Genome-Scale Metabolic Reconstruction and in Silico Perturbation Analysis of the Polar Diatom Fragilariopsis cylindrus Predicts High Metabolic Robustness

 , , ,
, , ,
Abstract
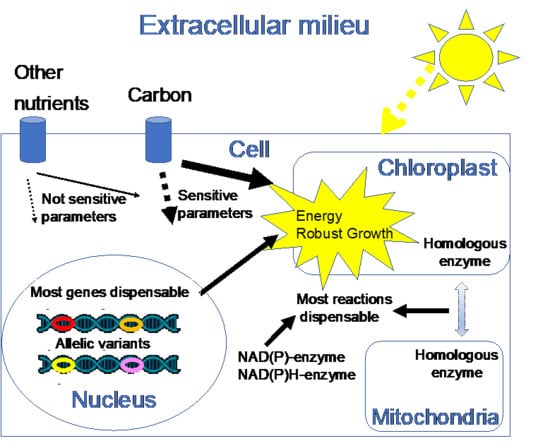
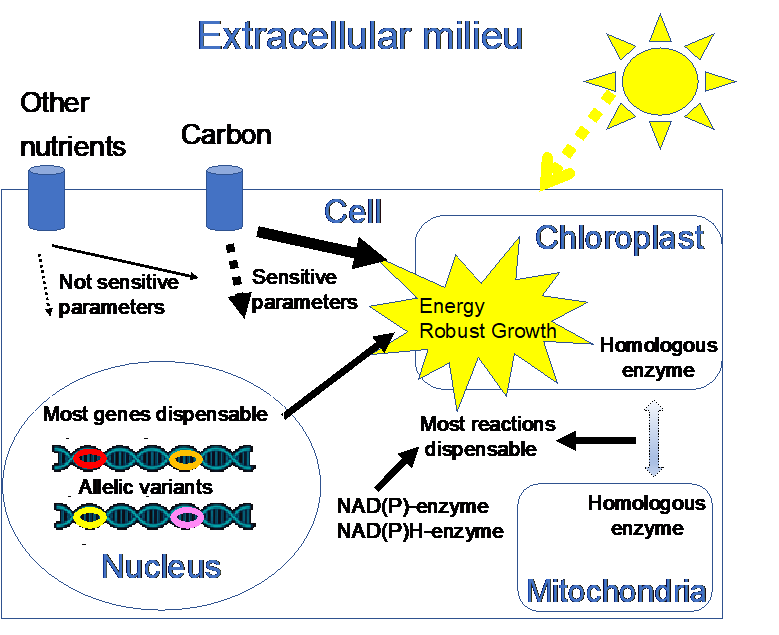
:
1. Introduction
2. Methods
2.1. Metabolic Network Reconstruction
2.2. Flux Balance Analysis (FBA)
2.3. Quality Control of the Genome-Scale Model
2.4. Sensitivity Analysis of Model Output
2.5. Model Implementation in MATLAB
2.6. Network Theory Analysis
3. Results and Discussions
3.1. Prediction of Energy Dissipation Pathways, Cell Bioenergetics, and Growth Rate with Flux Balance Analysis (FBA)
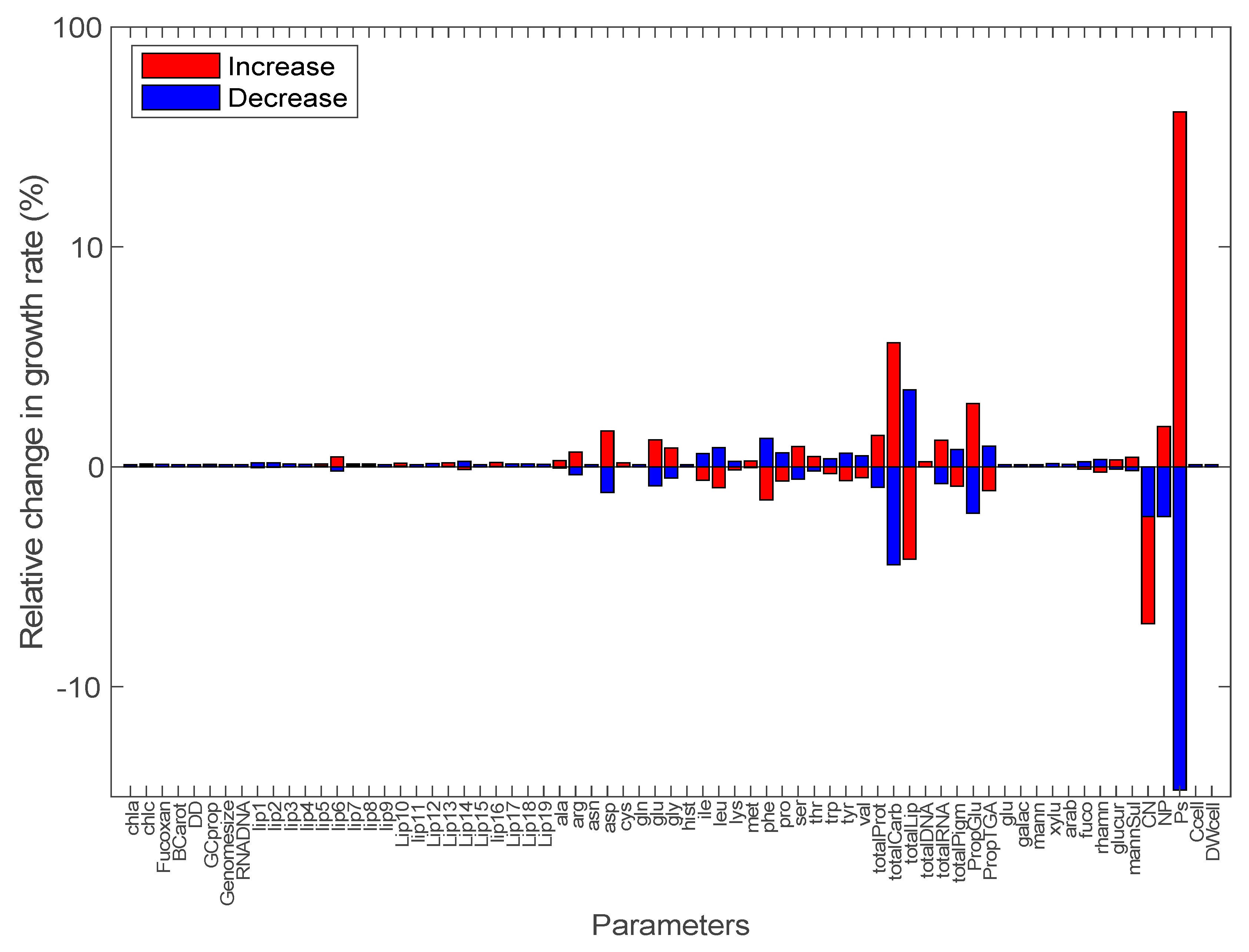
3.2. Sensitivity Analysis of Model Parameters
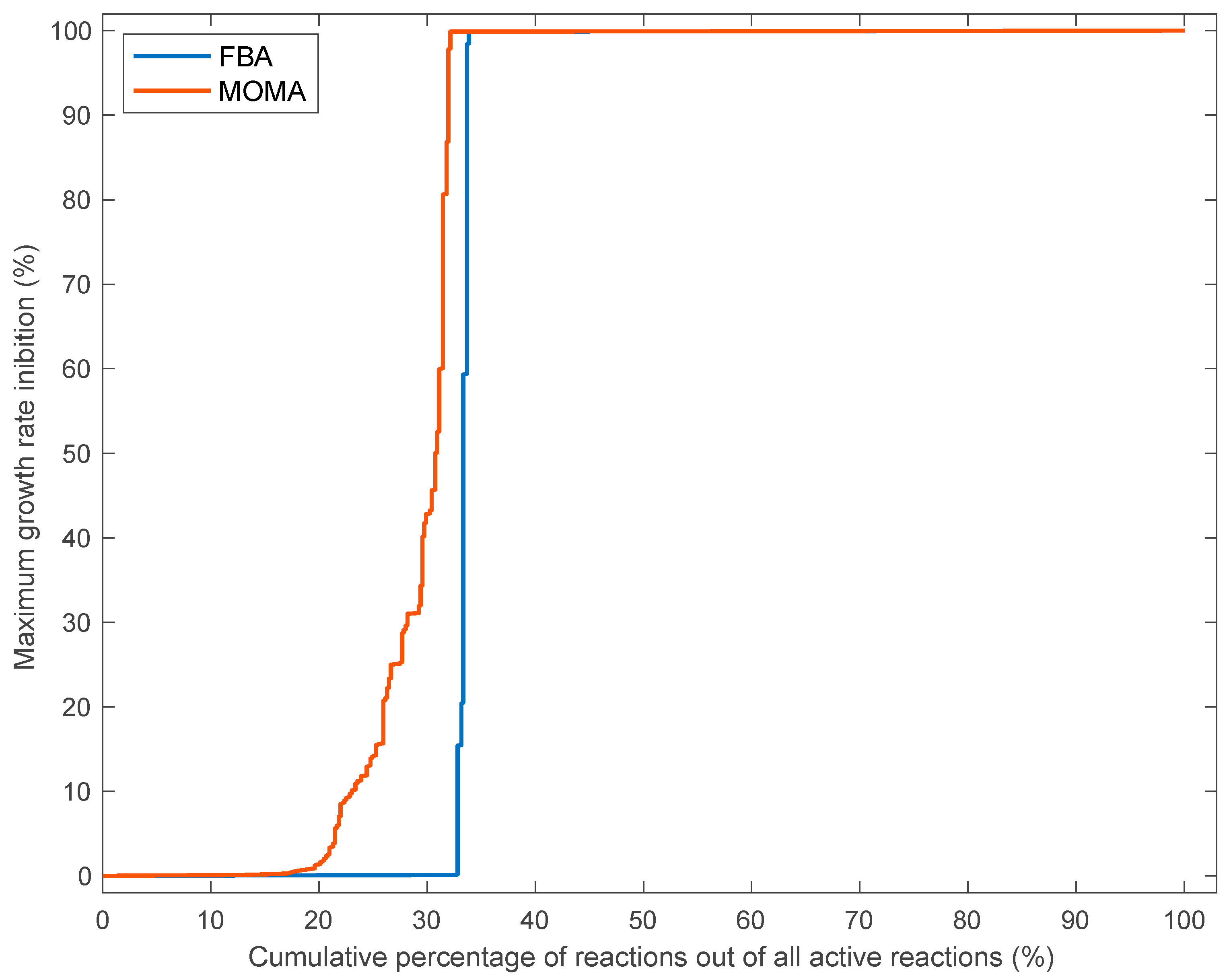
3.3. Effects of Single Reaction Deletions Calculated Using FBA and MOMA
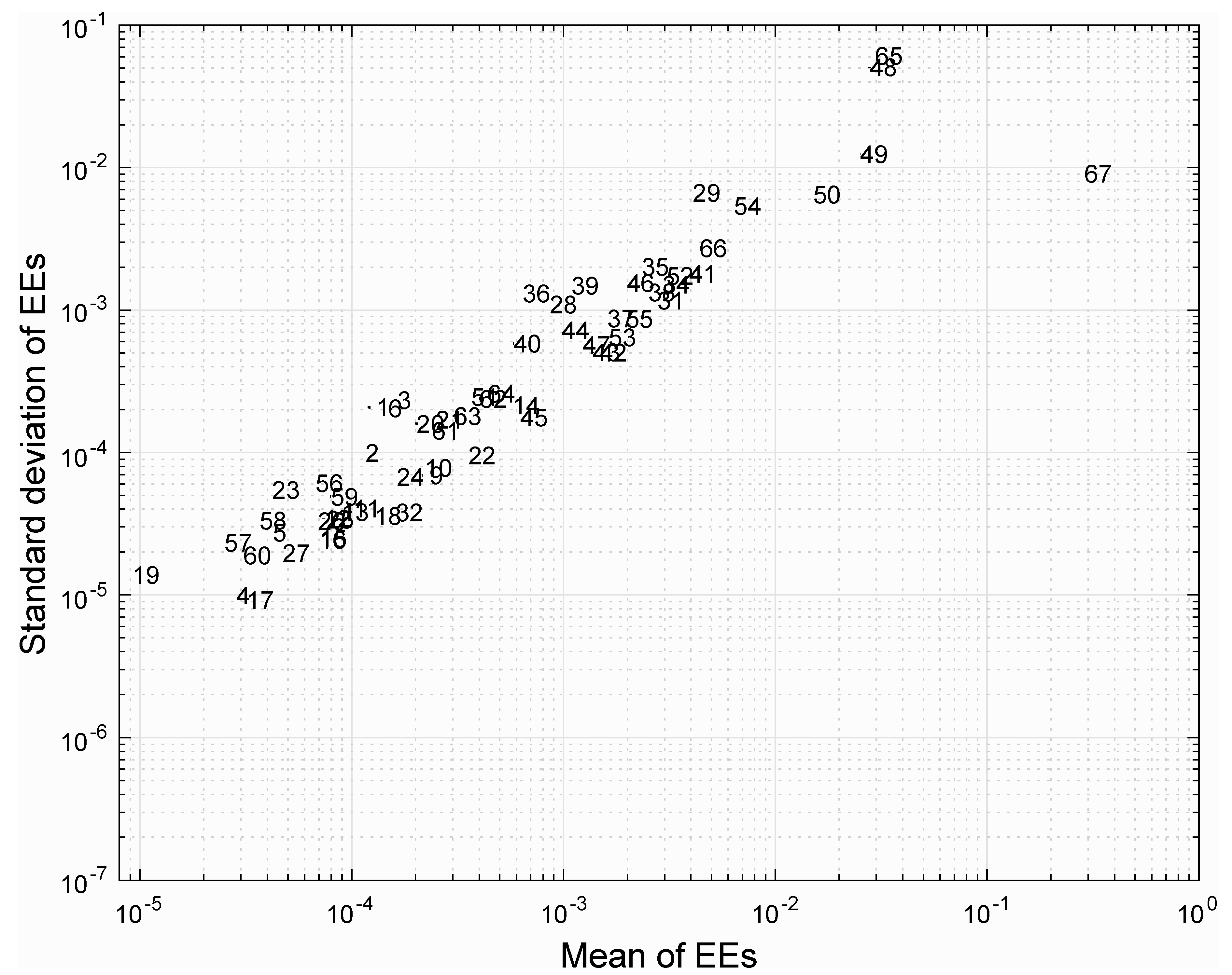
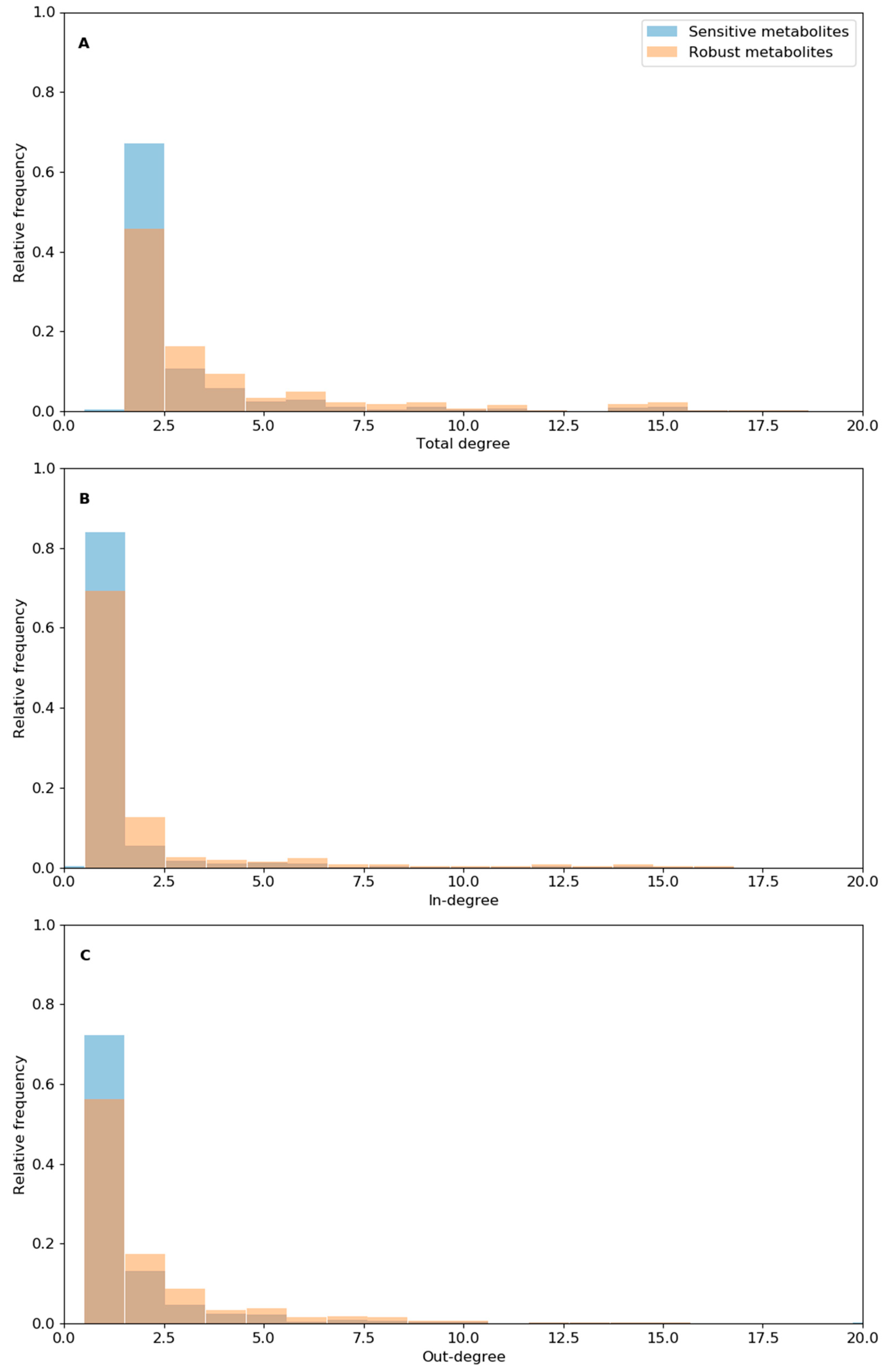
3.4. Analysis of Reaction Robustness Using Network Theory Metrics
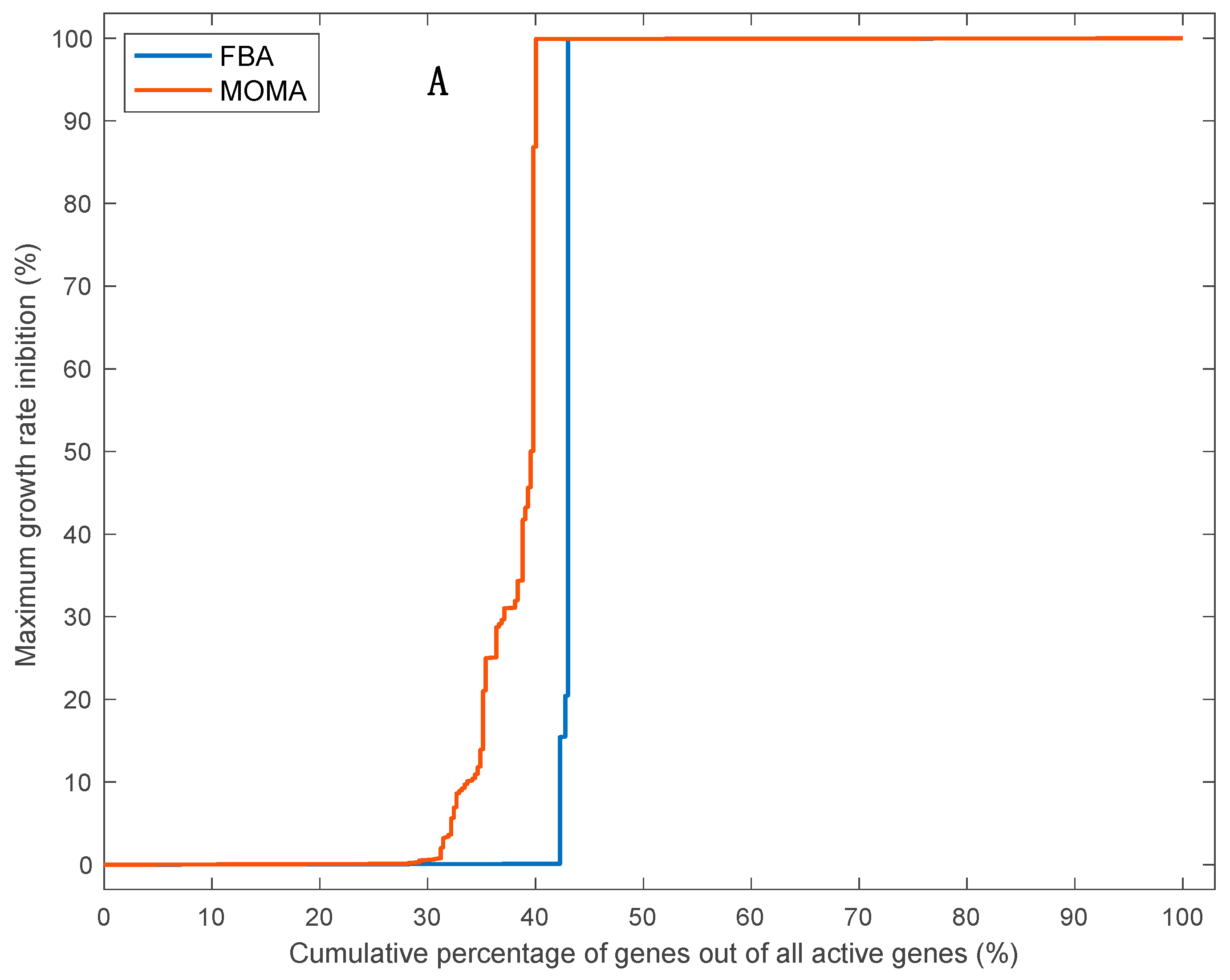
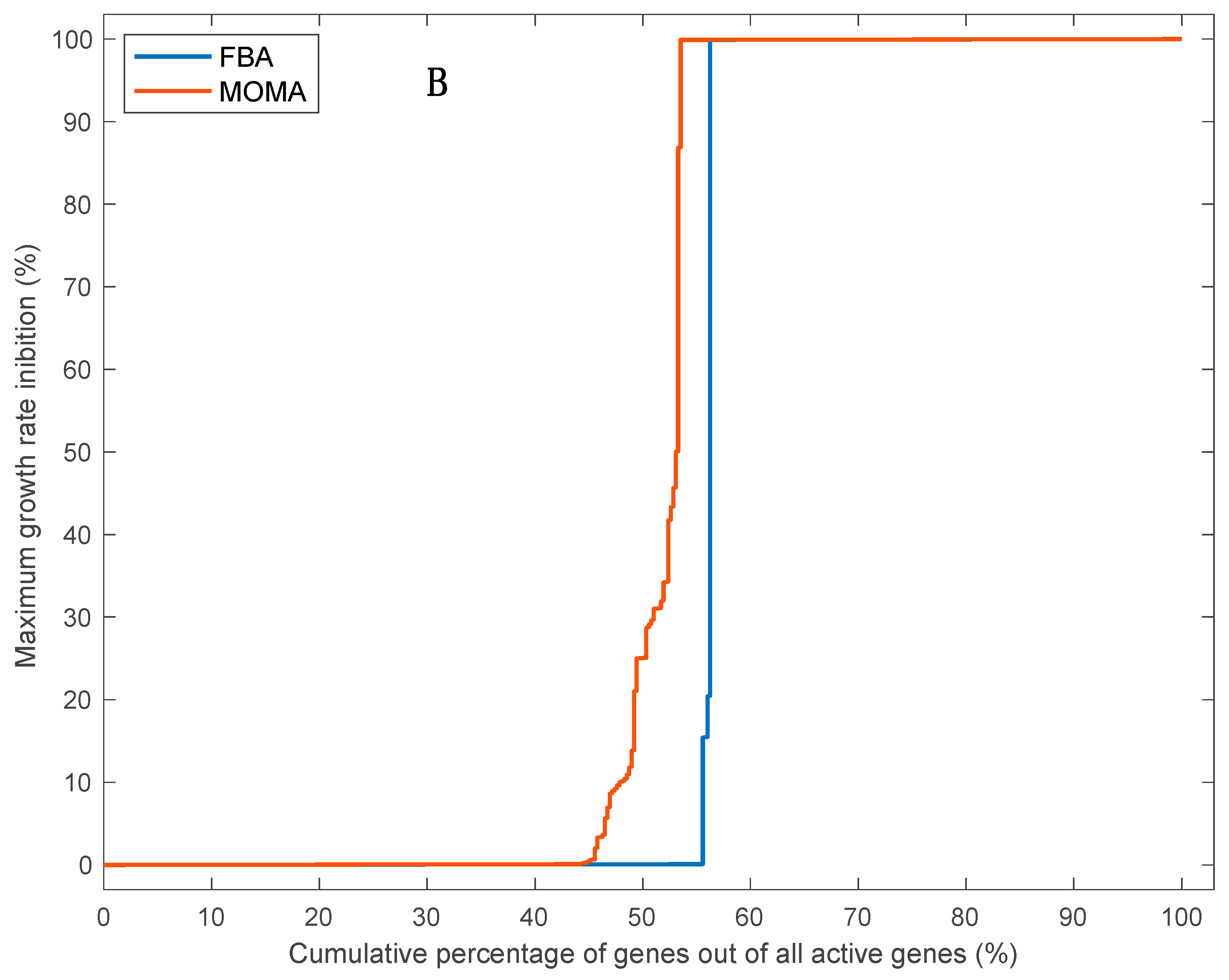
3.5. Effects of Single Gene Deletion Calculated Using FBA and MOMA
4. Conclusions
Data Availability
Computer Code and Software
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Uitz, J.; Claustre, H.; Gentili, B.; Stramski, D. Phytoplankton class-specific primary production in the world’s oceans: Seasonal and interannual variability from satellite observations. Glob. Biogeochem. Cycles 2010, 24, GB3016. [Google Scholar] [CrossRef]
- Arrigo, K.R. Sea ice ecosystems. Annu. Rev. Mar. Sci. 2014, 6, 439–467. [Google Scholar] [CrossRef] [PubMed]
- Lyon, B.R.; Mock, T. Polar microalgae: New approaches towards understanding adaptations to an extreme and changing environment. Biology (Basel) 2014, 3, 56–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopes, A.; Thomas, D.N.; Mock, T. Polar microalgae: Functional genomics, physiology, and the environment. In Psychrophiles: From Biodiversity to Biotechnology; Margesin, R., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 305–344. [Google Scholar] [CrossRef]
- Morgan-Kiss, R.M.; Priscu, J.C.; Pocock, T.; Gudynaite-Savitch, L.; Huner, N.P.A. Adaptation and Acclimation of Photosynthetic Microorganisms to Permanently Cold Environments. Microbiol. Mol. Biol. Rev. 2006, 70, 222–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teoh, M.L.; Phang, S.M.; Chu, W.L. Response of Antarctic, temperate, and tropical microalgae to temperature stress. J. Appl. Phycol. 2013, 25, 285–297. [Google Scholar] [CrossRef]
- Svenning, J.B.; Dalheim, L.; Eilersten, H.C.; Vasskog, T. Temperature dependent growth rate, lipid content and fatty acid composition of the marine cold-water diatom Porosira glacialis. Algal Res. 2019, 37, 11–16. [Google Scholar] [CrossRef]
- Young, J.N.; Goldman, J.A.L.; Kranz, S.A.; Tortell, P.D. Slow carboxylation of Rubisco constrains the rate of carbon fixation during Antarctic phytoplankton blooms. New Phytol. 2014, 205, 172–181. [Google Scholar] [CrossRef]
- Gerecht, A.C.; Eriksen, G.K.; Uradnikova, M.; Eilertsen, H.C. High ribulose-1,5-bisphosphate carboxylase/oxygenase content in northern diatom species. bioRxiv 2019, 569285. [Google Scholar] [CrossRef] [Green Version]
- Lacour, T.; Larivière, J.; Ferland, J.; Bruyant, F.; Lavaud, J.; Babin, M. The role of sustained photoprotective non-photochemical quenching in low temperature and high light acclimation in the bloom-forming arctic diatom Thalassiosira gravida. Front. Mar. Sci. 2018, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Lacour, T.; Larivière, J.; Babin, M. Growth, Chl a content, photosynthesis, and elemental composition in polar and temperate microalgae. Limnol. Oceanogr. 2017, 62, 43–58. [Google Scholar] [CrossRef]
- Gianchandani, E.P.; Chavali, A.K.; Papin, J.A. The application of flux balance analysis in systems biology. Wires. Syst. Biol. Med. 2010, 2, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Levering, J.; Broddrick, J.; Dupont, C.L.; Peers, G.; Beeri, K.; Mayers, J.; Gallina, A.A.; Allen, A.E.; Palsson, B.O.; Zengler, K. Genome-scale model reveals metabolic basis of biomass partitioning in a model diatom. PLoS ONE 2016, 11, e0155038. [Google Scholar] [CrossRef]
- Broddrick, J.T.; Du, N.; Smith, S.R.; Tsuji, Y.; Jallet, D.; Maxwell, A.W.; Peers, G.; Matsuda, Y.; Dupont, C.L.; Mitchell, G.; et al. Cross-compartment metabolic coupling enables flexible photoprotective mechanisms in the diatom Phaeodactylum tricornutum. New Phytol. 2019, 222, 1364–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitan, O.; Dinamarca, J.; Zelzion, E.; Lun, D.S.; Guerra, L.T.; Kim, M.K.; Kim, J.; Van Mooy, B.A.S.; Bhattacharya, D.; Falkowski, P.G. Remodeling of intermediate metabolism in the diatom Phaeodactylum tricornutum under nitrogen stress. PNAS 2015, 112, 412–417. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Fabris, M.; Baart, G.; Kim, M.K.; Goossens, A.; Vyverman, W.; Falkowski, P.G.; Lun, D.S. Flux balance analysis of primary metabolism in the diatom Phaeodactylum tricornutum. Plant J. 2016, 85, 161–176. [Google Scholar] [CrossRef] [Green Version]
- Deutscher, D.; Meilijson, I.; Kupiec, M.; Ruppin, E. Multiple knockout analysis of genetic robustness in the yeast metabolic network. Nat. Genet. 2006, 38, 993–998. [Google Scholar] [CrossRef]
- Behre, J.; Wilhelm, T.; von Kamp, A.; Ruppin, E.; Schuster, S. Structural robustness of metabolic networks with respect to multiple knockouts. J. Theor. Biol. 2008, 252, 433–441. [Google Scholar] [CrossRef]
- Phillips, P.C. Epistasis—The essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 2008, 9, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Rancati, G.; Moffat, J.; Typas, A.; Pavelka, N. Emerging and evolving concepts in gene essentiality. Nat. Rev. Genet. 2018, 19, 34–49. [Google Scholar] [CrossRef]
- Zuñiga, C.; Li, C.-T.; Huelsman, T.; Levering, J.; Zielinski, D.C.; McConnell, B.O.; Long, C.P.; Knoshaug, E.P.; Guarnieri, M.T.; Antoniewicz, M.R.; et al. Genome-scale metabolic model for the green alga Chlorella vulgaris UTEX 395 accurately predicts phenotypes under autotrophic, heterotrophic, and mixotrophic growth conditions. Plant. Physiol. 2016, 172, 589–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mock, T.; Otillar, R.P.; Strauss, J.; McMullan, M.; Paajanen, P.; Schmutz, J.; Salamov, A.; Sanges, R.; Toseland, A.; Ward, B.J.; et al. Evolutionary genomics of the cold-adapted diatom Fragilariopsis cylindrus. Nature 2017, 54, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, F.; Martin, A.; Bowman, J.P.; Wilson, R.; McMinn, A. Dark metabolism: a molecular insight into how the Antarctic sea-ice diatom Fragilariopsis cylindrus survives long-term darkness. New Phytol. 2019, 223, 675–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paajanen, P.; Strauss, J.; van Oosterhout, C.; McMullan, M.; Clark, M.D.; Mock, T. Building a locally diploid genome and transcriptome of the diatom Fragilariopsis cylindrus. Sci. Data 2017, 4, 170149. [Google Scholar] [CrossRef] [Green Version]
- Poulin, M.; Daugbjerg, N.; Gradinger, R.; Ilyash, L.; Ratkova, T.; von Quillfeldt, C.H. The pan-Arctic biodiversity of marine pelagic and sea-ice unicellular eukaryotes: a first-attempt assessment. Mar. Biodivers. 2011, 41, 13–28. [Google Scholar] [CrossRef]
- Lourenço, S.O.; Barbarino, E.; Marquez, U.M.L.; Aidar, E. Distribution of intracellular nitrogen in marine microalgae: Basis for the calculation of specific nitrogen-to-protein conversion factors. J. Phycol. 1998, 34, 798–811. [Google Scholar] [CrossRef]
- Brown, M.R. The amino-acid and sugar composition of 16 species of microalgae used in mariculture. J. Exp. Mar. Biol. Ecol. 1991, 145, 79–99. [Google Scholar] [CrossRef]
- Ibarra, R.U.; Edwards, J.S.; Palsson, B.O. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 2002, 420, 186–189. [Google Scholar] [CrossRef]
- Segré, D.; Vitkup, D.; Church, G.M. Analysis of optimality in natural and perturbed metabolic networks. PNAS 2002, 99, 15112–15117. [Google Scholar] [CrossRef] [Green Version]
- Lewis, N.E.; Hixson, K.K.; Conrad, T.M.; Lerman, J.A.; Charusanti, P.; Polpitiya, A.D.; Adkins, J.N.; Schramm, G.; Purvine, S.O.; Lopez-Ferrer, D.; et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 2010, 6, 390. [Google Scholar] [CrossRef]
- Labhsetwar, P.; Melo, M.C.R.; Cole, J.A.; Luthey-Schulten, Z. Population FBA predicts metabolic phenotypes in yeast. PLoS Comput. Biol. 2017, 13, e1005728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broddrick, J.T.; Welkie, D.G.; Jallet, D.; Goldena, S.S.; Peers, G.; Palsson, B.O. Predicting the metabolic capabilities of Synechococcus elongatus PCC 7942 adapted to different light regimes. Metab. Eng. 2019, 52, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Raven, J.A. Energetics and Transport in Aquatic Plants; A. R. Liss: New York, NY, USA, 1984. [Google Scholar]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models: The COBRA Toolbox v3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostromins, A.; Stalidzans, E. Paint4Net: COBRA Toolbox extension for visualization of stoichiometric models of metabolism. BioSystems 2012, 109, 233–239. [Google Scholar] [CrossRef]
- Pianosi, F.; Sarrazin, F.; Wagener, T. A Matlab toolbox for global sensitivity analysis. Environ. Model. Software 2015, 70, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Python Software Foundation. Python Language Reference, Version 2.7. Available online: http://www.python.org (accessed on 10 January 2020).
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy2008), Pasadena, CA, USA, 19–24 August 2008; Gäel Varoquaux, T.V., Millman, J., Eds.; New Mexico. pp. 11–15. [Google Scholar]
- Bailleul, B.; Berne, N.; Murik, O.; Petroutsos, D.; Prihoda, J.; Tanaka, A.; Villanova, V.; Bligny, R.; Flori, S.; Falconet, D.; et al. Energetic coupling between plastids and mitochondria drives CO2 assimilation in diatoms. Nature 2015, 524, 366–369. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, M.; Raven, J.A.; Levasseur, M. Energy cost and putative benefits of cellular mechanisms modulating buoyancy in aflagellate marine phytoplankton. J. Phycol. 2016, 52, 239–251. [Google Scholar] [CrossRef]
- Goldman, J.A.L.; Kranz, S.A.; Young, J.N.; Tortell, P.D.; Stanley, R.H.R.; Bender, M.L.; Morel, F.M.M. Gross and net production during the spring bloom along the Western Antarctic Peninsula. New Phytol. 2015, 205, 182–191. [Google Scholar] [CrossRef]
- Papp, B.; Pal, C.; Hurst, L. Metabolic network analysis of the causes and evolution of enzyme dispensability in yeast. Nature 2004, 429, 661–664. [Google Scholar] [CrossRef]
- Harrison, R.; Papp, B.; Pa, C.; Oliver, S.G.; Delner, D. Plasticity of genetic interactions in metabolic networks of yeast. PNAS 2007, 104, 2307–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Values | Units | Notes |
|---|---|---|---|
| Ccell | 7.5 | pg/cell | Measured in F. cylindrus (This study) |
| DWcell | 15 | pg/cell | Assuming a DWcell:Ccell ratio of 2 |
| HCO3- uptake | 0.78 | mmol g DW−1 h−1 | Measured in F. cylindrus (This study) |
| C:N | 5.7 | mol:mol | Calculated in F. cylindrus (Garcia et al., 2018) |
| N:P | 10 | mol:mol | Calculated in F. cylindrus (Garcia et al., 2018) |
| Total proteins | 0.46 | g/g DW | Assumed (see SI.1 Section) |
| Total carbohydrates | 0.31 | g/g DW | Assumed (see SI.1 Section) |
| Total lipids | 0.21 | g/g DW | Assumed (see SI.1 Section) |
| DNA | 0.0022 | g/g DW | Calculated in F. cylindrus (This study) |
| RNA | 0.018 | g/g DW | Calculated in F. cylindrus (This study) |
| Total pigments | 0.016 | g/g DW | Measured in F. cylindrus (This study) |
| Chlorophyll a | 0.14 | pg/cell | Measured in F. cylindrus (This study) |
| Chlorophyll c | 0.030 | pg/cell | Measured in F. cylindrus (This study) |
| Fucoxanthin | 0.060 | pg/cell | Measured in F. cylindrus (This study) |
| Beta-carotene | 0.0040 | pg/cell | Measured in F. cylindrus (This study) |
| Diadinoxanthin | 0.0090 | pg/cell | Measured in F. cylindrus (This study) |
| Glucan | 30 | % of total carbohydrates | Assumed (see SI.1 Section) |
| Triacylglycerol | 20 | % of total lipids | Assumed (see SI.1 Section) |
| GC proportion | 40 | % of nucleobases | Calculated in F. cylindrus (This study) |
| Genome size | 6.1 × 107 | number of bases | Calculated in F. cylindrus (This study) |
| RNA:DNA ratio | 8.0 | g/g DW | Based on other diatoms [27] |
| Glucose | 0 | mol/g DW | Free glucose assumed negligible |
| 8 sugars * | 0.10 to 0.32 * | mol/g DW | Measured in P. tricornutm [14] |
| 20 amino acids * | 0.0040 to 0.080 * | g/g DW | Measured in P. tricornutm [28] |
| 19 lipid species * | 0.010 to 0.12 * | mol/g DW | Measured in P. tricornutm [14] |
| Robust Reactions | Sensitive Reactions | |
|---|---|---|
| Nature of reactions | Same reaction with different energy sources | Reactions with no alternative routes |
| Same reaction in different cell compartments | ||
| Examples | 5-methyltetrahydrofolate oxidoreductase using NAD or NADPH | Uptake of nitrate, phosphate, and carbon |
| NADH- or ferredoxin-dependent nitrite reductase | Chrysolaminarin synthesis | |
| Cytosolic and mitochondrial aspartate transaminase or threonine aldolase | Biomass reactions | |
| Reaction activation | More activated reactions than inactivated reactions | More inactivated reactions than activated reactions |
| Total number of active reactions | Higher for the robust reaction set than for the sensitive reaction set | |
| Energy dissipation | Compensatory response higher for robust reactions than for sensitive reactions | |
| Activation of photorespiration | Yes, for two reactions (FBA) | No activation (FBA or MOMA) |
| Yes, for up to 37% of robust reactions (MOMA) | ||
| Activation of photon harvesting | Yes, for all reactions (FBA) | Yes, for 80% of reactions (FBA) |
| No activation (MOMA) | No activation (MOMA) | |
| Activation of gross chloroplastic ATP | Yes, for all reactions (FBA) | Yes, for 80% of reactions (FBA) |
| No activation (MOMA) | No activation (MOMA) | |
| Activation of gross mitochondrial ATP | Yes, for 57% of reactions (FBA) | Yes, for 40% of reactions (FBA) |
| Yes, for 100% of reactions (MOMA) | Yes, for all reactions (MOMA) but to a lesser extent | |
| Activation of cyclic PSI | Yes, for 40% of reactions (FBA) | Yes, for 46% of reactions (FBA) |
| Yes, for 60% of reactions (MOMA) | Yes, for 60% of reactions (MOMA) but to a lesser extent | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lavoie, M.; Saint-Béat, B.; Strauss, J.; Guérin, S.; Allard, A.; V. Hardy, S.; Falciatore, A.; Lavaud, J. Genome-Scale Metabolic Reconstruction and in Silico Perturbation Analysis of the Polar Diatom Fragilariopsis cylindrus Predicts High Metabolic Robustness. Biology 2020, 9, 30. https://doi.org/10.3390/biology9020030
Lavoie M, Saint-Béat B, Strauss J, Guérin S, Allard A, V. Hardy S, Falciatore A, Lavaud J. Genome-Scale Metabolic Reconstruction and in Silico Perturbation Analysis of the Polar Diatom Fragilariopsis cylindrus Predicts High Metabolic Robustness. Biology. 2020; 9(2):30. https://doi.org/10.3390/biology9020030
Chicago/Turabian StyleLavoie, Michel, Blanche Saint-Béat, Jan Strauss, Sébastien Guérin, Antoine Allard, Simon V. Hardy, Angela Falciatore, and Johann Lavaud. 2020. "Genome-Scale Metabolic Reconstruction and in Silico Perturbation Analysis of the Polar Diatom Fragilariopsis cylindrus Predicts High Metabolic Robustness" Biology 9, no. 2: 30. https://doi.org/10.3390/biology9020030
APA StyleLavoie, M., Saint-Béat, B., Strauss, J., Guérin, S., Allard, A., V. Hardy, S., Falciatore, A., & Lavaud, J. (2020). Genome-Scale Metabolic Reconstruction and in Silico Perturbation Analysis of the Polar Diatom Fragilariopsis cylindrus Predicts High Metabolic Robustness. Biology, 9(2), 30. https://doi.org/10.3390/biology9020030