Oncolytic Newcastle Disease Virus as Cutting Edge between Tumor and Host

Abstract
: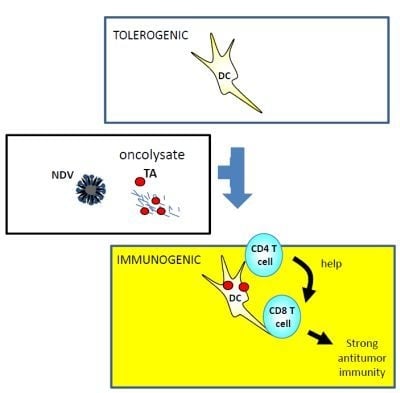
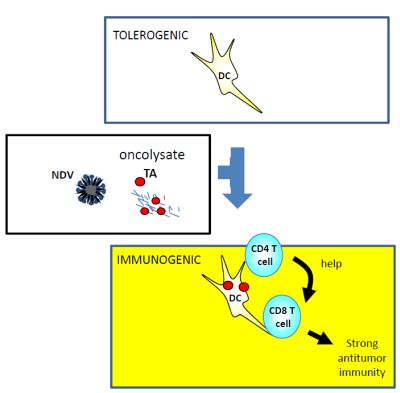{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. NDV for Cancer Therapy
- (i)
- Binding, fusion, transduction of the viral genome and transcription of viral genes: This first step involves the binding of the virus—via a lectin-like cell binding domain of the HN molecule—to ubiquitously expressed host cell surface receptors expressing distinct carbohydrate side chains (i.e., α2-3 and α2-6-N-linked sialic acids [6]). This is followed by the activation of the fusion protein F, which is synthesized as an inactive precursor (F0, 67 kDa). During fusion, F undergoes proteolytic cleavage to yield the biologically active protein consisting of the disulfide-linked chains F1 (55 kDa) and F2 (12.5 kDa). The concerted action of HN and F leads to fusion of the viral membrane with the host cell membrane. This involves two receptor binding sites of the globular head of HN and activation of the HN stalk and of the F protein [7]. This membrane fusion event allows the viral genome to enter the cytoplasm of the host cell. There, the negative strand RNA-genome is transcribed into messenger RNAs and translated into viral proteins. The three proteins NP, P and L, which are produced in infected cells, are then used to assemble the nucleocapsid as antigenome.
- (ii)
- Viral replication (second step): The anti-genome is then used as a template for amplification of the viral genome. Very interestingly, NDV can trigger, shortly after infection, autophagy to enhance virus replication [8]. The M protein and the envelope proteins HN and F, after post-translational modification, move to the membrane where virus assembly and budding occurs [9]. In this process, single copies of the NDV genome become wrapped into an outer coat envelope that is made from the host cells’ plasma membrane.

2.1. Tumor Selective Replication and Safety Profile
2.2. Oncolytic Potential
2.3. Immunostimulatory Properties

3. Experience with NDV in Preclinical and Clinical Studies
3.1. Pioneering Studies (1950s–1970s)
3.2. Regain of Interest in NDV (1990s–2010s)
4. NDV as Adjuvant of Tumor Vaccine

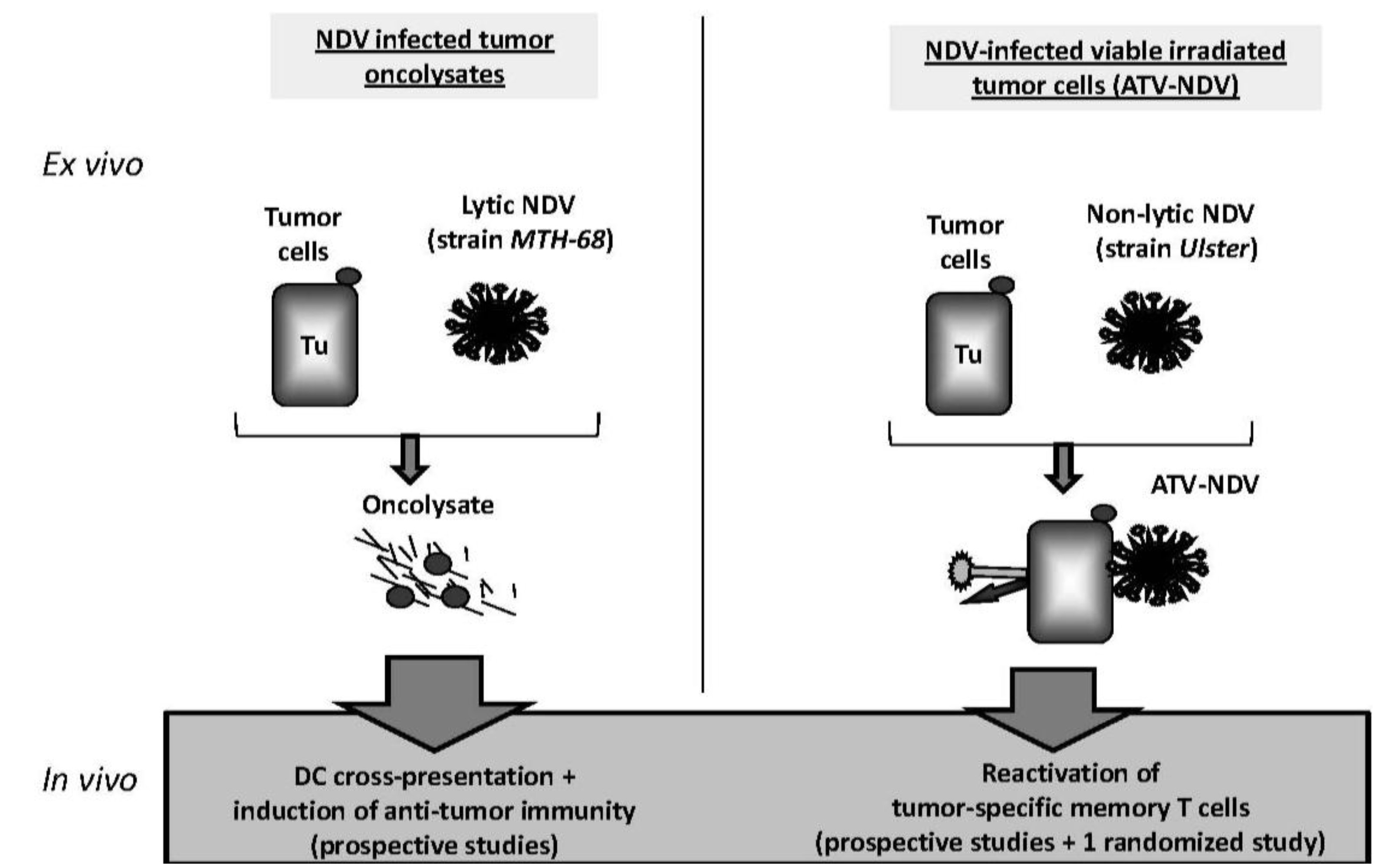
4.1. Immunotherapies with NDV Oncolysate
4.2. ATV-NDV: Live Autologous Virus Infected Tumor Cell Vaccine
5. DC Based Vaccines
5.1. Discovery of the DCs
5.2. DC-Based Vaccine
5.2.1. Disappointment about DC-Based Vaccine (1990–2010)
5.2.2. The Second Birth of DC Therapy (2010-today)
5.2.3. Future DC-Based Cancer Vaccine Therapy

6. Combining Systemic NDV Application with DC Vaccination
6.1. NDV-DC Therapy
6.2. Rationale for the Systemic Application of NDV
6.2.1. Production of High Levels of Type 1 IFN
6.2.2. Effect of Virus-Induced T Helper Cells on the Response to NDV-DC
6.2.3. Th1 Polarizing and Immune System Conditioning Effect
6.3. Rationale for the Use of DCs and NDV Oncolysate of Autologous Tumor Cells
6.3.1. Autologous DCs
6.3.2. Autologous Tumor Antigens
6.3.3. The Oncolytic and Immunostimulatory Properties of NDV
7. Conclusion


Acknowledgements
Conflicts of Interest
References
- Flanagan, A.D.; Love, R.; Tesar, W. Propagation of Newcastle disease virus in Ehrlich ascites cells in vitro and in vivo. Proc. Soc. Exp. Biol. Med. 1955, 90, 82–86. [Google Scholar] [CrossRef]
- The U.S. Food Drug Administration (FDA). Available online: http://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/ucm210012.htm/ (accessed on 15 March 2012).
- Alexander, D.J. Historical aspects. In Newcastle Disease; Alexander, D.J., Ed.; Kluwer Academic: Boston, MA, USA, 1988; pp. 1–10. [Google Scholar]
- Doyle, T.M. A hitherto unrecorded disease of fowls due to a filter-passing virus. J. Comp. Pathol. Ther. 1927, 40, 144–169. [Google Scholar]
- De Leeuw, O.; Peeters, B. Complete nucleotide sequence of Newcastle disease virus: Evidence for the existence of a new genus within the subfamily Paramyxovirinae. J. Gen. Virol. 1999, 80, 131–136. [Google Scholar]
- Sánchez-Felipe, L.; Villar, E.; Muñoz-Barroso, I. α2-3- and α2-6- N-linked sialic acids allow efficient interaction of Newcastle Disease Virus with target cells. Glycoconj. J. 2012, 29, 539–549. [Google Scholar] [CrossRef]
- Porotto, M.; Salah, Z.; DeVito, I.; Talekar, A.; Palmer, S.G.; Xu, R.; Wilson, I.A.; Moscona, A. The second receptor binding site of the globular head of the Newcastle disease virus hemagglutinin-neuraminidase activates the stalk of multiple paramyxovirus receptor binding proteins to trigger fusion. J. Virol. 2012, 86, 5730–5741. [Google Scholar]
- Meng, C.; Zhou, Z.; Jiang, K.; Yu, S.; Jia, L.; Wu, Y.; Liu, Y.; Meng, S.; Ding, C. Newcastle disease virus triggers autophagy in U251 glioma cells to enhance virus replication. Arch. Virol. 2012, 8, 1011–1018. [Google Scholar]
- Nagai, Y.; Hamaguchi, M.; Toyoda, T. Molecular biology of Newcastle disease virus. Prog. Vet. Microbiol. Immunol. 1989, 5, 16–64. [Google Scholar]
- Schirrmacher, V.; Fournier, P. Newcastle disease virus: A promising vector for viral therapy, immune therapy, and gene therapy of cancer. Methods Mol. Biol. 2009, 542, 565–605. [Google Scholar] [CrossRef]
- Russell, S.J. RNA viruses as virotherapy agents. Cancer Gene Ther. 2002, 9, 961–966. [Google Scholar] [CrossRef]
- Fiola, C.; Peeters, B.; Fournier, P.; Arnold, A.; Bucur, M.; Schirrmacher, V. Tumor-selective replication of Newcastle Disease Virus: Association with defects of tumor cells defence. Int. J. Cancer 2006, 119, 328–338. [Google Scholar] [CrossRef]
- Reichard, K.W.; Lorence, R.M.; Cascino, C.J.; Peeples, M.E.; Walter, R.J.; Fernando, M.B.; Reyes, H.M.; Greager, J.A. Newcastle disease virus selectively kills human tumor cells. J. Surg. Res. 1992, 52, 448–453. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Takimoto, T.; Scroggs, R.A.; Portner, A. Differentially regulated interferon response determines the outcome of Newcastle disease virus infection in normal and tumor cell lines. J. Virol. 2006, 80, 5145–5155. [Google Scholar] [CrossRef]
- Wilden, H.; Fournier, P.; Zawatzky, R.; Schirrmacher, V. Expression of RIG-I, IRF3, IFN-β and IRF7 determines resistance or susceptibility of cells to infection by Newcastle Disease Virus. Int. J. Oncol. 2009, 34, 971–982. [Google Scholar]
- Fournier, P.; Wilden, H.; Schirrmacher, V. Importance of retinoic acid-inducible gene I and of receptor for type I interferon for cellular resistance to infection by Newcastle disease virus. Int. J. Oncol. 2012, 40, 287–298. [Google Scholar]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Biyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate anti-viral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Schmidt, A.; Rothenfusser, S.; Hopfner, K.P. Sensing of viral nucleic acids by RIG-I: From translocation to translation. Eur. J. Cell Biol. 2012, 91, 78–85. [Google Scholar] [CrossRef]
- Melchjorsen, J.; Jensen, S.B.; Malmgaard, L.; Rasmussen, S.B.; Weber, F.; Bowie, A.G.; Matikainen, S.; Paludan, S.R. Activation of innate defense against a paramyxovirus is mediated by RIG-I and TLR7 and TLR8 in a cell-type-specific manner. J. Virol. 2005, 79, 12944–12951. [Google Scholar] [CrossRef]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of MDA-5 in type 1 IFN responses to polyriboinosinic: Polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; Endres, S.; Hartmann, G. 5'-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef]
- Bowie, A.G.; Fitzgerald, K.A. RIG-I: Tri-ing to discriminate between self and non-self RNA. Trends Immunol. 2007, 28, 147–150. [Google Scholar] [CrossRef]
- Taniguchi, T.; Takaoka, A. The interferon-α/β system in anti-viral responses: A multimodal machinery of gene regulation by the IRF family of transcription factors. Curr. Opin. Immunol. 2002, 14, 111–116. [Google Scholar] [CrossRef]
- Tailor, P.; Tamura, T.; Ozato, K. IRF family proteins and type 1 interferon induction in dendritic cells. Cell Res. 2006, 16, 134–140. [Google Scholar] [CrossRef]
- Lorence, R.M.; Roberts, M.S.; Groene, W.S.; Rabin, H. Replication-competent, oncolytic Newcastle disease virus for cancer therapy. In Replication-competent Viruses for Cancer Therapy. Monographs in Virology; Hern aiz Driever, P., Rabkin, S.D., Eds.; Karger: Basel, Switzerland, 2001; Volume 22, pp. 160–182. [Google Scholar]
- Lamb, R.A.; Parks, G.D. Paramyxoviridae: Their viruses and their replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1449–1496. [Google Scholar]
- Sinkovics, J.G.; Horvath, J.C. Newcastle disease virus (NDV): Brief history of its oncolytic strains. J. Clin. Virol. 2000, 16, 1–15. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Griesbach, A.; Ahlert, T. Anti-tumor effects of Newcastle Disease Virus in vivo: Local versus systemic effects. Int. J. Oncol. 2001, 18, 945–952. [Google Scholar]
- Apostolidis, L.; Schirrmacher, V.; Fournier, P. Host mediated anti-tumor effect of oncolytic Newcastle disease virus after locoregional application. Int. J. Oncol. 2007, 31, 1009–1019. [Google Scholar]
- Ravindra, P.V.; Tiwari, A.K.; Ratta, B.; Chaturvedi, U.; Palia, S.K.; Chauhan, R.S. Newcastle disease virus-induced cytopathic effect in infected cells is caused by apoptosis. Virus Res. 2009, 141, 13–20. [Google Scholar] [CrossRef]
- Elankumaran, S.; Rockemann, D.; Samal, S.K. Newcastle disease virus exerts oncolysis by both intrinsic and extrinsic caspase-dependent pathways of cell death. J. Virol. 2006, 80, 7522–7534. [Google Scholar] [CrossRef]
- Kumar, R.; Tiwari, A.K.; Chaturvedi, U.; Kumar, G.R.; Sahoo, A.P.; Rajmani, R.S.; Saxena, L.; Saxena, S.; Tiwari, S.; Kumar, S. Velogenic newcastle disease virus as an oncolytic virotherapeutics: in vitro characterization. Appl. Biochem. Biotechnol. 2012, 167, 2005–2022. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Molouki, A.; Yusoff, K. NDV-induced apoptosis in absence of Bax; evidence of involvement of apoptotic proteins upstream of mitochondria. Virol. J. 2012, 9, 179. [Google Scholar] [CrossRef]
- Ch'ng, W.C.; Stanbridge, E.J.; Yusoff, K.; Shafee, N. The oncolytic activity of newcastle disease virus in clear cell renal carcinoma cells in normoxic and hypoxic conditions: The interplay between VHL and interferon-β signaling. J. Interferon Cytokine Res. 2013. [Google Scholar] [CrossRef]
- Bian, H.; Wilden, H.; Fournier, P.; Peeters, B.; Schirrmacher, V. In vivo efficacy of systemic tumor targeting of a viral RNA vector with oncolytic properties using a bispecific adapter protein. Int. J. Oncol. 2006, 29, 1359–1369. [Google Scholar]
- Ahlert, T.; Schirrmacher, V. Isolation of a human melanoma adapted Newcastle disease virus mutant with highly selective replication patterns. Cancer Res. 1990, 50, 5962–5968. [Google Scholar]
- Schirrmacher, V.; Haas, C.; Bonifer, R.; Ahlert, T.; Gerhards, R.; Ertel, C. Human tumor cell modification by virus infection: An efficient and safe way to produce cancer vaccine with pleiotropic immune stimulatory properties when using Newcastle Disease Virus. Gene Ther. 1999, 6, 63–73. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Ahlert, T.; Pröbstle, T.; Steiner, H.H.; Herold-Mende, C.; Gerhards, R.; Hagmüller, E.; Steiner, H.H. Immunization with virus-modified tumor cells. Semin. Oncol. 1998, 25, 677–696. [Google Scholar]
- Ertel, C.; Millar, N.S.; Emmerson, P.T.; Schirrmacher, V.; von Hoegen, P. Viral hemagglutinin augments peptide-specific cytotoxic T cell responses. Eur. J. Immunol. 1993, 23, 2592–2596. [Google Scholar] [CrossRef]
- Washburn, B.; Schirrmacher, V. Human tumor cell infection by Newcastle Disease Virus leads to up-regulation of HLA and cell adhesion molecules and to induction of interferons, chemokines and finally apoptosis. Int. J. Oncol. 2002, 21, 85–93. [Google Scholar]
- Clemens, M.J.; Elia, A. The double-stranded RNA-dependent protein kinase PKR: Structure and function. J. Interferon Cytokine Res. 1997, 17, 503–524. [Google Scholar] [CrossRef]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell type-specific involvement of RIG-I in anti-viral response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Zeng, J.; Fournier, P.; Schirrmacher, V. Stimulation of human natural interferon-α response via paramyxovirus hemagglutinin lectin-cell interaction. J. Mol. Med. (Berl.) 2002, 80, 443–451. [Google Scholar] [CrossRef]
- Zeng, J.; Fournier, P.; Schirrmacher, V. Induction of interferon-α and tumor necrosis factor-related apoptosis-inducing ligand in human blood mononuclear cells by hemagglutinin-neuraminidase but not F protein of Newcastle disease virus. Virology 2002, 297, 19–30. [Google Scholar] [CrossRef]
- Janke, M.; Peeters, B.; de Leeuw, O.; Moorman, R.; Arnold, A.; Fournier, P.; Schirrmacher, V. Recombinant Newcastle Disease Virus (NDV) with inserted gene coding for GM-CSF as a new vector for cancer immunogene therapy. Gene Ther. 2007, 14, 1639–1649. [Google Scholar] [CrossRef]
- Washburn, B.; Weigand, M.A.; Grosse-Wilde, A.; Janke, M.; Stahl, H.; Rieser, E.; Sprick, M.R.; Schirrmacher, V.; Walczak, H. TNF-related apoptosis-inducing ligand mediated tumoricidal activity of human monocytes stimulated by Newcastle Disease Virus. J. Immunol. 2003, 170, 1814–1821. [Google Scholar]
- Bai, L.; Koopmann, J.; Fiola, C.; Fournier, P.; Schirrmacher, V. Dendritic cells pulsed with viral oncolysates potently stimulate autologous T cells from cancer patients. Int. J. Oncol. 2002, 21, 685–694. [Google Scholar]
- Jarahian, M.; Watzl, C.; Fournier, P.; Arnold, A.; Djandji, D.; Zahedi, S.; Cerwenka, A.; Paschen, A.; Schirrmacher, V.; Momburg, F. Activation of natural killer cells by Newcastle Disease Virus hemagglutinin-neuraminidase. J. Virol. 2009, 83, 8108–8121. [Google Scholar] [CrossRef]
- Von Hoegen, P.; Zawatzky, R.; Schirrmacher, V. Modifification of tumor cells by a low dose of Newcastle disease virus. III. Potentiation of tumor-specific cytolytic T cell activity via induction of interferon-α/beta. Cell. Immunol. 1990, 126, 80–90. [Google Scholar] [CrossRef]
- Schirrmacher, V. Antitumor immune memory and its activation for control of residual tumor cells and improvement of patient survival. In Virus Therapy of Human Cancers; Sinkovics, J., Horvath, J., Eds.; Marcel Decker: New York, NY, USA, 2004; pp. 481–531. [Google Scholar]
- Fujita, M.; Scheurer, M.E.; Decker, S.A.; McDonald, H.A.; Kohanbash, G.; Kastenhuber, E.R.; Kato, H.; Bondy, M.L.; Ohlfest, J.R.; Okada, H. Role of type 1 IFNs in antiglioma immunosurveillance—Using mouse studies to guide examination of novel prognostic markers in humans. Clin. Cancer Res. 2010, 16, 3409–3421. [Google Scholar] [CrossRef]
- Termeer, C.C.; Schirrmacher, V.; Bröcker, E.B.; Becker, J.C. Newcastle-Disease-Virus infection induces a B7-1/B7-2 independent T cell-co-stimulatory activity in human melanoma cells. Cancer Gene Ther. 2000, 7, 316–323. [Google Scholar]
- Goodbourn, L.D.; Randall, R.E. Interferons: Cell signalling, immune modulation, anti-viral response and virus countermeasures. J. Gen. Virol. 2000, 81, 2341–2364. [Google Scholar]
- Lindenmann, J. Viruses as immunological adjuvants in cancer. Biochim. Biophys. Acta. 1974, 355, 49–75. [Google Scholar]
- Sinkovics, J.; Horvath, J. New developments in the virus therapy of cancer: A historical review. Intervirology 1993, 36, 193–214. [Google Scholar]
- Goto, M.; Okazaki, M.; Yazaki, H. Oncolytic effect of Newcastle disease virus on Yoshida sarcoma (1). Jpn. J. Microbiol. 1959, 3, 171–181. [Google Scholar]
- Wheelock, E.F.; Dingle, J.H. Observations on the repeated administration of viruses to a patient with acute leukemia. A preliminary report. N. Engl. J. Med. 1964, 271, 645–651. [Google Scholar] [CrossRef]
- Cassel, W.A.; Garrett, R.E. Newcastle Disease Virus as an antineoplastic agent. Cancer 1965, 1, 863–888. [Google Scholar] [CrossRef]
- Jacotot, H. Oncolytic power in vivo of the Newcastle virus with regard to sarcoma ascitic Yoshida. C. R. Acad. Sci. Hebd. Seances Acad. Sci. D 1967, 26422, 2602–2603. [Google Scholar]
- Csatary, L.K. Viruses in the treatment of cancer. Lancet 1971, 2, 825. [Google Scholar] [CrossRef]
- Csatary, L.K.; Moss, R.W.; Beuth, J.; Torocsik, B.; Szeberenyi, J.; Bakacs, T. Beneficial treatment of patients with advanced cancer using a Newcastle disease virus vaccine (MTH-68/H). Anticancer Res. 1999, 19, 635–638. [Google Scholar]
- Csatary, L.K.; Eckhardt, S.; Bukosza, I.; Czegledi, F.; Fenyvesi, C.; Gergely, P.; Bodey, B.; Csatary, C.M. Attenuated veterinary virus vaccine for the treatment of cancer. Cancer Detect. Prev. 1993, 17, 619–627. [Google Scholar]
- Csatary, L.K.; Bakacs, T. Use of Newcastle disease virus vaccine (MTH-68/H) in a patient with high-grade glioblastoma. JAMA 1999, 281, 1588–1589. [Google Scholar] [CrossRef]
- Czegledi, A.; Wehmann, E.; Lomniczi, B. On the origins and relationships of Newcastle disease virus vaccine strains Hertfordshire and Mukteswar, and virulent strain Herts’33. Avian Pathol. 2003, 32, 271–276. [Google Scholar] [CrossRef]
- Nefedova, Y.; Huang, M.; Kusmartsev, S.; Bhattacharya, R.; Cheng, P.; Salup, R.; Jove, R.; Gabrilovich, D. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J. Immunol. 2004, 172, 464–474. [Google Scholar]
- Nelson, J. Scientific interest in newcastle disease virus is reviving. J. Natl. Cancer Inst. 1999, 91, 1708–1710. [Google Scholar] [CrossRef]
- Lorence, R.M.; Katubig, B.B.; Reichard, K.W.; Reyes, H.M.; Phuangsab, A.; Sassetti, M.D.; Walter, R.J.; Peeples, M.E. Complete regression of human fibrosarcoma xenografts after local Newcastle disease virus therapy. Cancer Res. 1994, 54, 6017–6021. [Google Scholar]
- Lorence, R.M.; Reichard, K.W.; Katubig, B.B.; Reyes, H.M.; Phuangsab, A.; Mitchell, B.R.; Cascino, C.J.; Walter, R.J.; Peeples, M.E. Complete regression of human neuroblastoma xenografts in athymic mice after local Newcastle disease virus therapy. J. Natl. Cancer Inst. 1994, 86, 1228–1233. [Google Scholar] [CrossRef]
- Lorence, R.M.; Roberts, M.S.; O'Neil, J.D.; Groene, W.S.; Miller, J.A.; Mueller, S.N.; Bamat, M.K. Phase 1 clinical experience using intravenous administration of PV701, an oncolytic Newcastle disease virus. Curr. Cancer Drug Targets 2007, 7, 157–167. [Google Scholar] [CrossRef]
- Pecora, A.L.; Rizvi, N.; Cohen, G.I.; Meropol, N.J.; Sterman, D.; Marshall, J.L.; Goldberg, S.; Gross, P.; O'Neil, J.D.; Groene, W.S.; et al. Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J. Clin. Oncol. 2002, 20, 2251–2266. [Google Scholar] [CrossRef]
- Laurie, S.A.; Bell, J.C.; Atkins, H.L.; Roach, J.; Bamat, M.K.; O'Neil, J.D.; Roberts, M.S.; Groene, W.S.; Lorence, R.M. A phase 1 clinical study of intravenous administration of PV701, an oncolytic virus, using two-step desensitization. Clin. Cancer Res. 2006, 12, 2555–2562. [Google Scholar] [CrossRef]
- Hotte, S.J.; Lorence, R.M.; Hirte, H.W.; Polawski, S.R.; Bamat, M.K.; O'Neil, J.D.; Roberts, M.S.; Groene, W.S.; Major, P.P. An optimized clinical regimen for the oncolytic virus PV701. Clin. Cancer Res. 2007, 13, 977–985. [Google Scholar] [CrossRef]
- Lorence, R.M.; Pecora, A.L.; Major, P.P.; Hotte, S.J.; Laurie, S.A.; Roberts, M.S.; Groene, W.S.; Bamat, M.K. Overview of phase I studies of intravenous administration of PV701, an oncolytic virus. Curr. Opin. Mol. Ther. 2003, 5, 618–624. [Google Scholar]
- Phuangsab, A.; Lorence, M.; Reichard, K.W.; Peeples, M.E.; Walter, R.J. Newcastle disease virus therapy of human tumor xenografts: Anti-tumor effects of local or systemic administration. Cancer Lett. 2001, 172, 27–36. [Google Scholar] [CrossRef]
- Csatary, L.K.; Gosztonyi, G.; Szeberenyi, J.; Fabian, Z.; Liszka, V.; Bodey, B.; Csatary, C.M. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J. Neurooncol. 2004, 67, 83–93. [Google Scholar] [CrossRef]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef]
- Austin, F.C.; Boone, C.W. Virus augmentation of the antigenicity of tumor cell extracts. Adv. Cancer Res. 1979, 30, 301–345. [Google Scholar] [CrossRef]
- Sinkovics, J.G.; Horvath, J.C. Virus therapy of human cancers. Melanoma Res. 2003, 13, 431–432. [Google Scholar] [CrossRef]
- Cassel, W.A.; Murray, D.R.; Torbin, A.H.; Olkowski, Z.L.; Moore, M.E. Viral oncolysate in the management of malignant melanoma. I. Preparation of the oncolysate and measurement of immunologic responses. Cancer 1977, 40, 672–679. [Google Scholar] [CrossRef]
- Murray, D.R.; Cassel, W.A.; Torbin, A.H.; Olkowski, Z.L.; Moore, M.E. Viral oncolysate in the management of malignant melanoma. II. Clinical studies. Cancer 1977, 40, 680–686. [Google Scholar] [CrossRef]
- Cassel, W.A.; Murray, D.R. A ten-year follow-up on stage II malignant melanoma patients treated postsurgically with Newcastle disease virus oncolysate. Med. Oncol. Tumor Pharmacother. 1992, 9, 169–171. [Google Scholar]
- Horvath, J.C. Newcastle disease virus: Its oncolytic properties oncolysate. In Viral Therapy of Human Cancers; Sinkovics, J.G., Horvath, J.C., Eds.; Marcel Dekker: New York, NY, USA, 2005; pp. 533–574. [Google Scholar]
- Schirrmacher, V. Clinical trials of antitumor vaccination with an autologous tumor cell vaccine modified by virus infection: Improvement of patient survival based on improved antitumor immune memory. Cancer Immunol. Immunother. 2005, 54, 587–598. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Ahlert, T.; Heicappell, R.; Appelhans, B.; von Hoegen, P. Successful application of non-oncogenic viruses for antimetastatic cancer immunotherapy. Cancer Rev. 1986, 5, 19–49. [Google Scholar]
- Heicappell, R.; Schirrmacher, V.; von Hoegen, P.; Ahlert, T.; Appelhans, B. Prevention of metastatic spread by post-operative immunotherapy with virally modified autologous tumor cells. I: Parameters for optimal therapeutic effects. Int. J. Cancer 1986, 37, 569–577. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Fournier, P. Newcastle Disease Virus: A Promising Vector for Viral Therapy of Cancer. Viral Ther. Cancer. 2009. [Google Scholar] [CrossRef]
- Ahlert, T.; Sauerbrei, W.; Bastert, G.; Ruhland, S.; Bartik, B.; Simiantonaki, N.; Schumacher, J.; Häcker, B.; Schumacher, M.; Schirrmacher, V. Tumor-cell number and viability as quality and efficacy parameters of autologous virus-modified cancer vaccines in patients with breast or ovarian cancer. J. Clin. Oncol. 1997, 15, 1354–1366. [Google Scholar]
- Ockert, D.; Schirrmacher, V.; Beck, N.; Stoelben, E.; Ahlert, T.; Flechtenmacher, J.; Hagmüller, E.; Buchcik, R.; Nagel, M.; Saeger, H.D. Newcastle Disease Virus infected intact autologous tumor cell vaccine for adjuvant active specific immunotherapy of resected colorectal carcinoma. Clin. Cancer Res. 1996, 2, 21–28. [Google Scholar]
- Schlag, P.; Manasterski, M.; Gerneth, T.; Hohenberger, P.; Dueck, M.; Herfarth, C.; Liebrich, W.; Schirrmacher, V. Active specific Immunotherapy with NDV modified autologous tumor cells following liver metastases resection in colorectal cancer: First evaluation of clinical response of a Phase II trial. Cancer Immunol. Immunother. 1992, 35, 325–330. [Google Scholar] [CrossRef]
- Steiner, H.H.; Bonsanto, M.M.; Beckhove, P.; Brysch, M.; Geletneky, K.; Ahmadi, R.; Schuele-Freyer, R.; Kremer, P.; Ranaie, G.; Matejic, D.; et al. Anti-tumor vaccination of patients with glioblastoma multiforme: A pilot study to assess: Feasibility, safety and clinical benefit. J. Clin. Oncol. 2004, 22, 4272–4281. [Google Scholar] [CrossRef]
- Karcher, J.; Dyckhoff, G.; Beckhove, P.; Reisser, C.; Brysch, M.; Ziouta, Y.; Helmke, B.H.; Weidauer, H.; Schirrmacher, V.; Herold-Mende, C. Anti-tumor vaccination with HNSCC with autologous virus-modified tumor cells. Cancer Res. 2004, 64, 8057–8061. [Google Scholar] [CrossRef]
- Schulze, T.; Kemmner, W.; Weitz, J.; Wernecke, K.D.; Schirrmacher, V.; Schlag, P.M. Efficiency of adjuvant active specific immunization with Newcastle disease virus modified tumor cells in colorectal cancer patients following resection of liver metastases: Results of a prospective randomized trial. Cancer Immunol. Immunother. 2009, 58, 61–69. [Google Scholar] [CrossRef]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type 1 peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Volchenkov, R.; Sprater, F.; Vogelsang, P.; Appel, S. The 2011 Nobel Prize in physiology or medicine. Scand. J. Immunol. 2012, 75, 1–4. [Google Scholar] [CrossRef]
- Nelson, D.J.; McMenamin, C.; McWilliam, A.S.; Brenan, M.; Holt, P.G. Development of the airway intraepithelial dendritic cell network in the rat from class-II major histocompatibility (Ia)-negative precursors: Differential regulation of Ia expression at different levels of the respiratory tract. J. Exp. Med. 1994, 179, 203–212. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Thompson, A.J.; Locarnini, S.A. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol. Cell. Biol. 2007, 85, 435–445. [Google Scholar] [CrossRef]
- Finkelman, F.D.; Lees, A.; Birnbaum, R.; Gause, W.C.; Morris, S.C. Dendritic cells can present antigen in vivo in a tolerogenic or immunogenic fashion. J. Immunol. 1996, 157, 1406–1414. [Google Scholar]
- Bevan, M.J. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 1976, 143, 1283–1288. [Google Scholar] [CrossRef]
- Albert, M.L.; Pearce, S.F.; Francisco, L.M.; Sauter, B.; Roy, P.; Silverstein, R.L.; Bhardwaj, N. Immature dendritic cells phagocytose apoptotic cells via αvbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J. Exp. Med. 1998, 188, 1359–1368. [Google Scholar] [CrossRef]
- Yamazaki, S.; Patel, M.; Harper, A.; Bonito, A.; Fukuyama, H.; Pack, M.; Tarbell, K.V.; Talmor, M.; Ravetch, J.V.; Inaba, K.; et al. Effective expansion of alloantigen-specific Foxp3+ CD25+ CD4+ regulatory T cells by dendritic cells during the mixed leukocyte reaction. Proc. Natl. Acad. Sci. USA 2006, 103, 2758–2763. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr. The immune system evolved to discriminate infectious nonself from non-infectious self. Immunol. Today 1992, 13, 11–16. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, danger and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Kalinski, P. Dendritic cells in immunotherapy of established cancer: Roles of signals 1, 2, 3 and 4. Curr. Opin. Invest. Drugs. 2009, 10, 526–535. [Google Scholar]
- Curtsinger, J.M.; Lins, D.C.; Mescher, M.F. Signal 3 determines tolerance versus full activation of naive CD8 T cells: Dissociating proliferation and development of effector function. J. Exp. Med. 2003, 197, 1141–1151. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Johnson, C.M.; Mescher, M.F. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J. Immunol. 2003, 171, 5165–5171. [Google Scholar]
- Curtsinger, J.M.; Mescher, M.F. Inflammatory cytokines as a third signal for T cell activation. Curr. Opin. Immunol. 2010, 22, 333–340. [Google Scholar] [CrossRef]
- Haring, J.S.; Badovinac, V.P.; Harty, J.T. Inflaming the CD8+ T cell response. Immunity 2006, 25, 19–29. [Google Scholar] [CrossRef]
- Dolan, B.P.; Gibbs, K.D., Jr.; Ostrand-Rosenberg, S. Tumor-specific CD4+ T cells are activated by “cross-dressed” dendritic cells presenting peptide-MHC class II complexes acquired from cell-based cancer vaccines. J. Immunol. 2006, 176, 1447–1455. [Google Scholar]
- Wakim, L.M.; Bevan, M.J. Cross-dressed dendritic cells drive memory CD8+ T cell activation after viral infection. Nature 2011, 471, 629–632. [Google Scholar] [CrossRef]
- Dolan, B.P.; Gibbs, K.D., Jr.; Ostrand-Rosenberg, S. Dendritic cells cross-dressed with peptide MHC class I complexes prime CD8+ T cells. J. Immunol. 2006, 177, 6018–6024. [Google Scholar]
- Kleindienst, P.; Brocker, T. Endogenous dendritic cells are required for amplification of T cell responses induced by dendritic cell vaccines in vivo. J. Immunol. 2003, 170, 2817–2823. [Google Scholar]
- Petersen, T.R.; Sika-Paotonu, D.; Knight, D.A.; Simkins, H.M.A.; Hermans, I.F. Exploiting the role of endogenous lymphoid-resident dendritic cells in the priming of NKT cells and CD8+ T cells to dendritic cell-based vaccines. PLoS One 2011, 6, e17657. [Google Scholar]
- Steinman, R.M.; Pope, M. Exploiting dendritic cells to improve vaccine efficacy. J. Clin. Invest. 2002, 109, 1519–1526. [Google Scholar]
- Inaba, K.; Metlay, J.P.; Crowley, M.T.; Steinman, R.M. Dendritic cells pulsed with protein antigens in vitro can prime antigen-specific, MHC-restricted T cells in situ. J. Exp. Med. 1990, 172, 631–640. [Google Scholar] [CrossRef]
- Inaba, K.; Inaba, M.; Romani, N.; Aya, H.; Deguchi, M.; Ikehara, S.; Muramatsu, S.; Steinman, R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992, 176, 1693–1702. [Google Scholar] [CrossRef]
- Hsu, F.J.; Benike, C.; Fagnoni, F.; Liles, T.M.; Czerwinski, D.; Taidi, B.; Engleman, E.G.; Levy, R. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat. Med. 1996, 2, 52–58. [Google Scholar] [CrossRef]
- Nestle, F.O.; Alijagic, S.; Gilliet, M.; Sun, Y.; Grabbe, S.; Dummer, R.; Burg, G.; Schadendorf, D. Vaccination of melanoma patients with peptide-or tumor lysate-pulsed dendritic cells. Nat. Med. 1998, 4, 328–332. [Google Scholar] [CrossRef]
- Small, E.J.; Schellhammer, P.F.; Higano, C.S.; Redfern, C.H.; Nemunaitis, J.J.; Valone, F.H.; Verjee, S.S.; Jones, L.A.; Hershberg, R.M. Placebo-controlled phase III trial of immunologic therapy with Sipuleucel-T (APC-8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J. Clin. Oncol. 2006, 24, 3089–3094. [Google Scholar] [CrossRef]
- Peoples, G.E.; Holmes, J.P.; Hueman, M.T.; Mittendorf, E.A.; Amin, A.; Khoo, S.; Dehqanzada, Z.A.; Gurney, J.M.; Woll, M.M.; Ryan, G.B.; et al. Combined clinical trial results of a HER2/neu (E-75) vaccine for the prevention of recurrence in high-risk breast cancer patients: US Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Clin. Cancer Res. 2008, 14, 797–803. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef]
- Tacken, P.J.; de Vries, I.J.; Torensma, R.; Figdor, C.G. Dendritic-cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 2007, 7, 790–802. [Google Scholar] [CrossRef]
- Brown, R.D.; Pope, B.; Murray, A.; Esdale, W.; Sze, D.M.; Gibson, J.; Ho, P.J.; Hart, D.; Joshua, D. Dendritic cells from patients with myeloma are numerically normal but functionally defective as they fail to up-regulate CD80 (B7–1) expression after huCD40LT stimulation because of inhibition by transforming growth factor-β1 and interleukin-10. Blood 2001, 98, 2992–2998. [Google Scholar] [CrossRef]
- Ratta, M.; Fagnoni, F.; Curti, A.; Vescovini, R.; Sansoni, P.; Oliviero, B.; Fogli, M.; Ferri, E.; Della Cuna, G.R.; Tura, S.; et al. Dendritic cells are functionally defective in multiple myeloma: The role of interleukin-6. Blood 2002, 100, 230–237. [Google Scholar] [CrossRef]
- Tucci, M.; Stucci, S.; Strippoli, S.; Dammacco, F.; Silvestris, F. Dendritic cells and malignant plasma cells: An alliance in multiple myeloma tumor progression? Oncologist 2011, 16, 1040–1048. [Google Scholar] [CrossRef]
- Beyer, M.; Kochanek, M.; Giese, T.; Endl, E.; Weihrauch, M.R.; Knolle, P.A.; Classen, S.; Schultze, J.L. In vivo peripheral expansion of naive CD4+CD25high FoxP3+ regulatory T cells in patients with multiple myeloma. Blood 2006, 107, 3940–3949. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- De Vries, I.J.; Krooshoop, D.J.; Scharenborg, N.M.; Lesterhuis, W.J.; Diepstra, J.H.; van Muijen, G.N.; Strijk, S.P.; Ruers, T.J.; Boerman, O.C.; Oyen, W.J.; et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003, 63, 12–17. [Google Scholar]
- Morse, M.A.; Coleman, R.E.; Akabani, G.; Niehaus, N.; Coleman, D.; Lyerly, H.K. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 1999, 59, 56–58. [Google Scholar]
- Yewdall, A.W.; Drutman, S.B.; Jinwala, F.; Bahjat, K.S.; Bhardwaj, N. CD8+ T cell priming by dendritic cell vaccines requires antigen transfer to endogenous antigen presenting cells. PLoS One 2010, 5, e11144. [Google Scholar]
- Schadendorf, D.; Ugurel, S.; Schuler-Thurner, B.; Nestle, F.O.; Enk, A.; Bröcker, E.B.; Grabbe, S.; Rittgen, W.; Edler, L.; Sucker, A.; et al. DCs study group of the DeCOG. Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed DCs (DCs) in first-line treatment of patients with metastatic melanoma: A randomized phase III trial of the DCs study group of the DeCOG. Ann. Oncol. 2006, 17, 563–570. [Google Scholar] [CrossRef]
- Small, E.J.; Fratesi, P.; Reese, D.M.; Strang, G.; Laus, R.; Peshwa, M.V.; Valone, F.H. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J. Clin. Oncol. 2000, 18, 3894–3903. [Google Scholar]
- Higano, C.S.; Schellhammer, P.F.; Small, E.J.; Burch, P.A.; Nemunaitis, J.; Yuh, L.; Provost, N.; Frohlich, M.W. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer 2009, 115, 3670–3679. [Google Scholar] [CrossRef]
- Liau, L.M.; Prins, R.M.; Kiertscher, S.M.; Odesa, S.K.; Kremen, T.J.; Giovannone, A.J.; Lin, J.W.; Chute, D.J.; Mischel, P.S.; Cloughesy, T.F.; et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T cell responses modulated by the local central nervous system tumor micro-environment. Clin. Cancer Res. 2005, 11, 5515–5525. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Corak, J.; Ciernik, I.F.; Kavanaugh, D.; Carbone, D.P. Decreased antigen presentation by dendritic cells in patients with breast cancer. Clin. Cancer Res. 1997, 3, 483–490. [Google Scholar]
- Satthaporn, S.; Robins, A.; Vassanasiri, W.; El-Sheemy, M.; Jibril, J.A.; Clark, D.; Valerio, D.; Eremin, O. Dendritic cells are dysfunctional in patients with operable breast cancer. Cancer Immunol. Immunother. 2004, 53, 510–518. [Google Scholar] [CrossRef]
- Kiladjian, J.J.; Mesa, R.A.; Hoffman, R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011, 117, 4706–4715. [Google Scholar] [CrossRef]
- Schlee, M.; Hartmann, G. The chase for the RIG-I ligand—Recent advances. Mol. Ther. 2010, 18, 1254–1262. [Google Scholar] [CrossRef]
- Mohty, M.; Vialle-Castellano, A.; Nunes, J.A.; Isnardon, D.; Olive, D.; Gaugler, B. IFN-α skews monocyte differentiation into Toll like receptor 7-expressing dendritic cells with potent functional activities. J. Immunol. 2003, 171, 3385–3393. [Google Scholar]
- Santini, S.M.; Lapenta, C.; Logozzi, M.; Parlato, S.; Spada, M.; di Pucchio, T.; Belardelli, F. Type 1 interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J. Exp. Med. 2000, 191, 1777–1788. [Google Scholar] [CrossRef]
- Nagano, Y.; Kojima, Y. Pouvoir immunisant du virus vaccinal inactivé par des rayons ultraviolets. C. R. Seances. Soc. Biol. Fil. 1954, 148, 1700–1702. [Google Scholar]
- Lindenmann, J.; Burke, D.C.; Isaacs, A. Studies on the production, mode of action and properties of interferon. Br. J. Exp. Pathol. 1957, 38, 551–562. [Google Scholar]
- Blanco, P.; Palucka, A.K.; Gill, M.; Pascual, V.; Banchereau, J. Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science 2001, 294, 1540–1543. [Google Scholar] [CrossRef]
- Gallucci, S.; Lolkema, M.; Matzinger, P. Natural adjuvants: Endogenous activators of dendritic cells. Nat. Med. 1999, 5, 1249–1255. [Google Scholar] [CrossRef]
- Ito, T.; Amakawa, R.; Inaba, M.; Ikehara, S.; Inaba, K.; Fukuhara, S. Differential regulation of human blood dendritic cell subsets by IFNs. J. Immunol. 2001, 166, 2961–2969. [Google Scholar]
- Paquette, R.L.; Hsu, N.C.; Kiertscher, S.M.; Park, A.N.; Tran, L.; Roth, M.D.; Glaspy, J.A. Interferon-α and granulocyte-macrophage colony-stimulating factor differentiate peripheral blood monocytes into potent antigen-presenting cells. J. Leukoc. Biol. 1998, 64, 358–367. [Google Scholar]
- Le Bon, A.; Etchart, N.; Rossmann, C.; Ashton, M.; Hou, S.; Gewert, D.; Borrow, P.; Tough, D.F. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 2003, 4, 1009–1015. [Google Scholar]
- Marrack, P.; Kappler, J.; Mitchell, T. Type I interferons keep activated T cells alive. J. Exp. Med. 1999, 189, 521–530. [Google Scholar] [CrossRef]
- Berenson, L.S.; Farrar, J.D.; Murphy, T.L.; Murphy, K.M. Frontline: Absence of functional STAT4 activation despite detectable tyrosine phosphorylation induced by murine IFN-α. Eur. J. Immunol. 2004, 34, 2365–2374. [Google Scholar] [CrossRef]
- Ramos, H.J.; Davis, A.M.; George, T.C.; Farrar, J.D. IFN-α is not sufficient to drive Th1 development due to lack of stable T-bet expression. J. Immunol. 2007, 179, 3792–3803. [Google Scholar]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef]
- De Visser, K.E.; Korets, L.V.; Coussens, L.M. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 2005, 7, 411–423. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Brinkmann, V.; Geiger, T.; Alkan, S.; Heusser, C.H. Interferon-α increases the frequency of interferon γ-producing human CD4+ T cells. J. Exp. Med. 1993, 178, 1655–1663. [Google Scholar] [CrossRef]
- Wenner, C.A.; Guler, M.L.; Macatonia, S.E.; O’Garra, A.; Murphy, K.M. Roles of IFN-α and IFN-β in IL-12-induced T helper cell-1 development. J. Immunol. 1996, 156, 1442–1447. [Google Scholar]
- Tough, D.F.; Borrow, P.; Sprent, J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science 1996, 272, 1947–1950. [Google Scholar]
- Lewis, J.J. Therapeutic cancer vaccines: Using unique antigens. Proc. Natl. Acad. Sci. USA 2004, 101, 14653–14656. [Google Scholar] [CrossRef]
- Parmiani, G.; de Filippo, A.; Novellino, L.; Castelli, C. Unique human tumor antigens: Immunobiology and use in clinical trials. J. Immunol. 2007, 178, 1975–1979. [Google Scholar]
- Appay, V.; Douek, D.C.; Price, D.A. CD8+ T cell efficacy in vaccination and disease. Nat. Med. 2008, 14, 623–628. [Google Scholar] [CrossRef]
- Minkis, K.; Kavanagh, D.G.; Alter, G.; Bogunovic, D.; O’Neill, D.; Adams, S.; Pavlick, A.B.; Walker, D.; Brockman, M.A.; Gandhi, R.T.; et al. Type 2 bias of T cells expanded from the blood of melanoma patients switched to type 1 by IL-12p70 mRNA-transfected dendritic cells. Cancer Res. 2008, 68, 9441–9450. [Google Scholar] [CrossRef]
- Aspord, C.; Pedroza-Gonzalez, A.; Gallegos, M.; Tindle, S.; Burton, E.C.; Su, D.; Marches, F.; Banchereau, J.; Palucka, A.K. Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. J. Exp. Med. 2007, 204, 1037–1047. [Google Scholar] [CrossRef]
- Gilboa, E. The makings of a tumor rejection antigen. Immunity 1999, 11, 263–270. [Google Scholar] [CrossRef]
- Magyarics, Z.; Rajnavölgyi, E. Professional type I interferon-producing cells—A unique subpopulation of dendritic cells. Acta. Microbiol. Immunol. Hung. 2005, 52, 443–462. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Schulz, O.; Diebold, S.S.; Chen, M.; Näslund, T.I.; Nolte, M.A.; Alexopoulou, L.; Azuma, Y.T.; Flavell, R.A.; Liljeström, P.; Reis e Sousa, C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005, 433, 887–892. [Google Scholar]
- Fournier, P.; Arnold, A.; Schirrmacher, V. Polarization of human monocyte-derived dendritic cells to DC1 by in vitro stimulation with Newcastle Disease Virus. J. BUON 2009, 14, 111–122. [Google Scholar]
- Kalinski, P.; Nakamura, Y.; Watchmaker, P.; Giermasz, A.; Muthuswamy, R.; Mailliard, R.B. Helper roles of NK and CD8+ T cells in the induction of tumor immunity. Polarized dendritic cells as cancer vaccines. Immunol. Res. 2006, 36, 137–146. [Google Scholar] [CrossRef]
- Di Pucchio, T.; Pilla, L.; Capone, I.; Ferrantini, M.; Montefiore, E.; Urbani, F.; Patuzzo, R.; Pennacchioli, E.; Santinami, M.; Cova, A.; et al. Immunization of stage IV melanoma patients with Melan-A/MART-1 and gp100 peptides plus IFN-α results in the activation of specific CD8+ T cells and monocyte/dendritic cell precursors. Cancer Res. 2006, 66, 4943–4951. [Google Scholar] [CrossRef]
- Kuwashima, N.; Nishimura, F.; Eguchi, J.; Sato, H.; Hatano, M.; Tsugawa, T.; Sakaida, T.; Dusak, J.E.; Fellows-Mayle, W.K.; Papworth, G.D.; et al. Delivery of dendritic cells engineered to secrete IFN-α into central nervous system tumors enhances the efficacy of peripheral tumor cell vaccines: Dependence on apoptotic pathways. J. Immunol. 2005, 175, 2730–2740. [Google Scholar]
- Kumar-Sinha, C.; Varambally, S.; Sreekumar, A.; Chinnaiyan, A.M. Molecular cross-talk between the TRAIL and interferon signalling pathways. J. Biol. Chem. 2002, 277, 575–585. [Google Scholar]
- Rogge, L.; Barberis-Maino, L.; Biffi, M.; Passini, N.; Presky, D.H.; Gubler Sinigaglia, E. Selective expression of an interleukin-12 receptor component by human T helper 1 cells. J. Exp. Med. 1997, 185, 825–831. [Google Scholar] [CrossRef]
- Tough, D.F. Type 1 interferon as a link between innate and adaptive immunity through dendritic cell stimulation. Leuk. Lymphoma 2004, 45, 257–264. [Google Scholar] [CrossRef]
- Gallucci, S.; Matzinger, P. Danger signals: SOS to the immune system. Curr. Opin. Immunol. 2001, 13, 114–119. [Google Scholar] [CrossRef]
- Cisco, R.M.; Abdel-Wahab, Z.; Dannull, J.; Nair, S.; Tyler, D.S.; Gilboa, E.; Vieweg, J.; Daaka, Y.; Pruitt, S.K. Induction of human dendritic cell maturation using transfection with RNA encoding a dominant positive toll-like receptor 4. J. Immunol. 2004, 172, 7162–7168. [Google Scholar]
- Bonehill, A.; Tuyaerts, S.; van Nuffel, A.M.; Heirman, C.; Bos, T.J.; Fostier, K.; Neyns, B.; Thielemans, K. Enhancing the T cell stimulatory capacity of human dendritic cells by coelectroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol. Ther. 2008, 16, 1170–1180. [Google Scholar] [CrossRef]
- Fournier, P.; Arnold, A.; Wilden, H.; Schirrmacher, V. Newcastle disease virus induces pro-inflammatory conditions and type 1 interferon for counter-acting Treg activity. Int. J. Oncol. 2012, 40, 840–850. [Google Scholar]
- Kurooka, M.; Kaneda, Y. Inactivated Sendai virus particles eradicate tumors by inducing immune responses through blocking regulatory T cells. Cancer Res. 2007, 67, 227–236. [Google Scholar] [CrossRef]
- Woo, C.Y.; Clay, T.M.; Lyerly, H.K.; Morse, M.A.; Osada, T. Role of natural killer cell function in dendritic cell-based vaccines. Expert Rev. Vaccines 2006, 5, 66–65. [Google Scholar]
- Van Tendeloo, V.F.; van de Velde, A.; van Driessche, A.; Cools, N.; Anguille, S.; Ladell, K.; Gostick, E.; Vermeulen, K.; Pieters, K.; Nijs, G.; et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc. Natl. Acad. Sci. USA 2010, 107, 13824–13829. [Google Scholar] [CrossRef]
- Ebihara, T.; Masuda, H.; Akazawa, T.; Shingai, M.; Kikuta, H.; Ariga, T.; Matsumoto, M.; Seya, T. Induction of NKG2D ligands on human dendritic cells by TLR ligand stimulation and RNA virus infection. Int. Immunol. 2007, 19, 1145–1155. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Fournier, P. Danger signals in tumor cells: A risk factor for autoimmune disease? Expert Rev. Vaccines 2010, 9, 347–350. [Google Scholar] [CrossRef]
- Lazar, I.; Yaacov, B.; Shiloach, T.; Eliahoo, E.; Kadouri, L.; Lotem, M.; Perlman, R.; Zakay-Rones, Z.; Panet, A.; Ben-Yehuda, D. The Oncolytic Activity of Newcastle Disease Virus NDV-HUJ on Chemoresistant Primary Melanoma Cells Is Dependent on the Proapoptotic Activity of the Inhibitor of Apoptosis Protein Livin. J. Virol. 2010, 84, 639–646. [Google Scholar] [CrossRef]
- Elankumaran, S.; Chavan, V.; Qiao, D.; Shobana, R.; Moorkanat, G.; Biswas, M.; Samal, S.K. Type I Interferon-Sensitive Recombinant Newcastle Disease Virus for Oncolytic Virotherapy. J. Virol. 2010, 84, 3835–3844. [Google Scholar] [CrossRef]
- Puhlmann, J.; Puehler, F.; Mumberg, D.; Boukamp, P.; Beier, R. Rac1 is required for oncolytic NDV replication in human cancer cells and establishes a link between tumorigenesis and sensitivity to oncolytic virus. Oncogene 2010, 29, 2205–2216. [Google Scholar] [CrossRef]
- Mansour, M.; Palese, P.; Zamarin, D. Oncolytic Specificity of Newcastle Disease Virus Is Mediated by Selectivity for Apoptosis-Resistant Cells. J. Virol. 2011, 85, 6015–6023. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fournier, P.; Schirrmacher, V. Oncolytic Newcastle Disease Virus as Cutting Edge between Tumor and Host. Biology 2013, 2, 936-975. https://doi.org/10.3390/biology2030936
Fournier P, Schirrmacher V. Oncolytic Newcastle Disease Virus as Cutting Edge between Tumor and Host. Biology. 2013; 2(3):936-975. https://doi.org/10.3390/biology2030936
Chicago/Turabian StyleFournier, Philippe, and Volker Schirrmacher. 2013. "Oncolytic Newcastle Disease Virus as Cutting Edge between Tumor and Host" Biology 2, no. 3: 936-975. https://doi.org/10.3390/biology2030936
APA StyleFournier, P., & Schirrmacher, V. (2013). Oncolytic Newcastle Disease Virus as Cutting Edge between Tumor and Host. Biology, 2(3), 936-975. https://doi.org/10.3390/biology2030936