 (ATF4) Which Has Implications for the Treatment of Proteasome Inhibitor-Resistant Multiple Myeloma. Oncolytic Virother. 2015, 4, 1–11. [Google Scholar] [CrossRef]
- Smith, J.A.; Schmechel, S.C.; Raghavan, A.; Abelson, M.; Reilly, C.; Katze, M.G.; Kaufman, R.J.; Bohjanen, P.R.; Schiff, L.A. Reovirus Induces and Benefits from an Integrated Cellular Stress Response. J. Virol. 2006, 80, 2019–2033. [Google Scholar] [CrossRef]
- Fros, J.J.; Major, L.D.; Scholte, F.E.M.; Gardner, J.; van Hemert, M.J.; Suhrbier, A.; Pijlman, G.P. Chikungunya Virus Non-Structural Protein 2-Mediated Host Shut-off Disables the Unfolded Protein Response. J. Gen. Virol. 2015, 96, 580–589. [Google Scholar] [CrossRef]
- Baer, A.; Lundberg, L.; Swales, D.; Waybright, N.; Pinkham, C.; Dinman, J.D.; Jacobs, J.L.; Kehn-Hall, K. Venezuelan Equine Encephalitis Virus Induces Apoptosis through the Unfolded Protein Response Activation of EGR1. J. Virol. 2016, 90, 3558–3572. [Google Scholar] [CrossRef] [PubMed]
- Estaquier, J.; Idziorek, T.; de Bels, F.; Barré-Sinoussi, F.; Hurtrel, B.; Aubertin, A.M.; Venet, A.; Mehtali, M.; Muchmore, E.; Michel, P.; et al. Programmed Cell Death and AIDS: Significance of T-Cell Apoptosis in Pathogenic and Nonpathogenic Primate Lentiviral Infections. Proc. Natl. Acad. Sci. USA 1994, 91, 9431–9435. [Google Scholar] [CrossRef] [PubMed]
- Laforge, M.; Silvestre, R.; Rodrigues, V.; Garibal, J.; Campillo-Gimenez, L.; Mouhamad, S.; Monceaux, V.; Cumont, M.-C.; Rabezanahary, H.; Pruvost, A.; et al. The Anti-Caspase Inhibitor Q-VD-OPH Prevents AIDS Disease Progression in SIV-Infected Rhesus Macaques. J. Clin. Investig. 2018, 128, 1627–1640. [Google Scholar] [CrossRef] [PubMed]
- Monceaux, V.; Estaquier, J.; Février, M.; Cumont, M.-C.; Rivière, Y.; Aubertin, A.-M.; Ameisen, J.C.; Hurtrel, B. Extensive Apoptosis in Lymphoid Organs during Primary SIV Infection Predicts Rapid Progression towards AIDS. AIDS 2003, 17, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Cumont, M.-C.; Diop, O.; Vaslin, B.; Elbim, C.; Viollet, L.; Monceaux, V.; Lay, S.; Silvestri, G.; Le Grand, R.; Müller-Trutwin, M.; et al. Early Divergence in Lymphoid Tissue Apoptosis between Pathogenic and Nonpathogenic Simian Immunodeficiency Virus Infections of Nonhuman Primates. J. Virol. 2008, 82, 1175–1184. [Google Scholar] [CrossRef]
- Viollet, L.; Monceaux, V.; Petit, F.; Ho Tsong Fang, R.; Cumont, M.-C.; Hurtrel, B.; Estaquier, J. Death of CD4+ T Cells from Lymph Nodes during Primary SIVmac251 Infection Predicts the Rate of AIDS Progression. J. Immunol. 2006, 177, 6685–6694. [Google Scholar] [CrossRef]
- Lu, Y.-N.; Kavianpour, S.; Zhang, T.; Zhang, X.; Nguyen, D.; Thombre, R.; He, L.; Wang, J. MARK2 Phosphorylates eIF2α in Response to Proteotoxic Stress. PLoS Biol. 2021, 19, e3001096. [Google Scholar] [CrossRef]
- Wu, Z.; Mei, F.; Gan, Y.; Liu, A.; Hu, J.; Jin, Y.; Yin, Y. FAM69C Functions as a Kinase for eIF2α and Promotes Stress Granule Assembly. EMBO Rep. 2023, 24, e55641. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; Zhang, S.; et al. The Role of Host eIF2α in Viral Infection. Virol. J. 2020, 17, 112. [Google Scholar] [CrossRef] [PubMed]
- Galabru, J.; Hovanessian, A. Autophosphorylation of the Protein Kinase Dependent on Double-Stranded RNA. J. Biol. Chem. 1987, 262, 15538–15544. [Google Scholar] [CrossRef]
- García, M.A.; Meurs, E.F.; Esteban, M. The dsRNA Protein Kinase PKR: Virus and Cell Control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef]
- Carpick, B.W.; Graziano, V.; Schneider, D.; Maitra, R.K.; Lee, X.; Williams, B.R. Characterization of the Solution Complex between the Interferon-Induced, Double-Stranded RNA-Activated Protein Kinase and HIV-I Trans-Activating Region RNA. J. Biol. Chem. 1997, 272, 9510–9516. [Google Scholar] [CrossRef]
- Kim, I.; Liu, C.W.; Puglisi, J.D. Specific Recognition of HIV TAR RNA by the dsRNA Binding Domains (dsRBD1-dsRBD2) of PKR. J. Mol. Biol. 2006, 358, 430–442. [Google Scholar] [CrossRef]
- Maitra, R.K.; McMillan, N.A.; Desai, S.; McSwiggen, J.; Hovanessian, A.G.; Sen, G.; Williams, B.R.; Silverman, R.H. HIV-1 TAR RNA Has an Intrinsic Ability to Activate Interferon-Inducible Enzymes. Virology 1994, 204, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Spanggord, R.J.; Vuyisich, M.; Beal, P.A. Identification of Binding Sites for Both dsRBMs of PKR on Kinase-Activating and Kinase-Inhibiting RNA Ligands. Biochemistry 2002, 41, 4511–4520. [Google Scholar] [CrossRef]
- Clerzius, G.; Gélinas, J.-F.; Gatignol, A. Multiple Levels of PKR Inhibition during HIV-1 Replication. Rev. Med. Virol. 2011, 21, 42–53. [Google Scholar] [CrossRef]
- Lee, E.-S.; Yoon, C.-H.; Kim, Y.-S.; Bae, Y.-S. The Double-Strand RNA-Dependent Protein Kinase PKR Plays a Significant Role in a Sustained ER Stress-Induced Apoptosis. FEBS Lett. 2007, 581, 4325–4332. [Google Scholar] [CrossRef]
- Benkirane, M.; Neuveut, C.; Chun, R.F.; Smith, S.M.; Samuel, C.E.; Gatignol, A.; Jeang, K.T. Oncogenic Potential of TAR RNA Binding Protein TRBP and Its Regulatory Interaction with RNA-Dependent Protein Kinase PKR. EMBO J. 1997, 16, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Blair, E.D.; Roberts, C.M.; Snowden, B.W.; Gatignol, A.; Benkirane, M.; Jeang, K.-T. Expression of TAR RNA-Binding Protein in Baculovirus and Co-Immunoprecipitation with Insect Cell Protein Kinase. J. Biomed. Sci. 1995, 2, 322–329. [Google Scholar] [CrossRef]
- Clerzius, G.; Gélinas, J.-F.; Daher, A.; Bonnet, M.; Meurs, E.F.; Gatignol, A. ADAR1 Interacts with PKR during Human Immunodeficiency Virus Infection of Lymphocytes and Contributes to Viral Replication. J. Virol. 2009, 83, 10119–10128. [Google Scholar] [CrossRef]
- Cole, J.L. Activation of PKR: An Open and Shut Case? Trends Biochem. Sci. 2007, 32, 57–62. [Google Scholar] [CrossRef]
- Daher, A.; Longuet, M.; Dorin, D.; Bois, F.; Segeral, E.; Bannwarth, S.; Battisti, P.L.; Purcell, D.F.; Benarous, R.; Vaquero, C.; et al. Two Dimerization Domains in the Trans-Activation Response RNA-Binding Protein (TRBP) Individually Reverse the Protein Kinase R Inhibition of HIV-1 Long Terminal Repeat Expression. J. Biol. Chem. 2001, 276, 33899–33905. [Google Scholar] [CrossRef]
- Duarte, M.; Graham, K.; Daher, A.; Battisti, P.L.; Bannwarth, S.; Segeral, E.; Jeang, K.T.; Gatignol, A. Characterization of TRBP1 and TRBP2. Stable Stem-Loop Structure at the 5′ End of TRBP2 mRNA Resembles HIV-1 TAR and Is Not Found in Its Processed Pseudogene. J. Biomed. Sci. 2000, 7, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, P.A.; Anderson, E.; Lary, J.; Cole, J.L. Mechanism of PKR Activation by dsRNA. J. Mol. Biol. 2008, 381, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.R.; Kobayashi, R.; Mathews, M.B. The Tat Protein of Human Immunodeficiency Virus Type 1 Is a Substrate and Inhibitor of the Interferon-Induced, Virally Activated Protein Kinase, PKR. J. Biol. Chem. 1997, 272, 8388–8395. [Google Scholar] [CrossRef]
- Cai, R.; Carpick, B.; Chun, R.F.; Jeang, K.T.; Williams, B.R. HIV-I TAT Inhibits PKR Activity by Both RNA-Dependent and RNA-Independent Mechanisms. Arch. Biochem. Biophys. 2000, 373, 361–367. [Google Scholar] [CrossRef]
- McMillan, N.A.; Chun, R.F.; Siderovski, D.P.; Galabru, J.; Toone, W.M.; Samuel, C.E.; Mak, T.W.; Hovanessian, A.G.; Jeang, K.T.; Williams, B.R. HIV-1 Tat Directly Interacts with the Interferon-Induced, Double-Stranded RNA-Dependent Kinase, PKR. Virology 1995, 213, 413–424. [Google Scholar] [CrossRef]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M.; et al. The Roles of Apoptosis, Autophagy and Unfolded Protein Response in Arbovirus, Influenza Virus, and HIV Infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Yang, L.; Niu, F.; Buch, S. HIV Tat-Mediated Induction of Human Brain Microvascular Endothelial Cell Apoptosis Involves Endoplasmic Reticulum Stress and Mitochondrial Dysfunction. Mol. Neurobiol. 2016, 53, 132–142. [Google Scholar] [CrossRef]
- Tripathi, A.; Iyer, K.; Mitra, D. HIV-1 Replication Requires Optimal Activation of the Unfolded Protein Response. FEBS Lett. 2023, 597, 2908–2930. [Google Scholar] [CrossRef]
- Hortin, G.L.; Landt, M.; Powderly, W.G. Changes in Plasma Amino Acid Concentrations in Response to HIV-1 Infection. Clin. Chem. 1994, 40, 785–789. [Google Scholar] [CrossRef]
- Dröge, W.; Murthy, K.K.; Stahl-Hennig, C.; Hartung, S.; Plesker, R.; Rouse, S.; Peterhans, E.; Kinscherf, R.; Fischbach, T.; Eck, H.P. Plasma Amino Acid Dysregulation after Lentiviral Infection. AIDS Res. Hum. Retroviruses 1993, 9, 807–809. [Google Scholar] [CrossRef]
- Mody, A.; Bartz, S.; Hornik, C.P.; Kiyimba, T.; Bain, J.; Muehlbauer, M.; Kiboneka, E.; Stevens, R.; St Peter, J.V.; Newgard, C.B.; et al. Effects of HIV Infection on the Metabolic and Hormonal Status of Children with Severe Acute Malnutrition. PLoS ONE 2014, 9, e102233. [Google Scholar] [CrossRef]
- Stone, J.D.; Heise, C.C.; Miller, C.J.; Halsted, C.H.; Dandekar, S. Development of Malabsorption and Nutritional Complications in Simian Immunodeficiency Virus-Infected Rhesus Macaques. AIDS 1994, 8, 1245–1256. [Google Scholar] [CrossRef]
- Cosnefroy, O.; Jaspart, A.; Calmels, C.; Parissi, V.; Fleury, H.; Ventura, M.; Reigadas, S.; Andréola, M.-L. Activation of GCN2 upon HIV-1 Infection and Inhibition of Translation. Cell Mol. Life Sci. 2013, 70, 2411–2421. [Google Scholar] [CrossRef]
- Jaspart, A.; Calmels, C.; Cosnefroy, O.; Bellecave, P.; Pinson, P.; Claverol, S.; Guyonnet-Dupérat, V.; Dartigues, B.; Benleulmi, M.S.; Mauro, E.; et al. GCN2 Phosphorylates HIV-1 Integrase and Decreases HIV-1 Replication by Limiting Viral Integration. Sci. Rep. 2017, 7, 2283. [Google Scholar] [CrossRef] [PubMed]
- del Pino, J.; Jiménez, J.L.; Ventoso, I.; Castelló, A.; Muñoz-Fernández, M.Á.; de Haro, C.; Berlanga, J.J. GCN2 Has Inhibitory Effect on Human Immunodeficiency Virus-1 Protein Synthesis and Is Cleaved upon Viral Infection. PLoS ONE 2012, 7, e47272. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.M.; Worth, M.P.; Hinchman, M.M.; Parker, J.S.L.; Ledgerwood, E.D. Mammalian Orthoreovirus Infection Is Enhanced in Cells Pre-Treated with Sodium Arsenite. Viruses 2019, 11, 563. [Google Scholar] [CrossRef]
- Lee, J.; Stone, J.; Desai, P.; Kosowicz, J.G.; Liu, J.O.; Ambinder, R.F. Arsenicals, the Integrated Stress Response, and Epstein-Barr Virus Lytic Gene Expression. Viruses 2021, 13, 812. [Google Scholar] [CrossRef] [PubMed]
- McEwen, E.; Kedersha, N.; Song, B.; Scheuner, D.; Gilks, N.; Han, A.; Chen, J.-J.; Anderson, P.; Kaufman, R.J. Heme-Regulated Inhibitor Kinase-Mediated Phosphorylation of Eukaryotic Translation Initiation Factor 2 Inhibits Translation, Induces Stress Granule Formation, and Mediates Survival upon Arsenite Exposure. J. Biol. Chem. 2005, 280, 16925–16933. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.; Ramaglia, V.; Abdel-Nour, M.; Bianchi, A.A.; Tsalikis, J.; Chau, H.N.; Kalia, S.K.; Kalia, L.V.; Chen, J.-J.; Arnoult, D.; et al. The eIF2α Kinase HRI Triggers the Autophagic Clearance of Cytosolic Protein Aggregates. J. Biol. Chem. 2021, 296, 100050. [Google Scholar] [CrossRef]
- Huang, P.; Peslak, S.A.; Lan, X.; Khandros, E.; Yano, J.A.; Sharma, M.; Keller, C.A.; Giardine, B.; Qin, K.; Abdulmalik, O.; et al. The HRI-Regulated Transcription Factor ATF4 Activates BCL11A Transcription to Silence Fetal Hemoglobin Expression. Blood 2020, 135, 2121–2132. [Google Scholar] [CrossRef]
- Suragani, R.N.V.S.; Zachariah, R.S.; Velazquez, J.G.; Liu, S.; Sun, C.-W.; Townes, T.M.; Chen, J.-J. Heme-Regulated eIF2α Kinase Activated Atf4 Signaling Pathway in Oxidative Stress and Erythropoiesis. Blood 2012, 119, 5276–5284. [Google Scholar] [CrossRef]
- Protzer, U.; Seyfried, S.; Quasdorff, M.; Sass, G.; Svorcova, M.; Webb, D.; Bohne, F.; Hösel, M.; Schirmacher, P.; Tiegs, G. Antiviral Activity and Hepatoprotection by Heme Oxygenase-1 in Hepatitis B Virus Infection. Gastroenterology 2007, 133, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wilson, A.T.; Mathahs, M.M.; Wen, F.; Brown, K.E.; Luxon, B.A.; Schmidt, W.N. Heme Oxygenase-1 Suppresses Hepatitis C Virus Replication and Increases Resistance of Hepatocytes to Oxidant Injury. Hepatology 2008, 48, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Devadas, K.; Dhawan, S. Hemin Activation Ameliorates HIV-1 Infection via Heme Oxygenase-1 Induction. J. Immunol. 2006, 176, 4252–4257. [Google Scholar] [CrossRef]
- Ambegaokar, S.S.; Kolson, D.L. Heme Oxygenase-1 Dysregulation in the Brain: Implications for HIV-Associated Neurocognitive Disorders. Curr. HIV Res. 2014, 12, 174–188. [Google Scholar] [CrossRef]
- Malikov, V.; Naghavi, M.H. Localized Phosphorylation of a Kinesin-1 Adaptor by a Capsid-Associated Kinase Regulates HIV-1 Motility and Uncoating. Cell Rep. 2017, 20, 2792–2799. [Google Scholar] [CrossRef]
- Cossarizza, A.; Mussini, C.; Mongiardo, N.; Borghi, V.; Sabbatini, A.; De Rienzo, B.; Franceschi, C. Mitochondria Alterations and Dramatic Tendency to Undergo Apoptosis in Peripheral Blood Lymphocytes during Acute HIV Syndrome. AIDS 1997, 11, 19–26. [Google Scholar] [CrossRef]
- Macho, A.; Castedo, M.; Marchetti, P.; Aguilar, J.J.; Decaudin, D.; Zamzami, N.; Girard, P.M.; Uriel, J.; Kroemer, G. Mitochondrial Dysfunctions in Circulating T Lymphocytes from Human Immunodeficiency Virus-1 Carriers. Blood 1995, 86, 2481–2487. [Google Scholar] [CrossRef] [PubMed]
- Petit, F.; Arnoult, D.; Lelièvre, J.-D.; Moutouh-de Parseval, L.; Hance, A.J.; Schneider, P.; Corbeil, J.; Ameisen, J.C.; Estaquier, J. Productive HIV-1 Infection of Primary CD4+ T Cells Induces Mitochondrial Membrane Permeabilization Leading to a Caspase-Independent Cell Death. J. Biol. Chem. 2002, 277, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Petit, F.; Lelièvre, J.-D.; Estaquier, J. Mitochondria in HIV-1-Induced Apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 561–574. [Google Scholar] [CrossRef]
- Arnoult, D.; Petit, F.; Lelièvre, J.D.; Lecossier, D.; Hance, A.; Monceaux, V.; Hurtrel, B.; Ho Tsong Fang, R.; Ameisen, J.C.; Estaquier, J. Caspase-Dependent and-Independent T-Cell Death Pathways in Pathogenic Simian Immunodeficiency Virus Infection: Relationship to Disease Progression. Cell Death Differ. 2003, 10, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Estaquier, J.; Vallette, F.; Vayssiere, J.-L.; Mignotte, B. The Mitochondrial Pathways of Apoptosis. Adv. Exp. Med. Biol. 2012, 942, 157–183. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, I.; Estaquier, J. Viral Manipulation of the Host Metabolic Network. Exp. Suppl. 2018, 109, 377–401. [Google Scholar] [CrossRef]
- Elbim, C.; Monceaux, V.; François, S.; Hurtrel, B.; Gougerot-Pocidalo, M.-A.; Estaquier, J. Increased Neutrophil Apoptosis in Chronically SIV-Infected Macaques. Retrovirology 2009, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Elbim, C.; Monceaux, V.; Mueller, Y.M.; Lewis, M.G.; François, S.; Diop, O.; Akarid, K.; Hurtrel, B.; Gougerot-Pocidalo, M.-A.; Lévy, Y.; et al. Early Divergence in Neutrophil Apoptosis between Pathogenic and Nonpathogenic Simian Immunodeficiency Virus Infections of Nonhuman Primates. J. Immunol. 2008, 181, 8613–8623. [Google Scholar] [CrossRef]
- Elbim, C.; Pillet, S.; Prevost, M.H.; Preira, A.; Girard, P.M.; Rogine, N.; Matusani, H.; Hakim, J.; Israel, N.; Gougerot-Pocidalo, M.A. Redox and Activation Status of Monocytes from Human Immunodeficiency Virus-Infected Patients: Relationship with Viral Load. J. Virol. 1999, 73, 4561–4566. [Google Scholar] [CrossRef]
- Cumont, M.C.; Monceaux, V.; Viollet, L.; Lay, S.; Parker, R.; Hurtrel, B.; Estaquier, J. TGF-Beta in Intestinal Lymphoid Organs Contributes to the Death of Armed Effector CD8 T Cells and Is Associated with the Absence of Virus Containment in Rhesus Macaques Infected with the Simian Immunodeficiency Virus. Cell Death Differ. 2007, 14, 1747–1758. [Google Scholar] [CrossRef]
- Kasai, S.; Yasumoto, K.-I.; Sogawa, K. Attenuation of Inhibitory PAS Domain Protein-Induced Cell Death by Synthetic Peptides Derived from Mcl-1 Transmenbrane Domain. Cell Death Discov. 2021, 7, 92. [Google Scholar] [CrossRef]
- Münch, C. The Different Axes of the Mammalian Mitochondrial Unfolded Protein Response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-Omics Analysis Identifies ATF4 as a Key Regulator of the Mitochondrial Stress Response in Mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Eckl, E.-M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.F.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A Pathway Coordinated by DELE1 Relays Mitochondrial Stress to the Cytosol. Nature 2020, 579, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.-H.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial Stress Is Relayed to the Cytosol by an OMA1-DELE1-HRI Pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef]
- Lawrence, R.E.; Zoncu, R. The Lysosome as a Cellular Centre for Signalling, Metabolism and Quality Control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef]
- Li, T.Y.; Wang, Q.; Gao, A.W.; Li, X.; Sun, Y.; Mottis, A.; Shong, M.; Auwerx, J. Lysosomes Mediate the Mitochondrial UPR via mTORC1-Dependent ATF4 Phosphorylation. Cell Discov. 2023, 9, 92. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 Senses Lysosomal Amino Acids through an Inside-out Mechanism That Requires the Vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Grodet, A.; Lee, Y.-J.; Estaquier, J.; Blackstone, C. Release of OPA1 during Apoptosis Participates in the Rapid and Complete Release of Cytochrome c and Subsequent Mitochondrial Fragmentation. J. Biol. Chem. 2005, 280, 35742–35750. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Rismanchi, N.; Grodet, A.; Roberts, R.G.; Seeburg, D.P.; Estaquier, J.; Sheng, M.; Blackstone, C. Bax/Bak-Dependent Release of DDP/TIMM8a Promotes Drp1-Mediated Mitochondrial Fission and Mitoptosis during Programmed Cell Death. Curr. Biol. 2005, 15, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389.e6. [Google Scholar] [CrossRef]
- Estaquier, J.; Arnoult, D. Inhibiting Drp1-Mediated Mitochondrial Fission Selectively Prevents the Release of Cytochrome c during Apoptosis. Cell Death Differ. 2007, 14, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Ngo, J.; Wang, S.-P.; Williams, K.; Kramer, H.F.; Ho, G.; Rodriguez, C.; Yekkala, K.; Amuzie, C.; Bialecki, R.; et al. The Mitochondrial Fission Protein Drp1 in Liver Is Required to Mitigate NASH and Prevents the Activation of the Mitochondrial ISR. Mol. Metab. 2022, 64, 101566. [Google Scholar] [CrossRef]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.-C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 Processing and Mitochondrial Fusion by M-AAA Protease Isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible Proteolytic Inactivation of OPA1 Mediated by the OMA1 Protease in Mammalian Cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef]
- Yang, J.; Baron, K.R.; Pride, D.E.; Schneemann, A.; Guo, X.; Chen, W.; Song, A.S.; Aviles, G.; Kampmann, M.; Luke Wiseman, R.; et al. DELE1 Oligomerization Promotes Integrated Stress Response Activation. Nat. Struct. Mol. Biol. 2023, 30, 1295–1302. [Google Scholar] [CrossRef]
- Schubert, U.; Antón, L.C.; Bacík, I.; Cox, J.H.; Bour, S.; Bennink, J.R.; Orlowski, M.; Strebel, K.; Yewdell, J.W. CD4 Glycoprotein Degradation Induced by Human Immunodeficiency Virus Type 1 Vpu Protein Requires the Function of Proteasomes and the Ubiquitin-Conjugating Pathway. J. Virol. 1998, 72, 2280–2288. [Google Scholar] [CrossRef]
- Butticaz, C.; Michielin, O.; Wyniger, J.; Telenti, A.; Rothenberger, S. Silencing of Both Beta-TrCP1 and HOS (Beta-TrCP2) Is Required to Suppress Human Immunodeficiency Virus Type 1 Vpu-Mediated CD4 down-Modulation. J. Virol. 2007, 81, 1502–1505. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Früh, K.; Moses, A.V. Vpu Directs the Degradation of the Human Immunodeficiency Virus Restriction Factor BST-2/Tetherin via a {beta}TrCP-Dependent Mechanism. J. Virol. 2009, 83, 7931–7947. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Gers-Huber, G.; Lehmann, M.; Zufferey, M.; Luban, J.; Piguet, V. HIV-1 Vpu Neutralizes the Antiviral Factor Tetherin/BST-2 by Binding It and Directing Its Beta-TrCP2-Dependent Degradation. PLoS Pathog. 2009, 5, e1000574. [Google Scholar] [CrossRef] [PubMed]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A Novel Human WD Protein, h-Beta TrCp, That Interacts with HIV-1 Vpu Connects CD4 to the ER Degradation Pathway through an F-Box Motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Iwabu, Y.; Fujita, H.; Kinomoto, M.; Kaneko, K.; Ishizaka, Y.; Tanaka, Y.; Sata, T.; Tokunaga, K. HIV-1 Accessory Protein Vpu Internalizes Cell-Surface BST-2/Tetherin through Transmembrane Interactions Leading to Lysosomes. J. Biol. Chem. 2009, 284, 35060–35072. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu Antagonizes BST-2-Mediated Restriction of HIV-1 Release via Beta-TrCP and Endo-Lysosomal Trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef]
- Stoneham, C.A.; Singh, R.; Jia, X.; Xiong, Y.; Guatelli, J. Endocytic Activity of HIV-1 Vpu: Phosphoserine-Dependent Interactions with Clathrin Adaptors. Traffic 2017, 18, 545–561. [Google Scholar] [CrossRef]
- Goffinet, C.; Allespach, I.; Homann, S.; Tervo, H.-M.; Habermann, A.; Rupp, D.; Oberbremer, L.; Kern, C.; Tibroni, N.; Welsch, S.; et al. HIV-1 Antagonism of CD317 Is Species Specific and Involves Vpu-Mediated Proteasomal Degradation of the Restriction Factor. Cell Host Microbe 2009, 5, 285–297. [Google Scholar] [CrossRef]
- Lassot, I.; Ségéral, E.; Berlioz-Torrent, C.; Durand, H.; Groussin, L.; Hai, T.; Benarous, R.; Margottin-Goguet, F. ATF4 Degradation Relies on a Phosphorylation-Dependent Interaction with the SCF(betaTrCP) Ubiquitin Ligase. Mol. Cell Biol. 2001, 21, 2192–2202. [Google Scholar] [CrossRef]
- Besnard-Guerin, C.; Belaïdouni, N.; Lassot, I.; Segeral, E.; Jobart, A.; Marchal, C.; Benarous, R. HIV-1 Vpu Sequesters Beta-Transducin Repeat-Containing Protein (betaTrCP) in the Cytoplasm and Provokes the Accumulation of Beta-Catenin and Other SCFbetaTrCP Substrates. J. Biol. Chem. 2004, 279, 788–795. [Google Scholar] [CrossRef]
- Putters, J.; Slotman, J.A.; Gerlach, J.P.; Strous, G.J. Specificity, Location and Function of βTrCP Isoforms and Their Splice Variants. Cell Signal 2011, 23, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Pickering, S.; Sumner, J.; Kerridge, C.; Perera, M.; Neil, S. Differential Dysregulation of β-TrCP1 and -2 by HIV-1 Vpu Leads to Inhibition of Canonical and Non-Canonical NF-κB Pathways in Infected Cells. mBio 2023, 14, e0329322. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Illa, I.; Pezeshkpour, G.H.; Laukaitis, J.P.; Cohen, B.; Griffin, J.L. Mitochondrial Myopathy Caused by Long-Term Zidovudine Therapy. N. Engl. J. Med. 1990, 322, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.; Dalakas, M.C. Mitochondrial Toxicity of Antiviral Drugs. Nat. Med. 1995, 1, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Gerschenson, M.; Poirier, M.C. Fetal Patas Monkeys Sustain Mitochondrial Toxicity as a Result of in Utero Zidovudine Exposure. Ann. N. Y. Acad. Sci. 2000, 918, 269–281. [Google Scholar] [CrossRef]
- Petit, F.; Fromenty, B.; Owen, A.; Estaquier, J. Mitochondria Are Sensors for HIV Drugs. Trends Pharmacol. Sci. 2005, 26, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Estaquier, J.; Lelièvre, J.-D.; Petit, F.; Brunner, T.; Moutouh-De Parseval, L.; Richman, D.D.; Ameisen, J.C.; Corbeil, J. Effects of Antiretroviral Drugs on Human Immunodeficiency Virus Type 1-Induced CD4(+) T-Cell Death. J. Virol. 2002, 76, 5966–5973. [Google Scholar] [CrossRef]
- De Gassart, A.; Bujisic, B.; Zaffalon, L.; Decosterd, L.A.; Di Micco, A.; Frera, G.; Tallant, R.; Martinon, F. An Inhibitor of HIV-1 Protease Modulates Constitutive eIF2α Dephosphorylation to Trigger a Specific Integrated Stress Response. Proc. Natl. Acad. Sci. USA 2016, 113, E117–E126. [Google Scholar] [CrossRef]
- Subeha, M.R.; Goyeneche, A.A.; Bustamante, P.; Lisio, M.A.; Burnier, J.V.; Telleria, C.M. Nelfinavir Induces Cytotoxicity towards High-Grade Serous Ovarian Cancer Cells, Involving Induction of the Unfolded Protein Response, Modulation of Protein Synthesis, DNA Damage, Lysosomal Impairment, and Potentiation of Toxicity Caused by Proteasome Inhibition. Cancers 2021, 14, 99. [Google Scholar] [CrossRef]
- Brüning, A.; Rahmeh, M.; Friese, K. Nelfinavir and Bortezomib Inhibit mTOR Activity via ATF4-Mediated Sestrin-2 Regulation. Mol. Oncol. 2013, 7, 1012–1018. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, L.; Yang, J.; Zhang, X.; Mikkelsen, R.; Song, S.; Zhou, H. HIV Protease Inhibitors Sensitize Human Head and Neck Squamous Carcinoma Cells to Radiation by Activating Endoplasmic Reticulum Stress. PLoS ONE 2015, 10, e0125928. [Google Scholar] [CrossRef]
- Fraichard, C.; Bonnet-Serrano, F.; Laguillier-Morizot, C.; Hebert-Schuster, M.; Lai-Kuen, R.; Sibiude, J.; Fournier, T.; Cohen, M.; Guibourdenche, J. Protease Inhibitor Anti-HIV, Lopinavir, Impairs Placental Endocrine Function. Int. J. Mol. Sci. 2021, 22, 683. [Google Scholar] [CrossRef]
- McIntyre, R.L.; Molenaars, M.; Schomakers, B.V.; Gao, A.W.; Kamble, R.; Jongejan, A.; van Weeghel, M.; van Kuilenburg, A.B.P.; Possemato, R.; Houtkooper, R.H.; et al. Anti-Retroviral Treatment with Zidovudine Alters Pyrimidine Metabolism, Reduces Translation, and Extends Healthy Longevity via ATF-4. Cell Rep. 2023, 42, 111928. [Google Scholar] [CrossRef]
- Barbieri, A.M.; Chiodini, I.; Ragni, E.; Colaianni, G.; Gadda, F.; Locatelli, M.; Lampertico, P.; Spada, A.; Eller-Vainicher, C. Suppressive Effects of Tenofovir Disoproxil Fumarate, an Antiretroviral Prodrug, on Mineralization and Type II and Type III Sodium-Dependent Phosphate Transporters Expression in Primary Human Osteoblasts. J. Cell Biochem. 2018, 119, 4855–4866. [Google Scholar] [CrossRef]
- Ikebe, E.; Matsuoka, S.; Tezuka, K.; Kuramitsu, M.; Okuma, K.; Nakashima, M.; Kobayashi, S.; Makiyama, J.; Yamagishi, M.; Oyadomari, S.; et al. Activation of PERK-ATF4-CHOP Pathway as a Novel Therapeutic Approach for Efficient Elimination of HTLV-1-Infected Cells. Blood Adv. 2020, 4, 1845–1858. [Google Scholar] [CrossRef]
- Xu, Y.; Weatherall, C.; Bailey, M.; Alcantara, S.; De Rose, R.; Estaquier, J.; Wilson, K.; Suzuki, K.; Corbeil, J.; Cooper, D.A.; et al. Simian Immunodeficiency Virus Infects Follicular Helper CD4 T Cells in Lymphoid Tissues during Pathogenic Infection of Pigtail Macaques. J. Virol. 2013, 87, 3760–3773. [Google Scholar] [CrossRef]
- Moukambi, F.; Rabezanahary, H.; Rodrigues, V.; Racine, G.; Robitaille, L.; Krust, B.; Andreani, G.; Soundaramourty, C.; Silvestre, R.; Laforge, M.; et al. Early Loss of Splenic Tfh Cells in SIV-Infected Rhesus Macaques. PLoS Pathog. 2015, 11, e1005287. [Google Scholar] [CrossRef] [PubMed]
- Rabezanahary, H.; Moukambi, F.; Palesch, D.; Clain, J.; Racine, G.; Andreani, G.; Benmadid-Laktout, G.; Zghidi-Abouzid, O.; Soundaramourty, C.; Tremblay, C.; et al. Despite Early Antiretroviral Therapy Effector Memory and Follicular Helper CD4 T Cells Are Major Reservoirs in Visceral Lymphoid Tissues of SIV-Infected Macaques. Mucosal Immunol. 2020, 13, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Banga, R.; Procopio, F.A.; Noto, A.; Pollakis, G.; Cavassini, M.; Ohmiti, K.; Corpataux, J.-M.; de Leval, L.; Pantaleo, G.; Perreau, M. PD-1(+) and Follicular Helper T Cells Are Responsible for Persistent HIV-1 Transcription in Treated Aviremic Individuals. Nat. Med. 2016, 22, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Perreau, M.; Savoye, A.-L.; De Crignis, E.; Corpataux, J.-M.; Cubas, R.; Haddad, E.K.; De Leval, L.; Graziosi, C.; Pantaleo, G. Follicular Helper T Cells Serve as the Major CD4 T Cell Compartment for HIV-1 Infection, Replication, and Production. J. Exp. Med. 2013, 210, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Pluta, A.; Jaworski, J.P.; Cortés-Rubio, C.N. Balance between Retroviral Latency and Transcription: Based on HIV Model. Pathogens 2020, 10, 16. [Google Scholar] [CrossRef]
- Reddy, T.R.; Tang, H.; Li, X.; Wong-Staal, F. Functional Interaction of the HTLV-1 Transactivator Tax with Activating Transcription Factor-4 (ATF4). Oncogene 1997, 14, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.A.; Bentley, K.; Peeters, A.; Churchill, M.J.; Deacon, N.J. A Compilation of Cellular Transcription Factor Interactions with the HIV-1 LTR Promoter. Nucleic Acids Res. 2000, 28, 663–668. [Google Scholar] [CrossRef]
- Rabbi, M.F.; Saifuddin, M.; Gu, D.S.; Kagnoff, M.F.; Roebuck, K.A. U5 Region of the Human Immunodeficiency Virus Type 1 Long Terminal Repeat Contains TRE-like cAMP-Responsive Elements That Bind Both AP-1 and CREB/ATF Proteins. Virology 1997, 233, 235–245. [Google Scholar] [CrossRef]
- Neill, G.; Masson, G.R. A Stay of Execution: ATF4 Regulation and Potential Outcomes for the Integrated Stress Response. Front. Mol. Neurosci. 2023, 16, 1112253. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Martínez, J.A.; Reinke, A.W.; Bhimsaria, D.; Keating, A.E.; Ansari, A.Z. Combinatorial bZIP Dimers Display Complex DNA-Binding Specificity Landscapes. eLife 2017, 6, e19272. [Google Scholar] [CrossRef] [PubMed]
- Hai, T.; Curran, T. Cross-Family Dimerization of Transcription Factors Fos/Jun and ATF/CREB Alters DNA Binding Specificity. Proc. Natl. Acad. Sci. USA 1991, 88, 3720–3724. [Google Scholar] [CrossRef]
- Mu, Y.; Yu, Y.; Yue, X.; Musarat, I.; Gong, R.; Zhu, C.; Liu, Y.; Liu, F.; Zhu, Y.; Wu, J. The X Protein of HBV Induces HIV-1 Long Terminal Repeat Transcription by Enhancing the Binding of C/EBPβ and CREB1/2 Regulatory Proteins to the Long Terminal Repeat of HIV-1. Virus Res. 2011, 156, 81–90. [Google Scholar] [CrossRef]
- Gachon, F.; Thebault, S.; Peleraux, A.; Devaux, C.; Mesnard, J.M. Molecular Interactions Involved in the Transactivation of the Human T-Cell Leukemia Virus Type 1 Promoter Mediated by Tax and CREB-2 (ATF-4). Mol. Cell Biol. 2000, 20, 3470–3481. [Google Scholar] [CrossRef]
- Ameisen, J.C.; Estaquier, J.; Idziorek, T. From AIDS to Parasite Infection: Pathogen-Mediated Subversion of Programmed Cell Death as a Mechanism for Immune Dysregulation. Immunol. Rev. 1994, 142, 9–51. [Google Scholar] [CrossRef]
- Clerici, M.; Sarin, A.; Coffman, R.L.; Wynn, T.A.; Blatt, S.P.; Hendrix, C.W.; Wolf, S.F.; Shearer, G.M.; Henkart, P.A. Type 1/Type 2 Cytokine Modulation of T-Cell Programmed Cell Death as a Model for Human Immunodeficiency Virus Pathogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 11811–11815. [Google Scholar] [CrossRef]
- Finkel, T.H.; Tudor-Williams, G.; Banda, N.K.; Cotton, M.F.; Curiel, T.; Monks, C.; Baba, T.W.; Ruprecht, R.M.; Kupfer, A. Apoptosis Occurs Predominantly in Bystander Cells and Not in Productively Infected Cells of HIV- and SIV-Infected Lymph Nodes. Nat. Med. 1995, 1, 129–134. [Google Scholar] [CrossRef]
- Arnoult, D.; Viollet, L.; Petit, F.; Lelièvre, J.-D.; Estaquier, J. HIV-1 Triggers Mitochondrion Death. Mitochondrion 2004, 4, 255–269. [Google Scholar] [CrossRef]
- Estaquier, J.; Tanaka, M.; Suda, T.; Nagata, S.; Golstein, P.; Ameisen, J.C. Fas-Mediated Apoptosis of CD4+ and CD8+ T Cells from Human Immunodeficiency Virus-Infected Persons: Differential in Vitro Preventive Effect of Cytokines and Protease Antagonists. Blood 1996, 87, 4959–4966. [Google Scholar] [CrossRef]
- Estaquier, J.; Idziorek, T.; Zou, W.; Emilie, D.; Farber, C.M.; Bourez, J.M.; Ameisen, J.C. T Helper Type 1/T Helper Type 2 Cytokines and T Cell Death: Preventive Effect of Interleukin 12 on Activation-Induced and CD95 (FAS/APO-1)-Mediated Apoptosis of CD4+ T Cells from Human Immunodeficiency Virus-Infected Persons. J. Exp. Med. 1995, 182, 1759–1767. [Google Scholar] [CrossRef]
- Katsikis, P.D.; Wunderlich, E.S.; Smith, C.A.; Herzenberg, L.A.; Herzenberg, L.A. Fas Antigen Stimulation Induces Marked Apoptosis of T Lymphocytes in Human Immunodeficiency Virus-Infected Individuals. J. Exp. Med. 1995, 181, 2029–2036. [Google Scholar] [CrossRef]
- Sloand, E.M.; Young, N.S.; Kumar, P.; Weichold, F.F.; Sato, T.; Maciejewski, J.P. Role of Fas Ligand and Receptor in the Mechanism of T-Cell Depletion in Acquired Immunodeficiency Syndrome: Effect on CD4+ Lymphocyte Depletion and Human Immunodeficiency Virus Replication. Blood 1997, 89, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal Apoptosis Induced by Endoplasmic Reticulum Stress Is Regulated by ATF4-CHOP-Mediated Induction of the Bcl-2 Homology 3-Only Member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- Bagheri-Yarmand, R.; Sinha, K.M.; Gururaj, A.E.; Ahmed, Z.; Rizvi, Y.Q.; Huang, S.-C.; Ladbury, J.E.; Bogler, O.; Williams, M.D.; Cote, G.J.; et al. A Novel Dual Kinase Function of the RET Proto-Oncogene Negatively Regulates Activating Transcription Factor 4-Mediated Apoptosis. J. Biol. Chem. 2015, 290, 11749–11761. [Google Scholar] [CrossRef]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP Induces Activating Transcription Factor 5 (ATF5) to Trigger Apoptosis in Response to Perturbations in Protein Homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A.; et al. ERAD Inhibitors Integrate ER Stress with an Epigenetic Mechanism to Activate BH3-Only Protein NOXA in Cancer Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2200–2205. [Google Scholar] [CrossRef]
- Hiramatsu, N.; Messah, C.; Han, J.; LaVail, M.M.; Kaufman, R.J.; Lin, J.H. Translational and Posttranslational Regulation of XIAP by eIF2α and ATF4 Promotes ER Stress-Induced Cell Death during the Unfolded Protein Response. Mol. Biol. Cell 2014, 25, 1411–1420. [Google Scholar] [CrossRef]
- Chen, D.; Wang, M.; Zhou, S.; Zhou, Q. HIV-1 Tat Targets Microtubules to Induce Apoptosis, a Process Promoted by the pro-Apoptotic Bcl-2 Relative Bim. EMBO J. 2002, 21, 6801–6810. [Google Scholar] [CrossRef] [PubMed]
- Castellano, P.; Prevedel, L.; Eugenin, E.A. HIV-Infected Macrophages and Microglia That Survive Acute Infection Become Viral Reservoirs by a Mechanism Involving Bim. Sci. Rep. 2017, 7, 12866. [Google Scholar] [CrossRef]
- Dong, H.; Ye, X.; Zhong, L.; Xu, J.; Qiu, J.; Wang, J.; Shao, Y.; Xing, H. Role of FOXO3 Activated by HIV-1 Tat in HIV-Associated Neurocognitive Disorder Neuronal Apoptosis. Front. Neurosci. 2019, 13, 44. [Google Scholar] [CrossRef] [PubMed]
- Galvão-Lima, L.J.; Zambuzi, F.A.; Soares, L.S.; Fontanari, C.; Meireles, A.F.G.; Brauer, V.S.; Faccioli, L.H.; Gama, L.; Figueiredo, L.T.M.; Bou-Habib, D.C.; et al. HIV-1 Gag and Vpr Impair the Inflammasome Activation and Contribute to the Establishment of Chronic Infection in Human Primary Macrophages. Mol. Immunol. 2022, 148, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Chand, K.; Iyer, K.; Mitra, D. Comparative Analysis of Differential Gene Expression of HSP40 and HSP70 Family Isoforms during Heat Stress and HIV-1 Infection in T-Cells. Cell Stress Chaperones 2021, 26, 403–416. [Google Scholar] [CrossRef]
- Chand, K.; Barman, M.K.; Ghosh, P.; Mitra, D. DNAJB8 Facilitates Autophagic-Lysosomal Degradation of Viral Vif Protein and Restricts HIV-1 Virion Infectivity by Rescuing APOBEC3G Expression in Host Cells. FASEB J. 2023, 37, e22793. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Monaco, A.; Aricò, E.; Wang, E.; Tornesello, M.L.; Lewis, G.K.; Marincola, F.M.; Buonaguro, F.M. Gene Expression Profile of Peripheral Blood Mononuclear Cells in Response to HIV-VLPs Stimulation. BMC Bioinform. 2008, 9 (Suppl. 2), S5. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.L.; Moos, P.; Pond, C.D.; Larson, E.C.; Martins, L.J.; Szaniawski, M.A.; Planelles, V.; Barrows, L.R. HIV-1 Provirus Transcription and Translation in Macrophages Differs from Pre-Integrated cDNA Complexes and Requires E2F Transcriptional Programs. Virulence 2022, 13, 386–413. [Google Scholar] [CrossRef] [PubMed]
- Grelli, S.; Balestrieri, E.; Matteucci, C.; Minutolo, A.; D’Ettorre, G.; Lauria, F.; Montella, F.; Vullo, V.; Vella, S.; Favalli, C.; et al. Apoptotic Cell Signaling in Lymphocytes from HIV+ Patients during Successful Therapy. Ann. N. Y. Acad. Sci. 2006, 1090, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Swingler, S.; Mann, A.M.; Zhou, J.; Swingler, C.; Stevenson, M. Apoptotic Killing of HIV-1-Infected Macrophages Is Subverted by the Viral Envelope Glycoprotein. PLoS Pathog. 2007, 3, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, E.; Grelli, S.; Matteucci, C.; Minutolo, A.; d’Ettorre, G.; Di Sora, F.; Montella, F.; Vullo, V.; Vella, S.; Favalli, C.; et al. Apoptosis-Associated Gene Expression in HIV-Infected Patients in Response to Successful Antiretroviral Therapy. J. Med. Virol. 2007, 79, 111–117. [Google Scholar] [CrossRef]
- Laforge, M.; Campillo-Gimenez, L.; Monceaux, V.; Cumont, M.-C.; Hurtrel, B.; Corbeil, J.; Zaunders, J.; Elbim, C.; Estaquier, J. HIV/SIV Infection Primes Monocytes and Dendritic Cells for Apoptosis. PLoS Pathog. 2011, 7, e1002087. [Google Scholar] [CrossRef] [PubMed]
- Busca, A.; Saxena, M.; Kumar, A. Critical Role for Antiapoptotic Bcl-xL and Mcl-1 in Human Macrophage Survival and Cellular IAP1/2 (cIAP1/2) in Resistance to HIV-Vpr-Induced Apoptosis. J. Biol. Chem. 2012, 287, 15118–15133. [Google Scholar] [CrossRef]
- Xue, J.; Fu, C.; Cong, Z.; Peng, L.; Peng, Z.; Chen, T.; Wang, W.; Jiang, H.; Wei, Q.; Qin, C. Galectin-3 Promotes Caspase-Independent Cell Death of HIV-1-Infected Macrophages. FEBS J. 2017, 284, 97–113. [Google Scholar] [CrossRef]
- Serrano, A.; El Haddad, S.; Moal, F.; Prazuck, T.; Legac, E.; Robin, C.; Brulé, F.; Charpentier, S.; Normand, T.; Legrand, A.; et al. Dysregulation of Apoptosis and Autophagy Gene Expression in Peripheral Blood Mononuclear Cells of Efficiently Treated HIV-Infected Patients. AIDS 2018, 32, 1579–1587. [Google Scholar] [CrossRef]
- Muraduzzaman, A.K.M.; Islam, N.M.; Tabassum, S.; Munshi, S.U. Intrinsic Apoptotic Pathway Genes of Circulating Blood Neutrophils Triggered during HIV Infection and Remained Stimulated in ART Patients. Curr. HIV Res. 2023, 21, 122–127. [Google Scholar] [CrossRef]
- Dabrowska, A.; Kim, N.; Aldovini, A. Tat-Induced FOXO3a Is a Key Mediator of Apoptosis in HIV-1-Infected Human CD4+ T Lymphocytes. J. Immunol. 2008, 181, 8460–8477. [Google Scholar] [CrossRef]
- Perfettini, J.-L.; Roumier, T.; Castedo, M.; Larochette, N.; Boya, P.; Raynal, B.; Lazar, V.; Ciccosanti, F.; Nardacci, R.; Penninger, J.; et al. NF-kappaB and P53 Are the Dominant Apoptosis-Inducing Transcription Factors Elicited by the HIV-1 Envelope. J. Exp. Med. 2004, 199, 629–640. [Google Scholar] [CrossRef]
- Nardacci, R.; Antinori, A.; Larocca, L.M.; Arena, V.; Amendola, A.; Perfettini, J.-L.; Kroemer, G.; Piacentini, M. Characterization of Cell Death Pathways in Human Immunodeficiency Virus-Associated Encephalitis. Am. J. Pathol. 2005, 167, 695–704. [Google Scholar] [CrossRef]
- Shimizu, S.; Khan, M.Z.; Hippensteel, R.L.; Parkar, A.; Raghupathi, R.; Meucci, O. Role of the Transcription Factor E2F1 in CXCR4-Mediated Neurotoxicity and HIV Neuropathology. Neurobiol. Dis. 2007, 25, 17–26. [Google Scholar] [CrossRef]
- Moni, M.A.; Liò, P. Network-Based Analysis of Comorbidities Risk during an Infection: SARS and HIV Case Studies. BMC Bioinform. 2014, 15, 333. [Google Scholar] [CrossRef] [PubMed]
- Petralia, M.C.; Nicoletti, F.; Tancheva, L.; Kalfin, R.; Fagone, P.; Mangano, K. Gene Co-Expression Network Modular Analysis Reveals Altered Immune Mechanisms in HIV-HAND. Brain Sci. 2022, 12, 1378. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zang, Y.; Qiao, L.; Liu, K.; Ouyang, Y.; Zhang, Y.; Chen, D. ASPP2 Involvement in P53-Mediated HIV-1 Envelope Glycoprotein Gp120 Neurotoxicity in Mice Cerebrocortical Neurons. Sci. Rep. 2016, 6, 33378. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xiao, Y.; Torresilla, C.; Rassart, É.; Barbeau, B. Implication of Different HIV-1 Genes in the Modulation of Autophagy. Viruses 2017, 9, 389. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.Y.; Trinh, D.L.N.; Zajchowski, L.D.; Lee, B.; Elwi, A.N.; Kim, S.-W. Tid1 Is a New Regulator of P53 Mitochondrial Translocation and Apoptosis in Cancer. Oncogene 2010, 29, 1155–1166. [Google Scholar] [CrossRef]
- Genini, D.; Sheeter, D.; Rought, S.; Zaunders, J.J.; Susin, S.A.; Kroemer, G.; Richman, D.D.; Carson, D.A.; Corbeil, J.; Leoni, L.M. HIV Induces Lymphocyte Apoptosis by a P53-Initiated, Mitochondrial-Mediated Mechanism. FASEB J. 2001, 15, 5–6. [Google Scholar] [CrossRef]
- Laforge, M.; Limou, S.; Harper, F.; Casartelli, N.; Rodrigues, V.; Silvestre, R.; Haloui, H.; Zagury, J.-F.; Senik, A.; Estaquier, J. DRAM Triggers Lysosomal Membrane Permeabilization and Cell Death in CD4(+) T Cells Infected with HIV. PLoS Pathog. 2013, 9, e1003328. [Google Scholar] [CrossRef]
- Niu, G.; Zhang, H.; Liu, D.; Chen, L.; Belani, C.; Wang, H.-G.; Cheng, H. Tid1, the Mammalian Homologue of Drosophila Tumor Suppressor Tid56, Mediates Macroautophagy by Interacting with Beclin1-Containing Autophagy Protein Complex. J. Biol. Chem. 2015, 290, 18102–18110. [Google Scholar] [CrossRef]
- Samuels-Lev, Y.; O’Connor, D.J.; Bergamaschi, D.; Trigiante, G.; Hsieh, J.K.; Zhong, S.; Campargue, I.; Naumovski, L.; Crook, T.; Lu, X. ASPP Proteins Specifically Stimulate the Apoptotic Function of P53. Mol. Cell 2001, 8, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yang, X.; Shu, S.; Wang, S.; Guo, F.; Yin, Y.; Zhou, W.; Han, H.; Chai, X. Sufentanil Protects the Liver from Ischemia/Reperfusion-Induced Inflammation and Apoptosis by Inhibiting ATF4-Induced TP53BP2 Expression. Inflammation 2021, 44, 1160–1174. [Google Scholar] [CrossRef]
- Liu, Z.; Qiao, L.; Zhang, Y.; Zang, Y.; Shi, Y.; Liu, K.; Zhang, X.; Lu, X.; Yuan, L.; Su, B.; et al. ASPP2 Plays a Dual Role in Gp120-Induced Autophagy and Apoptosis of Neuroblastoma Cells. Front. Neurosci. 2017, 11, 150. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Sayano, T.; Tamai, S.; Yokota, S.; Kato, H.; Fujii, G.; Mihara, K. Identification of a Novel Protein MICS1 That Is Involved in Maintenance of Mitochondrial Morphology and Apoptotic Release of Cytochrome c. Mol. Biol. Cell 2008, 19, 2597–2608. [Google Scholar] [CrossRef]
- Seitaj, B.; Maull, F.; Zhang, L.; Wüllner, V.; Wolf, C.; Schippers, P.; La Rovere, R.; Distler, U.; Tenzer, S.; Parys, J.B.; et al. Transmembrane BAX Inhibitor-1 Motif Containing Protein 5 (TMBIM5) Sustains Mitochondrial Structure, Shape, and Function by Impacting the Mitochondrial Protein Synthesis Machinery. Cells 2020, 9, 2147. [Google Scholar] [CrossRef]
- Austin, S.; Mekis, R.; Mohammed, S.E.M.; Scalise, M.; Wang, W.-A.; Galluccio, M.; Pfeiffer, C.; Borovec, T.; Parapatics, K.; Vitko, D.; et al. TMBIM5 Is the Ca2+/H+ Antiporter of Mammalian Mitochondria. EMBO Rep. 2022, 23, e54978. [Google Scholar] [CrossRef]
- Cui, E.; Zhang, L.; Pan, X.; Zhang, Q.; Zhang, L.; Wu, F.; Chen, N.; Lv, L.; Chen, W.; Chen, H.; et al. RNA-Sequencing Approach for Exploring the Therapeutic Effect of Umbilical Cord Mesenchymal Stem/Stromal Cells on Lipopolysaccharide-Induced Acute Lung Injury. Front. Immunol. 2022, 13, 1021102. [Google Scholar] [CrossRef]
- Zhang, L.; Dietsche, F.; Seitaj, B.; Rojas-Charry, L.; Latchman, N.; Tomar, D.; Wüst, R.C.; Nickel, A.; Frauenknecht, K.B.; Schoser, B.; et al. TMBIM5 Loss of Function Alters Mitochondrial Matrix Ion Homeostasis and Causes a Skeletal Myopathy. Life Sci. Alliance 2022, 5, e202201478. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, M.; Yu, H.; Lu, J.; Cheng, K.K.Y.; Zhou, J.; Chen, H.; Jia, W. Activation of G0/G1 Switch Gene 2 by Endoplasmic Reticulum Stress Enhances Hepatic Steatosis. Metabolism 2019, 99, 32–44. [Google Scholar] [CrossRef]
- Russell, L.; Forsdyke, D.R. A Human Putative Lymphocyte G0/G1 Switch Gene Containing a CpG-Rich Island Encodes a Small Basic Protein with the Potential to Be Phosphorylated. DNA Cell Biol. 1991, 10, 581–591. [Google Scholar] [CrossRef]
- Siderovski, D.P.; Blum, S.; Forsdyke, R.E.; Forsdyke, D.R. A Set of Human Putative Lymphocyte G0/G1 Switch Genes Includes Genes Homologous to Rodent Cytokine and Zinc Finger Protein-Encoding Genes. DNA Cell Biol. 1990, 9, 579–587. [Google Scholar] [CrossRef]
- Zhang, X.; Heckmann, B.L.; Campbell, L.E.; Liu, J. G0S2: A Small Giant Controller of Lipolysis and Adipose-Liver Fatty Acid Flux. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1146–1154. [Google Scholar] [CrossRef]
- Kioka, H.; Kato, H.; Fujikawa, M.; Tsukamoto, O.; Suzuki, T.; Imamura, H.; Nakano, A.; Higo, S.; Yamazaki, S.; Matsuzaki, T.; et al. Evaluation of Intramitochondrial ATP Levels Identifies G0/G1 Switch Gene 2 as a Positive Regulator of Oxidative Phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Zhu, Y.; Zhang, P. Lipolytic Inhibitor G0/G1 Switch Gene 2 Inhibits Reactive Oxygen Species Production and Apoptosis in Endothelial Cells. Am. J. Physiol. Cell Physiol. 2015, 308, C496–C504. [Google Scholar] [CrossRef] [PubMed]
- B’chir, W.; Maurin, A.-C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 Pathway Is Essential for Stress-Induced Autophagy Gene Expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef]
- Whitney, M.L.; Jefferson, L.S.; Kimball, S.R. ATF4 Is Necessary and Sufficient for ER Stress-Induced Upregulation of REDD1 Expression. Biochem. Biophys. Res. Commun. 2009, 379, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.C.; Vega-Rubin-de-Celis, S.; Xie, X.-J.; Castrillon, D.H.; Kabbani, W.; Brugarolas, J. Cell-Type-Dependent Regulation of mTORC1 by REDD1 and the Tumor Suppressors TSC1/TSC2 and LKB1 in Response to Hypoxia. Mol. Cell Biol. 2011, 31, 1870–1884. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Dai, W.; Kutzler, L.; Lacko, H.A.; Jefferson, L.S.; Dennis, M.D.; Kimball, S.R. ATF4-Mediated Upregulation of REDD1 and Sestrin2 Suppresses mTORC1 Activity during Prolonged Leucine Deprivation. J. Nutr. 2020, 150, 1022–1030. [Google Scholar] [CrossRef]
- Cabrera-Rodríguez, R.; Pérez-Yanes, S.; Lorenzo-Sánchez, I.; Trujillo-González, R.; Estévez-Herrera, J.; García-Luis, J.; Valenzuela-Fernández, A. HIV Infection: Shaping the Complex, Dynamic, and Interconnected Network of the Cytoskeleton. Int. J. Mol. Sci. 2023, 24, 13104. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, C.; Pilli, M.; Kehrl, J.H. Roles of Autophagy in HIV Infection. Immunol. Cell Biol. 2015, 93, 11–17. [Google Scholar] [CrossRef]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy Is Involved in T Cell Death after Binding of HIV-1 Envelope Proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef]
- García-Expósito, L.; Barroso-González, J.; Puigdomènech, I.; Machado, J.-D.; Blanco, J.; Valenzuela-Fernández, A. HIV-1 Requires Arf6-Mediated Membrane Dynamics to Efficiently Enter and Infect T Lymphocytes. Mol. Biol. Cell 2011, 22, 1148–1166. [Google Scholar] [CrossRef]
- Moreau, K.; Ravikumar, B.; Puri, C.; Rubinsztein, D.C. Arf6 Promotes Autophagosome Formation via Effects on Phosphatidylinositol 4,5-Bisphosphate and Phospholipase D. J. Cell Biol. 2012, 196, 483–496. [Google Scholar] [CrossRef]
- Van Grol, J.; Subauste, C.; Andrade, R.M.; Fujinaga, K.; Nelson, J.; Subauste, C.S. HIV-1 Inhibits Autophagy in Bystander Macrophage/Monocytic Cells through Src-Akt and STAT3. PLoS ONE 2010, 5, e11733. [Google Scholar] [CrossRef]
- Zhou, D.; Spector, S.A. Human Immunodeficiency Virus Type-1 Infection Inhibits Autophagy. AIDS 2008, 22, 695–699. [Google Scholar] [CrossRef]
- Alfaisal, J.; Machado, A.; Galais, M.; Robert-Hebmann, V.; Arnauné-Pelloquin, L.; Espert, L.; Biard-Piechaczyk, M. HIV-1 Vpr Inhibits Autophagy during the Early Steps of Infection of CD4 T Cells. Biol. Cell 2019, 111, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Rozman, M.; Zidovec-Lepej, S.; Jambrosic, K.; Babić, M.; Drmić Hofman, I. Role of TLRs in HIV-1 Infection and Potential of TLR Agonists in HIV-1 Vaccine Development and Treatment Strategies. Pathogens 2023, 12, 92. [Google Scholar] [CrossRef]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, Regulation, Modification and Function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Farook, J.M.; Shields, J.; Tawfik, A.; Markand, S.; Sen, T.; Smith, S.B.; Brann, D.; Dhandapani, K.M.; Sen, N. GADD34 Induces Cell Death through Inactivation of Akt Following Traumatic Brain Injury. Cell Death Dis. 2013, 4, e754. [Google Scholar] [CrossRef] [PubMed]
- Imbeault, M.; Ouellet, M.; Tremblay, M.J. Microarray Study Reveals That HIV-1 Induces Rapid Type-I Interferon-Dependent P53 mRNA up-Regulation in Human Primary CD4+ T Cells. Retrovirology 2009, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Sirois, M.; Robitaille, L.; Allary, R.; Shah, M.; Woelk, C.H.; Estaquier, J.; Corbeil, J. TRAF6 and IRF7 Control HIV Replication in Macrophages. PLoS ONE 2011, 6, e28125. [Google Scholar] [CrossRef] [PubMed]
- Ishaq, M.; Marshall, H.; Natarajan, V. GADD34 Attenuates HIV-1 Replication by Viral 5′-UTR TAR RNA-Mediated Translational Inhibition. Virology 2020, 540, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Hernández, J.C.; Stevenson, M.; Latz, E.; Urcuqui-Inchima, S. HIV Type 1 Infection Up-Regulates TLR2 and TLR4 Expression and Function in Vivo and in Vitro. AIDS Res. Hum. Retroviruses 2012, 28, 1313–1328. [Google Scholar] [CrossRef]
- Henrick, B.M.; Yao, X.-D.; Rosenthal, K.L. HIV-1 Structural Proteins Serve as PAMPs for TLR2 Heterodimers Significantly Increasing Infection and Innate Immune Activation. Front. Immunol. 2015, 6, 426. [Google Scholar] [CrossRef]
- Shimasaki, S.; Koga, T.; Shuto, T.; Suico, M.A.; Sato, T.; Watanabe, K.; Morino-Koga, S.; Taura, M.; Okada, S.; Mori, K.; et al. Endoplasmic Reticulum Stress Increases the Expression and Function of Toll-like Receptor-2 in Epithelial Cells. Biochem. Biophys. Res. Commun. 2010, 402, 235–240. [Google Scholar] [CrossRef]
- Liao, K.; Guo, M.; Niu, F.; Yang, L.; Callen, S.E.; Buch, S. Cocaine-Mediated Induction of Microglial Activation Involves the ER Stress-TLR2 Axis. J. Neuroinflamm. 2016, 13, 33. [Google Scholar] [CrossRef]
- Zhang, C.; Bai, N.; Chang, A.; Zhang, Z.; Yin, J.; Shen, W.; Tian, Y.; Xiang, R.; Liu, C. ATF4 Is Directly Recruited by TLR4 Signaling and Positively Regulates TLR4-Trigged Cytokine Production in Human Monocytes. Cell Mol. Immunol. 2013, 10, 84–94. [Google Scholar] [CrossRef]
- Solomon, I.H.; Chettimada, S.; Misra, V.; Lorenz, D.R.; Gorelick, R.J.; Gelman, B.B.; Morgello, S.; Gabuzda, D. White Matter Abnormalities Linked to Interferon, Stress Response, and Energy Metabolism Gene Expression Changes in Older HIV-Positive Patients on Antiretroviral Therapy. Mol. Neurobiol. 2020, 57, 1115–1130. [Google Scholar] [CrossRef]
- Akay, C.; Lindl, K.A.; Shyam, N.; Nabet, B.; Goenaga-Vazquez, Y.; Ruzbarsky, J.; Wang, Y.; Kolson, D.L.; Jordan-Sciutto, K.L. Activation Status of Integrated Stress Response Pathways in Neurones and Astrocytes of HIV-Associated Neurocognitive Disorders (HAND) Cortex. Neuropathol. Appl. Neurobiol. 2012, 38, 175–200. [Google Scholar] [CrossRef]
- Guo, H.; Wang, Q.; Ghneim, K.; Wang, L.; Rampanelli, E.; Holley-Guthrie, E.; Cheng, L.; Garrido, C.; Margolis, D.M.; Eller, L.A.; et al. Multi-Omics Analyses Reveal That HIV-1 Alters CD4+ T Cell Immunometabolism to Fuel Virus Replication. Nat. Immunol. 2021, 22, 423–433. [Google Scholar] [CrossRef]
- Palmer, C.S.; Ostrowski, M.; Gouillou, M.; Tsai, L.; Yu, D.; Zhou, J.; Henstridge, D.C.; Maisa, A.; Hearps, A.C.; Lewin, S.R.; et al. Increased Glucose Metabolic Activity Is Associated with CD4+ T-Cell Activation and Depletion during Chronic HIV Infection. AIDS 2014, 28, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Valle-Casuso, J.C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M.; et al. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4+ T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 2019, 29, 611–626.e5. [Google Scholar] [CrossRef]
- Loisel-Meyer, S.; Swainson, L.; Craveiro, M.; Oburoglu, L.; Mongellaz, C.; Costa, C.; Martinez, M.; Cosset, F.-L.; Battini, J.-L.; Herzenberg, L.A.; et al. Glut1-Mediated Glucose Transport Regulates HIV Infection. Proc. Natl. Acad. Sci. USA 2012, 109, 2549–2554. [Google Scholar] [CrossRef]
- Yang, X.; Xia, R.; Yue, C.; Zhai, W.; Du, W.; Yang, Q.; Cao, H.; Chen, X.; Obando, D.; Zhu, Y.; et al. ATF4 Regulates CD4+ T Cell Immune Responses through Metabolic Reprogramming. Cell Rep. 2018, 23, 1754–1766. [Google Scholar] [CrossRef]
- Clerc, I.; Moussa, D.A.; Vahlas, Z.; Tardito, S.; Oburoglu, L.; Hope, T.J.; Sitbon, M.; Dardalhon, V.; Mongellaz, C.; Taylor, N. Entry of Glucose- and Glutamine-Derived Carbons into the Citric Acid Cycle Supports Early Steps of HIV-1 Infection in CD4 T Cells. Nat. Metab. 2019, 1, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Yero, A.; Bouassa, R.-S.M.; Ancuta, P.; Estaquier, J.; Jenabian, M.-A. Immuno-Metabolic Control of the Balance between Th17-Polarized and Regulatory T-Cells during HIV Infection. Cytokine Growth Factor Rev. 2023, 69, 1–13. [Google Scholar] [CrossRef]
- Sundrud, M.S.; Koralov, S.B.; Feuerer, M.; Calado, D.P.; Kozhaya, A.E.; Rhule-Smith, A.; Lefebvre, R.E.; Unutmaz, D.; Mazitschek, R.; Waldner, H.; et al. Halofuginone Inhibits TH17 Cell Differentiation by Activating the Amino Acid Starvation Response. Science 2009, 324, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, A.; Monteiro, P.; Chomont, N.; Diaz-Griffero, F.; Said, E.A.; Fonseca, S.; Wacleche, V.; El-Far, M.; Boulassel, M.-R.; Routy, J.-P.; et al. Peripheral Blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T Cells Are Highly Permissive to HIV-1 Infection. J. Immunol. 2010, 184, 1604–1616. [Google Scholar] [CrossRef]
- Gosselin, A.; Wiche Salinas, T.R.; Planas, D.; Wacleche, V.S.; Zhang, Y.; Fromentin, R.; Chomont, N.; Cohen, É.A.; Shacklett, B.; Mehraj, V.; et al. HIV Persists in CCR6+CD4+ T Cells from Colon and Blood during Antiretroviral Therapy. AIDS 2017, 31, 35–48. [Google Scholar] [CrossRef]
- Estes, J.D.; Harris, L.D.; Klatt, N.R.; Tabb, B.; Pittaluga, S.; Paiardini, M.; Barclay, G.R.; Smedley, J.; Pung, R.; Oliveira, K.M.; et al. Damaged Intestinal Epithelial Integrity Linked to Microbial Translocation in Pathogenic Simian Immunodeficiency Virus Infections. PLoS Pathog. 2010, 6, e1001052. [Google Scholar] [CrossRef]
- Favre, D.; Lederer, S.; Kanwar, B.; Ma, Z.-M.; Proll, S.; Kasakow, Z.; Mold, J.; Swainson, L.; Barbour, J.D.; Baskin, C.R.; et al. Critical Loss of the Balance between Th17 and T Regulatory Cell Populations in Pathogenic SIV Infection. PLoS Pathog. 2009, 5, e1000295. [Google Scholar] [CrossRef] [PubMed]
- McGary, C.S.; Alvarez, X.; Harrington, S.; Cervasi, B.; Ryan, E.S.; Iriele, R.I.; Paganini, S.; Harper, J.L.; Easley, K.; Silvestri, G.; et al. The Loss of CCR6+ and CD161+ CD4+ T-Cell Homeostasis Contributes to Disease Progression in SIV-Infected Rhesus Macaques. Mucosal Immunol. 2017, 10, 1082–1096. [Google Scholar] [CrossRef]
- Raffatellu, M.; Santos, R.L.; Verhoeven, D.E.; George, M.D.; Wilson, R.P.; Winter, S.E.; Godinez, I.; Sankaran, S.; Paixao, T.A.; Gordon, M.A.; et al. Simian Immunodeficiency Virus-Induced Mucosal Interleukin-17 Deficiency Promotes Salmonella Dissemination from the Gut. Nat. Med. 2008, 14, 421–428. [Google Scholar] [CrossRef]
- Yero, A.; Farnos, O.; Rabezanahary, H.; Racine, G.; Estaquier, J.; Jenabian, M.-A. Differential Dynamics of Regulatory T-Cell and Th17 Cell Balance in Mesenteric Lymph Nodes and Blood Following Early Antiretroviral Initiation during Acute Simian Immunodeficiency Virus Infection. J. Virol. 2019, 93, e00371-19. [Google Scholar] [CrossRef]
- Sears, T.K.; Angelastro, J.M. The Transcription Factor ATF5: Role in Cellular Differentiation, Stress Responses, and Cancer. Oncotarget 2017, 8, 84595–84609. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.B.; Mitchelmore, C.; Kjaerulff, K.M.; Rasmussen, T.E.; Pedersen, K.M.; Jensen, N.A. Mouse Atf5: Molecular Cloning of Two Novel mRNAs, Genomic Organization, and Odorant Sensory Neuron Localization. Genomics 2002, 80, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Vinson, C.; Myakishev, M.; Acharya, A.; Mir, A.A.; Moll, J.R.; Bonovich, M. Classification of Human B-ZIP Proteins Based on Dimerization Properties. Mol. Cell Biol. 2002, 22, 6321–6335. [Google Scholar] [CrossRef]
- Chen, M.; Liu, Y.; Yang, Y.; Qiu, Y.; Wang, Z.; Li, X.; Zhang, W. Emerging Roles of Activating Transcription Factor (ATF) Family Members in Tumourigenesis and Immunity: Implications in Cancer Immunotherapy. Genes Dis. 2022, 9, 981–999. [Google Scholar] [CrossRef]
- Paerhati, P.; Liu, J.; Jin, Z.; Jakoš, T.; Zhu, S.; Qian, L.; Zhu, J.; Yuan, Y. Advancements in Activating Transcription Factor 5 Function in Regulating Cell Stress and Survival. Int. J. Mol. Sci. 2022, 23, 7129. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Shaban, M.S.; Albert, B.V.; Gökçen, A.; Kracht, M. The Crosstalk of Endoplasmic Reticulum (ER) Stress Pathways with NF-κB: Complex Mechanisms Relevant for Cancer, Inflammation and Infection. Biomedicines 2018, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.-C.; Wang, J.-M.; Hsieh, W.-C.; Chang, Y.; Su, I.-J. Up-Regulation of Activating Transcription Factor-5 Suppresses SAP Expression to Activate T Cells in Hemophagocytic Syndrome Associated with Epstein-Barr Virus Infection and Immune Disorders. Am. J. Pathol. 2008, 173, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, X.; Che, Y.; Zhang, Y.; Tang, S.; Liao, Y.; Na, R.; Xiong, X.; Liu, L.; Li, Q. A Cellular Response Protein Induced during HSV-1 Infection Inhibits Viral Replication by Interacting with ATF5. Sci. China Life Sci. 2013, 56, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, B.; Qian, D.; Wang, M.; Huang, R.; Wei, L.; Li, L.; Zhang, L.; Liu, D.X. Human Cytomegalovirus Immediate-Early Protein Promotes Survival of Glioma Cells through Interacting and Acetylating ATF5. Oncotarget 2017, 8, 32157–32170. [Google Scholar] [CrossRef]
- Badu, P.; Pager, C.T. Activation of ATF3 via the Integrated Stress Response Pathway Regulates Innate Immune and Autophagy Processes to Restrict Zika Virus. bioRxiv 2023. bioRxiv:2023.07.26.550716. [Google Scholar] [CrossRef]
- Bernier, A.; Cleret-Buhot, A.; Zhang, Y.; Goulet, J.-P.; Monteiro, P.; Gosselin, A.; DaFonseca, S.; Wacleche, V.S.; Jenabian, M.-A.; Routy, J.-P.; et al. Transcriptional Profiling Reveals Molecular Signatures Associated with HIV Permissiveness in Th1Th17 Cells and Identifies Peroxisome Proliferator-Activated Receptor Gamma as an Intrinsic Negative Regulator of Viral Replication. Retrovirology 2013, 10, 160. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Family | Virus | Regulation of ATF4 (*) | Effect of ATF4 Regulation on Viral Replication (**) | Other Major Findings Related to ATF4 and Viral Infection. | Refs. |
|---|---|---|---|---|---|
| Adenoviridae | Adenovirus type 2 (AdV-2) | + (t) | ND | ATF4 transcript is transiently increased before being down-regulated after the onset of the adenovirus early gene expression. | [12] |
| Arteriviridae | Porcine reproductive and respiratory syndrome virus (PRRSV) | + (p) | [+] | ATF4 localizes to cytoplasmic viral replication complexes by the viral non-structural proteins nsp2/3. | [13] |
| Asfaviridae | African swine fever virus (ASFV) | − (p) | [+] | The viral protein DP71L inhibits the induction of ATF4 and its downstream target, CHOP, by promoting eIF2α dephosphorylation. | [14] |
| Bornaviridae | Borna disease virus (BDV) | + (p, n) | ND | ATF4 nuclear localization increases in cerebellar cells but not in the hippocampus of infected animals. | [15] |
| Caliciviridae | Rabbit hemorrhagic disease virus (RHDV) | + (t) | ND | ATF4 and CHOP mRNA levels increase are associated with apoptosis induction. | [16] |
| Circoviridae | Porcine circovirus type 2 (PCV2) | + (p) | [+] | The infection activates the PERK/eIF2α/ATF4/CHOP axis. | [17] |
| + (p) | ND | The viral proteins Replicase and Capsid induce the PERK/eIF2α/ATF4/CHOP axis. | [18] | ||
| + (t, p) | [+] | The viral protein ORF5 induces autophagy via the PERK/eIF2α/ATF4 and mTOR/ERK1/2/AMPK signaling pathways. | [19] | ||
| Coronaviridae | Coronavirus infectious bronchitis virus (IBV) | + (p) | [+] | ATF4 is up-regulated through PERK- and PKR-mediated eIF2α phosphorylation. | [20] |
| Nephropathogenic infectious bronchitis virus (NIBV) | + (t, p) | ND | Upon infection, the BiP/PERK/ATF4 signaling pathway is activated and induction of renal apoptosis is observed. | [21] | |
| Porcine deltacoronavirus (PDCoV) | + (t) | [−] | The infection activates the PERK/eIF2α/ATF4 axis and induces host translation attenuation. | [22] | |
| Porcine epidemic diarrhea virus (PEDV) | + (t, p, n) | ND | The ATF4 protein is present in apoptotic cells. | [23] | |
| Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) | − (p) | ND | Despite ISR activation and translational arrest, ATF4 and CHOP protein levels are not increased in infected cells. | [24] | |
| Flaviviridae | Bovine viral diarrhea virus (BVDV) | + (p) +/− (n) | [+] | Cytopathic BVDV induces ATF4 nuclear translocation and activates autophagy. Non-cytopathic BVDV induces ATF4 perinuclear localization but no autophagy. | [25] |
| Dengue virus (DENV) | + (n) | ND | None. | [26] | |
| + (p) | [+] | None. | [27] | ||
| Hepatitis C virus (HCV) | + (p) | ND | ATF4 and ATF6 pathways contribute to the induction of CHOP in HCV replicon cells that showed an increased vulnerability to oxidant injury. | [28] | |
| + (t) | ND | HCV induces chronic ER stress. | [29] | ||
| + (p) | ND | The viral core protein induces the PERK/ATF4 branch of the UPR which up-regulates the autophagy gene ATG12. | [30] | ||
| + (t) | ND | ATF4 may contribute to autophagy regulation during infection. | [31] | ||
| + (t, p) | ND | Cells expressing HCV proteins and exposed to oxidative stress adapt to cellular stress through eIF2α/ATF4 activation. | [32] | ||
| Japanese encephalitis virus (JEV) | + (t) | ND | None. | [33] | |
| + (t) | ND | None. | [34] | ||
| + (p) | [−] | The viral protein NS4B activates PERK, which induces apoptosis via the PERK/ATF4/CHOP pathway. | [35] | ||
| Tembusu virus (TMUV) | + (t, p) | ND | CHOP induction leads to caspase-3 activation. | [36] | |
| West Nile virus (WNV) | + (p, n) | [+] | ATF4 is involved in the up-regulation of GSH levels and the inhibition of stress granule formation induced by infection. | [37] | |
| Zika virus (ZIKV) | + (t) | ND | Upon infection, ATF4 transcript level is weakly increased. | [38] | |
| − (t) | ND | None. | [34] | ||
| + (p) | ND | The infection transiently activates ATF4 but phosphorylation of PERK and eIF2α is sustained. | [39] | ||
| Hepadnaviridae | Hepatitis B virus (HBV) | + (p) | ND | The reduction in intracellular ATP levels by the viral protein HBx induces ATF4 binding to the promoter of the COX2 gene and its transcription. | [40] |
| − (p) | ND | The viral HBx protein localizes in the ER lumen and directly interacts with BiP. This interaction results in suppression of eIF2α phosphorylation, which decreases the levels of ATF4/CHOP/Bcl-2. | [41] | ||
| + (t, n) | ND | HBV, with viral polymerase carrying the rt269L polymorphism, improves mitochondrial dynamics and enhances the autophagic flux, mainly thanks to the activation of the PERK/eIF2α/ATF4 signaling. | [42] | ||
| Herpesviridae | Epstein–Barr virus (EBV) | + (p) | ND | LMP1 increases the ATF4 protein level through PERK/eIF2α phosphorylation. ATF4 transactivates LMP1. | [43] |
| Human cytomegalovirus (HCMV) | + (t,p) | ND | The infection activates PERK, but the amount of phosphorylated eIF2α is limited and no translation attenuation is detected. | [44] | |
| + (p) | ND | The viral protein pUL38 induces phosphorylation of PERK and eIF2α, resulting in the accumulation of the ATF4 protein and cell protection against ER stress. | [45] | ||
| + (p) | ND | The viral protein UL148 activates ATF4 mainly through the PERK/eIF2α pathway. | [46] | ||
| Human herpes virus 6A (HHV-6A) | + (p) | ND | Induction of the PKR/eIF2α pathway results in a moderate increase in the ATF4 protein level, which peaks at the final stages of infection. | [47] | |
| Human herpes virus-8 (HHV-8) | + (t, p) | [+] | ATF4 induces MCP-1 production and pro-angiogenic properties in endothelial cells. | [48] | |
| + (p) | [+] | The viral protein ORF45 increases eIF2α phosphorylation and ATF4 translation, which in turn up-regulates the expression of lysosome-associated membrane protein 3 (LAMP3). | [49] | ||
| Herpes simplex virus-1 (HSV-1) | + (t, p) | ND | HSV-1 disarms the ER UPR in the early stages of viral infection. The activity of the eIF2α/ATF4 signaling increases at the final stage of HSV-1 replication. | [50] | |
| Murine cytomegalovirus (MCMV) | + (p) | [+] | MCMV activates the PERK/ATF4 pathway but only induces a subset of ATF4 targets. ATF4 is required for efficient viral DNA synthesis and late gene expression during a low-multiplicity infection. | [51] | |
| Murine gamma herpes virus 68 (MHV68) | + (p) | [−] | In response to ER stress, ATF4 inhibits B-cell receptor (BCR)-mediated MHV68 lytic gene expression by directly inhibiting the transcription of RTA, the MHV68 lytic switch transactivator. In a negative feedback loop, UPR-induced CHOP is required for and promotes BCR-mediated MHV68 lytic replication by decreasing upstream BiP and ATF4 protein levels. | [52] | |
| Pseudorabies virus (PRV) | + (t) | [+] | The eIF2α/ATF4 pathway is activated during infection. PRV-induces apoptosis in later stages of infection through the CHOP/Bcl-2 axis. Overexpression of BiP or ER stress-inducing treatment can enhance PRV production. | [53] | |
| + (t, p) | [+] | Infection-induced ER stress leads to PERK activation and up-regulation of ATF4, CHOP, and GADD34. | [54] | ||
| Paramyxoviridae | Newcastle disease virus (NDV) | + (p, n) | [+] | The PKR/eIF2α/ATF4 pathway leads to an increase in the GADD34 protein level. GADD34, in conjunction with PP1, dephosphorylates eIF2α and restores global protein translation, benefiting virus protein synthesis. | [55] |
| + (p) | [+] | Induction of the PERK/eIF-2α/ATF4/CHOP signaling pathway is involved in the cyclin D1-dependent G0/G1 phase cell cycle arrest. | [56] | ||
| Sendai Virus (SV) | + (p) | ND | IRF7 up-regulates ATF4 activity and protein level, whereas ATF4 in return inhibits IRF7 activation. | [57] | |
| Parvoviridae | Porcine parvovirus (PPV) | + (t) | [−] | CHOP inhibits PPV replication by promoting apoptosis. ATF4 knockdown promotes PPV replication. | [58] |
| Picornaviridae | Foot-and-mouth disease virus (FMDV) | + (p) | [+] | The capsid protein VP2 induces autophagy through the eIF2α/ATF4/AKT/mTOR cascade, and interacts with HSPB1. | [59] |
| Group B coxsackievirus (CVB) | − (p) | [+] | PERK is activated and eIF2α is phosphorylated, but ATF4 protein levels do not increase. The ATF4/CHOP branch is blunted, thus inhibiting cell death. | [60] | |
| Poxviridae | Myxoma virus (MYXV) | + (t) − (p) | ND | PERK is activated and eIF2α is phosphorylated, but ATF4 translation is inhibited, which prevents MCL1 and CHOP transactivation. | [61] |
| Reoviridae | Reovirus | + (p) | [+] | The relative impact of ATF4 on viral replication depends on the infecting viral strain. | [62] |
| Rhabdoviridae | Vesicular stomatitis virus (VSV) | ND | [+] | None. | [57] |
| Togaviridae | Chikungunya virus (CHIKV) | − (t) | ND | ER UPR induction is primed since the phosphorylation of eIF2α and partial splicing of the XBP1 mRNA are detected, but the viral protein nsP2 inhibits the transcription of a reporter gene under the control of the ATF4 promoter. | [63] |
| Venezuelan equine encephalitis virus (VEEV) | + (p) | ND | None. | [64] |
| Models | Major Findings | Ref. | |
|---|---|---|---|
| Replication | HIV-1 infected CD4+ Jurkat T cells. | Cell transfection with an ATF4-encoding plasmid up-regulates the HIV-1 proviral genome levels (qPCR of gag gene) and increases viral release (ELISA of p24). | [6] |
| 293 T cells transiently transfected with a plasmid encoding the HIV-1 genome and GFP gene. | siRNA directed against ATF4 transcripts decreases the viral release (ELISA of p24) and Gag protein level (WB). | [7] | |
| Reactivation | U1 cells * | Cell nucleofection with an ATF4-encoding plasmid increases the viral production in the cell culture supernatant (qPCR of gag gene and p24 levels by WB). | [6] |
| J-Lat A1 ** and U1 cells * treated by a GCN2 inhibitor or supplemented with amino acids. | Inhibition of GCN2/ATF4 signaling represses the transcription of HIV-1 (real time qPCR with LTR primers). | [8] | |
| J-Lat cells and CD4+ T cells from HIV-1 infected individuals | FOXO1 inhibitor-induced reactivation of HIV-1 is reduced by pharmacological inhibition of PERK/ATF4 (GFP reporter gene or dddPCR with LTR primers). | [9] | |
| J-Lat A1, 2D10 *** cells and primary CD4+ T cells | Induction of the ISR/ATF4 signaling with a specific agonist of BiP, induces HIV-1 transcriptional activity (real time qPCR with LTR primers). | [10] | |
| ATF4 Target Genes | Model Related to HIV-1 Infection | Major Findings | Refs. |
|---|---|---|---|
| BIM/BCL2L11 | T cells derived from BIM−/− knockout mice treated with Tat. | BIM facilitates Tat-induced apoptosis. | [194] |
| CD4+ T cells from pathogenic SIVmac251-infected rhesus macaques. | Infection by SIV up-regulates death ligand CD95L and proapoptotic BIM and BAK but not BAX protein levels. | [116] | |
| Latently HIV-1-infected macrophages and lymph nodes, and brain of HIV-1-infected individuals without detectable viral replication. | BIM is up-regulated and recruited into mitochondria both in vitro and in vivo in latently infected cells that are protected from apoptosis. | [195] | |
| SH-SY5Y cells treated with Tat. | FOXO3 down-regulates BCL2 transcript and protein levels and up-regulates BIM transcript and protein levels after entering the nucleus, eventually causing cellular apoptosis. | [196] | |
| Monocytes-derived macrophages purified from PBMCs *. | Immunofluorescence analysis shows structural alterations in the mitochondrial architecture and an increase in BIM protein levels in the cytoplasm of infected cells. | [197] | |
| TID1/DNAJA3 | CEM-GFP cells transfected with a plasmid encoding the HIV-1 genome and GFP gene. | HIV-1 infection increases TID1 transcript levels. | [198] |
| HEK-293T cells transfected with either a Luciferase-encoding reporter vector or a plasmid encoding the HIV-1 genome and GFP gene. | TID1 increases HIV-1 LTR-driven gene expression and the viral p24 antigen release. | [199] | |
| G0S2 | PBMCs and myeloid monocyte-derived dendritic cells treated with virus-like particles containing the HIV-1 Pr55gag precursor protein and gp120 molecule anchored through the trans-membrane portion of the Epstein–Barr virus gp220/350. | G0S2 transcript levels are increased in dendritic cells. | [200] |
| THP-1 cells infected with a replication- deficient HIV-1 encoding the envelope glycoproteins from the vesicular stomatitis virus (VSV-G). | G0S2 transcript levels are down-regulated in cells containing an integrated provirus, compared to bystander uninfected cells or cells harboring pre-integration viral complexes. | [201] | |
| MCL1 | PBMCs of HIV-1-infected individuals. | Apoptosis and viral load are inversely correlated with MCL1 mRNA levels. | [202] |
| Monocyte-derived macrophages purified from PBMCs. | The expression of the MCL1 gene is up-regulated in macrophages infected with wild-type HIV-1 and in mock-infected macrophages that had been stimulated with M-CSF. However, MCL1 is not up-regulated in macrophages infected with a Δenv HIV-1. | [203] | |
| PBMCs of HIV-1-infected patients before and during successful ART. | After 12 months of therapy, the expression of MCL1 appears significantly up-regulated. | [204] | |
| Monocyte-derived macrophages or monocyte-derived dendritic cells incubated with R5 HIV-1 Bal. | HIV-1 infection decreases the Mcl-1 protein level but increases Bax and Bak. | [205] | |
| Vpr-treated monocyte-derived macrophages. | Resistance to Vpr-induced apoptosis is specifically mediated by cIAP1/2 genes independently from Bcl-xL and Mcl-1, which play a key role in maintaining cell viability independently of the viral protein. | [206] | |
| HIV-1-infected macrophages and microglia. | Cells become viral reservoirs in response to acute infection through a BIM-dependent mechanism. | [195] | |
| THP-1-derived macrophages. | HIV-1 infection increases expression of the anti-apoptotic genes MCL1, BCL2 and BCL2L1 that encodes Bcl-xL. | [207] | |
| PBMCs of uninfected donors and HIV-1-positive patients treated by cART *. | Overexpression of MCL1 is detected in PBMCs of cART-treated patients. | [208] | |
| Neutrophils from either healthy individuals, or HIV-1 patients whether asymptomatic, symptomatic, or ART receivers. | HIV-1 infection increases MCL1 transcript levels in vivo, and ART partially reduces this increase. | [209] | |
| NOXA/PMAIP1 | Human CD4+ T cells infected with HIV-1 viruses lacking Env, Vpr, or Nef. Human PBMCs infected with wild-type HIV-1 viruses of different tropisms. | HIV-1 infection increases NOXA transcript levels, which is associated with cell death. | [210] |
| PUMA/BBC3 | Circulating CD4+ lymphocytes from untreated HIV-1 infected donors. | HIV-1 infection increases Puma protein levels, which drop upon ART. | [211] |
| HIV-1-associated encephalitis brain sections. | HIV-1 infection increases the Puma protein level in dying syncytia and neurons. | [212] | |
| Murine cortical neuron culture treated with gp120 III. | Gp120 III is sufficient to increase Puma protein levels and induce cell death. | [213] | |
| CD4+ primary T cells infected with HIV-1 lacking Env, Vpr, or Nef genes. | The Env, Vpr and Nef are not necessary for HIV-1-induced PUMA transcript levels increase and HIV-1-mediated cell death. | [210] | |
| TMBIM5/GHITM | Monocytes from control and HIV-1 patients. | TMBIM5 transcript levels are decreased in HIV-1-infected monocytes. | [214] |
| Brain from HIV-1-HAND * patients. | TMBIM5 transcript levels are increased mainly in HIV-1-HAND patient astrocytes. | [215] | |
| TP53BP2/ASPP2 | Primary cortical neuron cultures treated with gp120 protein. | A high dose of gp120 stimulates the interaction of TP53BP2 with p53, which induces BAX transcription and contributes to caspase-3 cleavage. | [216] |
| SH-SY5Y neuroblastoma cells treated with gp120 protein. | TP53BP2 regulates autophagy and apoptosis differently depending on the dose of gp120. | [217] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corne, A.; Adolphe, F.; Estaquier, J.; Gaumer, S.; Corsi, J.-M. ATF4 Signaling in HIV-1 Infection: Viral Subversion of a Stress Response Transcription Factor. Biology 2024, 13, 146. https://doi.org/10.3390/biology13030146
Corne A, Adolphe F, Estaquier J, Gaumer S, Corsi J-M. ATF4 Signaling in HIV-1 Infection: Viral Subversion of a Stress Response Transcription Factor. Biology. 2024; 13(3):146. https://doi.org/10.3390/biology13030146
Chicago/Turabian StyleCorne, Adrien, Florine Adolphe, Jérôme Estaquier, Sébastien Gaumer, and Jean-Marc Corsi. 2024. "ATF4 Signaling in HIV-1 Infection: Viral Subversion of a Stress Response Transcription Factor" Biology 13, no. 3: 146. https://doi.org/10.3390/biology13030146
APA StyleCorne, A., Adolphe, F., Estaquier, J., Gaumer, S., & Corsi, J. -M. (2024). ATF4 Signaling in HIV-1 Infection: Viral Subversion of a Stress Response Transcription Factor. Biology, 13(3), 146. https://doi.org/10.3390/biology13030146