Effectiveness of Molecules for Quantum Cellular Automata as Computing Devices



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
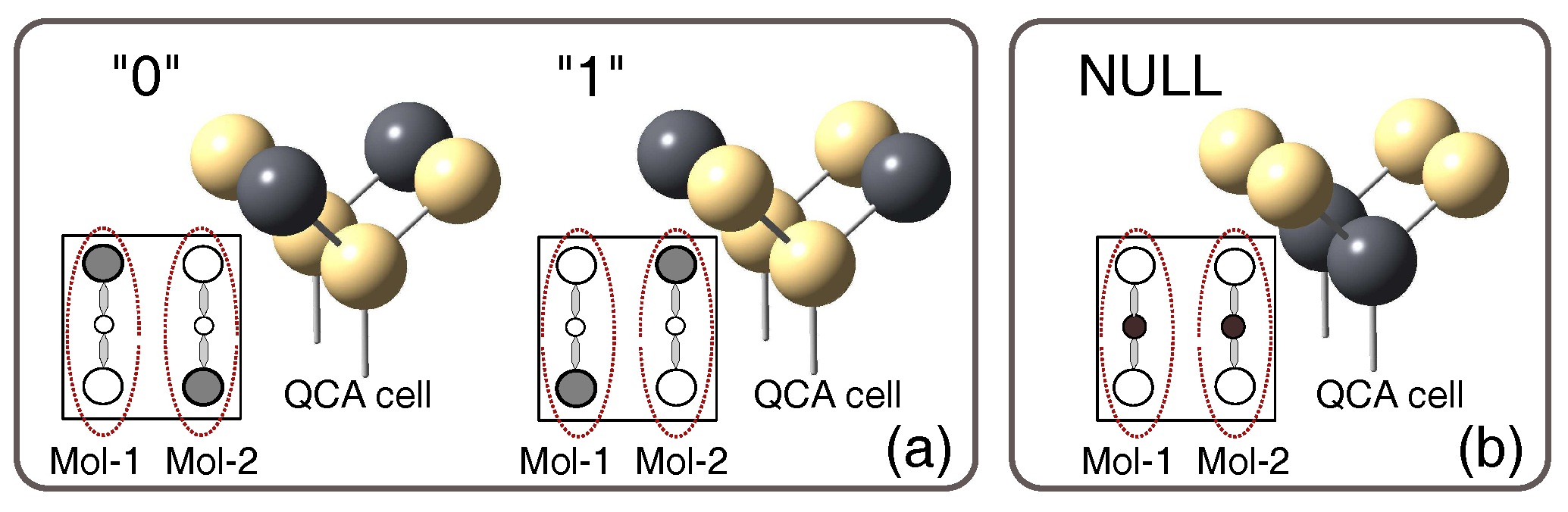
2. Background: Molecular QCA
3. Methodology: A Quantitative Approach for MQCA Characterization
- Ab initio simulation: accurate ab initio simulations based on quantum chemistry, are performed to analyze the electronic structure and the physical properties of the molecule. External stimuli are inserted in the simulation to study the molecule in different conditions: electric fields and point charges emulate the synchronizing clock, switching fields and driver.
- Post processing of ab initio simulation: from ab initio simulations we derive the key features, as figures of merit, describing the electrostatic behavior of the molecule which are used to describe the interaction with other molecules and to provide the quantitative information necessary for the analysis of molecular circuits. The figures of merit encase all the physical-chemical properties of the molecules in quantities which can be used by engineers to understand, simulate, design and fabricate MQCA devices.
- System-level analysis: the figures of merit defined in the second step of MosQuiTo enable the study of molecular circuits at the system level. The information propagation in MQCA circuits can be evaluated considering parameters which are closer to the electronic engineering techniques: the engineer does not have to deal with the physics of the device and can analyze it from a higher level of abstraction. An algorithm for the evaluation of molecular propagation in large MQCA circuits, which exploits the figures of merit, has been developed and published in [29].
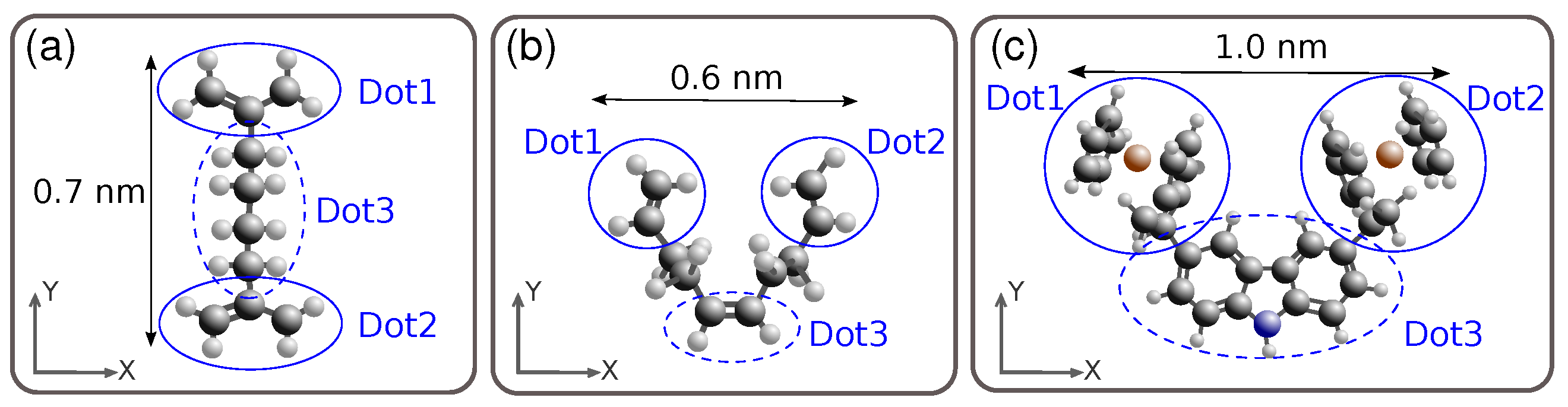
3.1. Aggregated Charge (AC)
3.2. Electric-Field Generated at the Receiver (EFGR)
3.3. Vin–Vout Transcharacteristics (VVT) and Vin–AC Transcharacteristics (VACT)
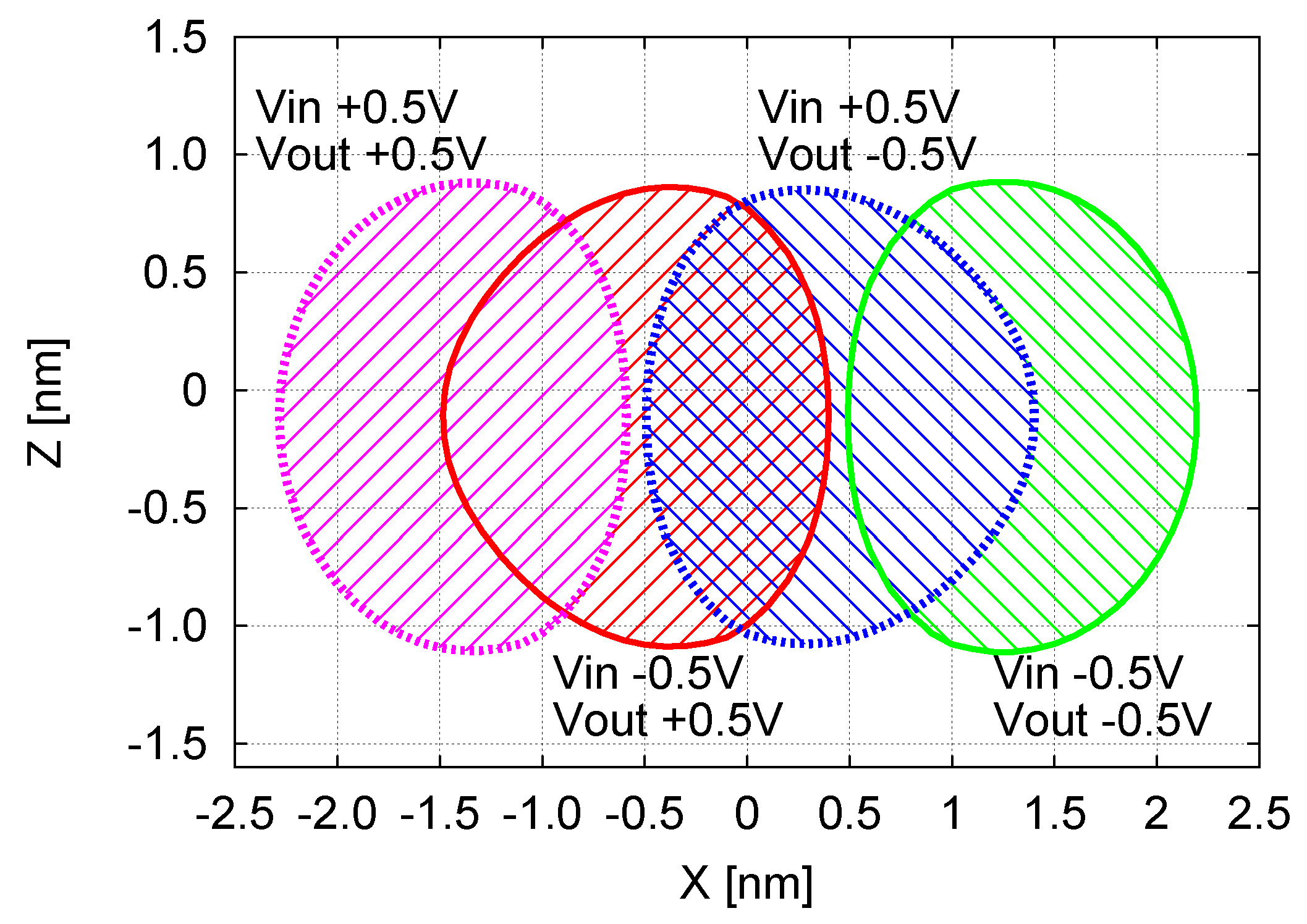
3.4. Vout Maps (VOM) and MQCA Cell Working Zone (CWZ)
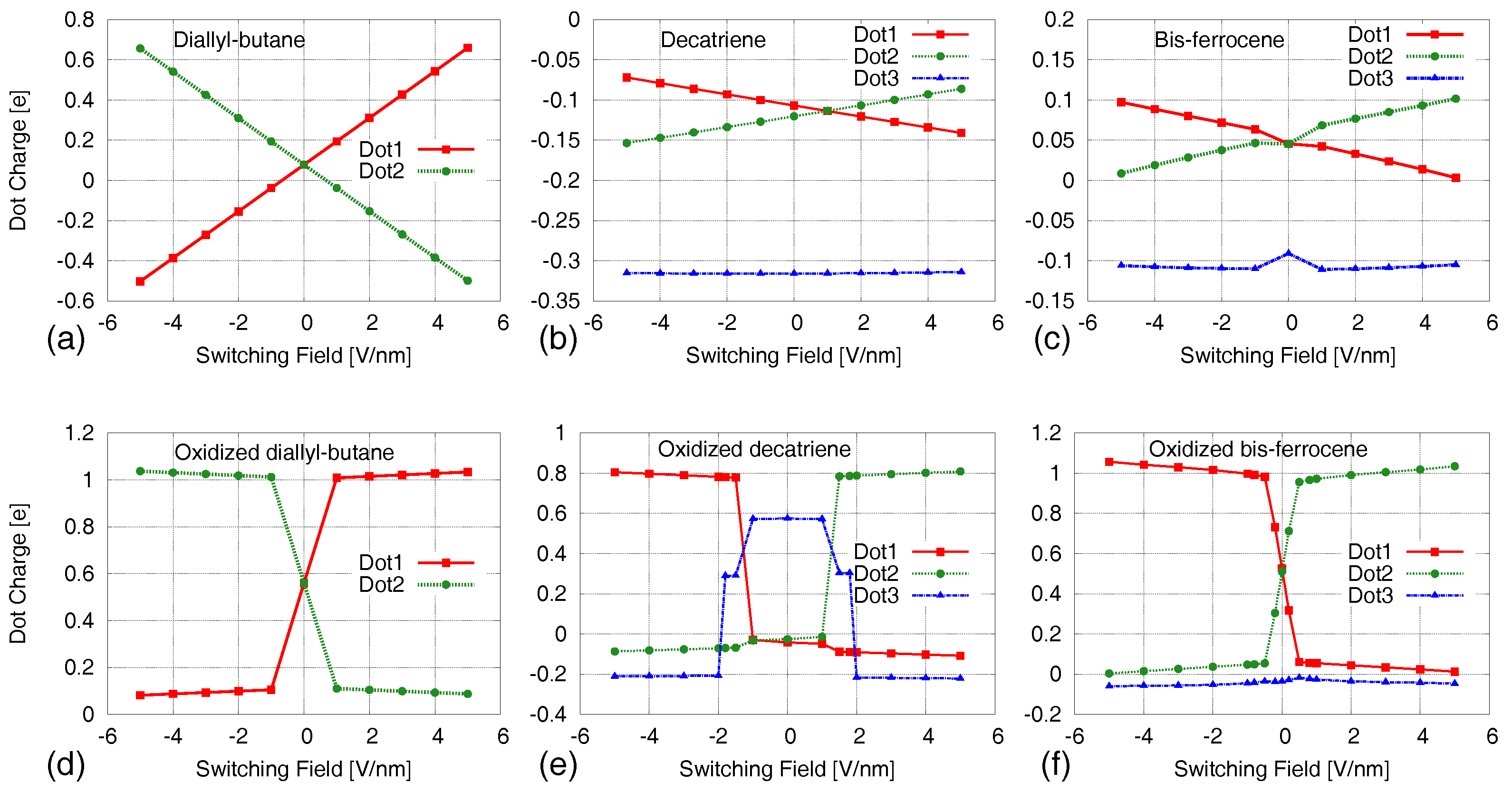
4. Results: Neutral Molecules
5. Results: Oxidized Molecules
5.1. Aggregated Charge–AC/VACT
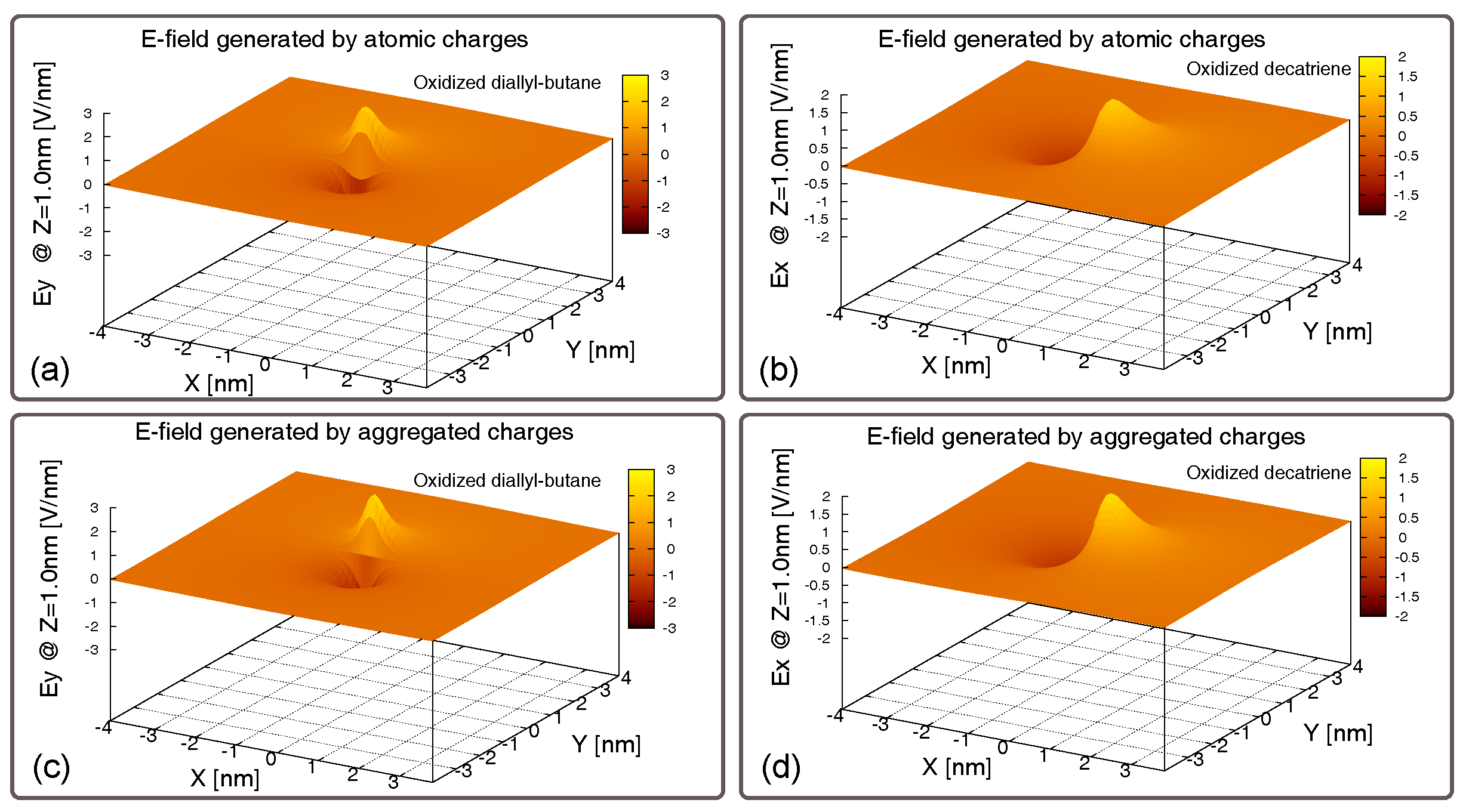
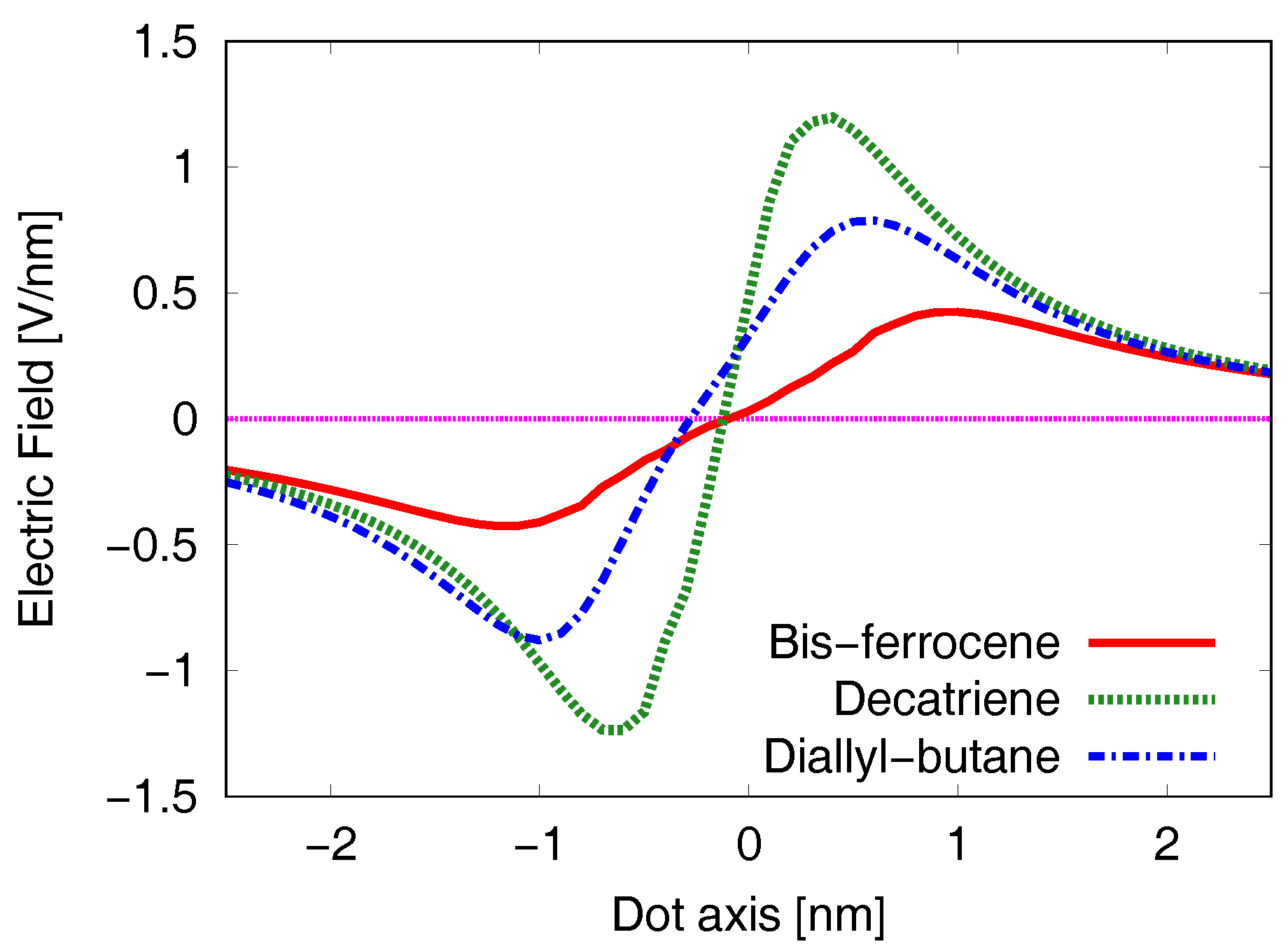
5.2. Generated Field
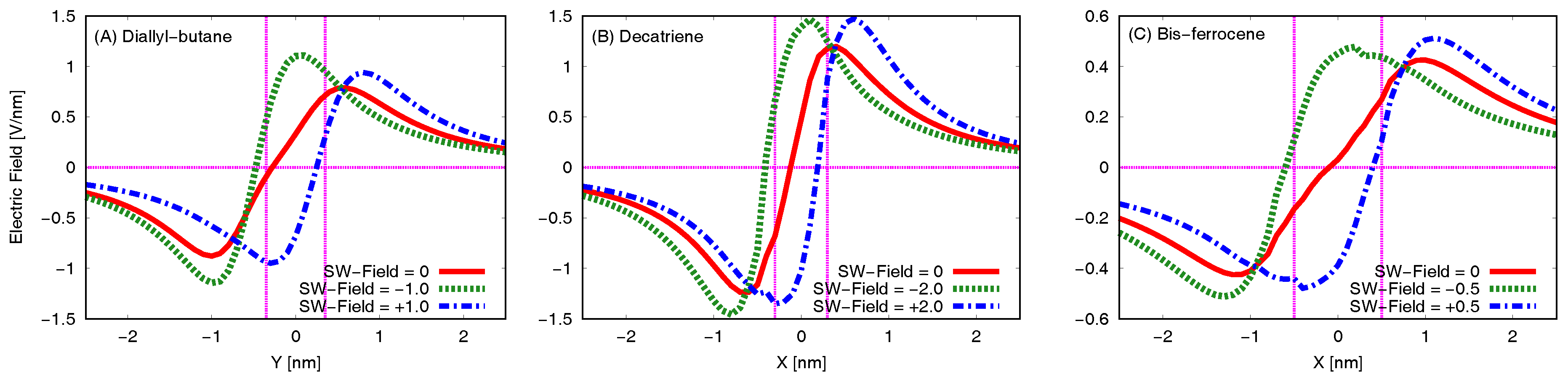
5.3. Generated Field at the Receiver–EFGR
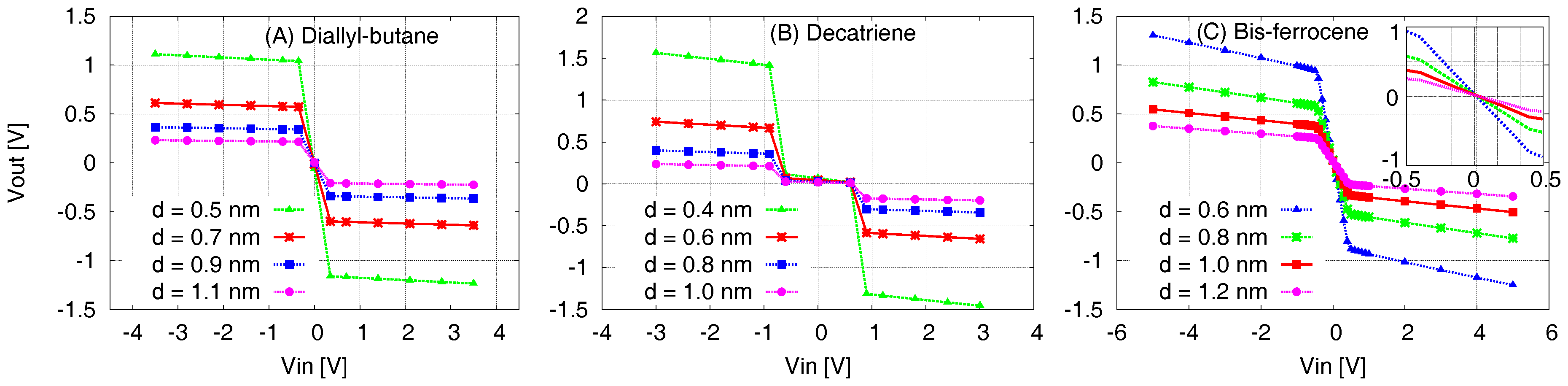
5.4. Molecule Transcharacteristics–VVT
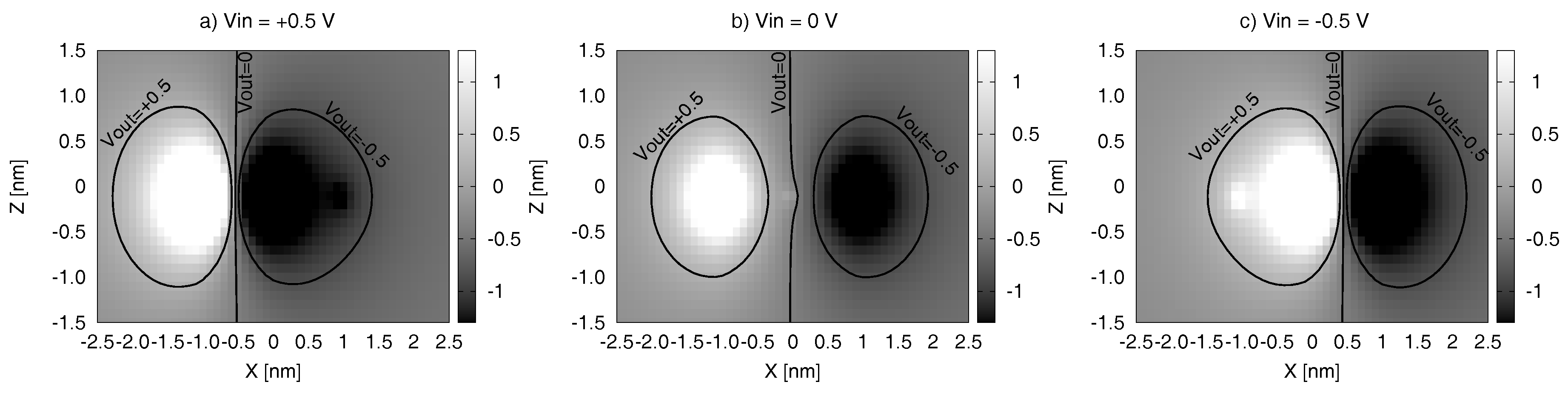
5.5. Output Voltage Maps–VOM
5.6. MQCA Cell Working Zone–CWZ
6. Conclusions and Future Works
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Aggregated Charge |
| CMOS | Complementary Metal-Oxide Semiconductor |
| CV | Cyclic-Voltammetry |
| CWZ | Cell Working Zone |
| DFT | Density Functional Theory |
| EF | Electric Field |
| EFGR | Electric Field Generated at the Receiver molecule |
| HF | Hartree-Fock |
| MQCA | Molecular Quantum-dot Cellular Automata |
| MUT | Molecule Under Test |
| QCA | Quantum-dot Cellular Automata |
| TSA | Two-State Approximation |
| VACT | Vin–Aggregated Charge Transcharacteristics |
| VOT | VOuT Map |
| VVT | Vin–Vout Transcharacteristics |
References
- Lent, C.S.; Tougaw, P.D.; Porod, W. Bistable saturation in coupled quantum dots for quantum cellular automata. Appl. Phys. Lett. 1993, 62, 714–716. [Google Scholar] [CrossRef]
- Lent, C.S.; Tougaw, P.D.; Porod, W.; Bernstein, G.H. Quantum cellular automata. Nanotechnology 1993, 4, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Vacca, M.; Graziano, M.; Zamboni, M. Nanomagnetic Logic Microprocessor: Hierarchical Power Model. IEEE Trans. VLSI Syst. 2012, 21, 1410–1420. [Google Scholar] [CrossRef]
- Lent, C.S.; Tougaw, P.D. Lines of interacting quantum-dot cells: A binary wire. J. Appl. Phys. 1993, 74, 6227–6233. [Google Scholar] [CrossRef]
- Pulimeno, A.; Graziano, M.; Wang, R.; Demarchi, D.; Piccinini, G. Charge distribution in a molecular QCA wire based on bis-ferrocene molecule. In Proceedings of the IEEE/ACM International Symposium on Nanoscale Architectures, New York, NY, USA, 15–17 July 2013. [Google Scholar]
- Pulimeno, A.; Graziano, M.; Piccinini, G. UDSM Trends Comparison: From Technology Roadmap to UltraSparc Niagara2. IEEE Trans. VLSI Syst. 2012, 20, 1341–1346. [Google Scholar] [CrossRef]
- Lu, Y.; Lent, C.S. A metric for characterizing the bistability of molecular quantum-dot cellular automata. Nanotechnology 2008, 19, 155703. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, M.; Lent, C.S. Molecular quantum-dot cellular automata: From molecular structure to circuit dynamics. J. Appl. Phys. 2007, 102, 034311. [Google Scholar] [CrossRef]
- Timler, J.; Lent, C.S. Maxwell’s demon and quantum-dot cellular automata. J. Appl. Phys. 2003, 94, 1050–1060. [Google Scholar] [CrossRef]
- Orlov, A.; Imre, A.; Csaba, G.; Ji, L.; Porod, W.; Bernstein, G.H. Magnetic quantum-dot cellular automata: Recent developments and prospects. J. Nanoelectron. Optoelectron. 2008, 3, 55–68. [Google Scholar] [CrossRef]
- Vacca, M.; Graziano, M.; Zamboni, M. Majority Voter Full Characterization for NanoMagnet Logic Circuits. IEEE Trans. Nanotechnol. 2012, 11, 940–947. [Google Scholar] [CrossRef]
- Timler, J.; Lent, C.S. Power gain and dissipation in quantum-dot cellular automata. J. Appl. Phys. 2002, 91, 823–831. [Google Scholar] [CrossRef]
- Lent, C.S.; Isaksen, B. Clocked molecular quantum-dot cellular automata. IEEE Trans. Electron Devices 2003, 50, 1890–1896. [Google Scholar] [CrossRef]
- Graziano, M.; Vacca, M.; Chiolerio, A.; Zamboni, M. A NCL-HDL Snake-Clock Based Magnetic QCA Architecture. IEEE Trans. Nanotechnol. 2011, 10, 1141–1149. [Google Scholar] [CrossRef]
- Lent, C.S.; Isaksen, B.; Lieberman, M. Molecular quantum-dot cellular automata. J. Am. Chem. Soc. 2003, 125, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Lent, C.S. Theoretical Study of Molecular Quantum-Dot Cellular Automata. J. Comput. Electron. 2005, 4, 115–118. [Google Scholar] [CrossRef]
- Wang, X.; Yu, L.; Inakollu, V.S.S.; Pan, X.; Ma, J.; Yu, H. Molecular Quantum Dot Cellular Automata Based on Diboryl Monoradical Anions. J. Phys. Chem. C 2018, 122, 2454–2460. [Google Scholar] [CrossRef]
- Siccardi, S.; Adamatzky, A. Actin quantum automata: Communication and computation in molecular networks. J. Appl. Phys. 2015, 6, 15–27. [Google Scholar] [CrossRef]
- Sadhu, T.; Das, B.; De1, D.; Das, J.C. Design of binary subtractor using actin quantum cellular automata. IET Nanobiotechnol. 2018, 12, 32–39. [Google Scholar] [CrossRef]
- Qi, H.; Sharma, S.; Li, Z.; Snider, G.L.; Orlov, A.O.; Lent, C.S.; Fehlner, T.P. Molecular quantum cellular automata cells. Electric field driven switching of a silicon surface bound array of vertically oriented two-dot molecular quantum cellular automata. J. Am. Chem. Soc. 2003, 125, 15250–15259. [Google Scholar] [CrossRef] [PubMed]
- Zoli, L. Active Bis-Ferrocene Molecules as Unit for Molecular Computation. Ph.D. Thesis, Universitad́i Bologna, Bologna, Italy, 2010. [Google Scholar]
- Arima, V.; Iurlo, M.; Zoli, L.; Kumar, S.; Piacenza, M.; della Sala, F.; Matino, F.; Maruccio, G.; Rinaldi, R.; Paolucci, F.; et al. Toward quantum-dot cellular automata units: Thiolated-carbazole linked bisferrocenes. Nanoscale 2012, 4, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Pulimeno, A.; Graziano, M.; Abrardi, C.; Demarchi, D.; Piccinini, G. Molecular QCA: A write-in system based on electric fields. In Proceedings of the IEEE International NanoElectronics Conference, Tao-Yuan, Taiwan, 21–24 June 2011. [Google Scholar]
- Pulimeno, A.; Graziano, M.; Demarchi, D.; Piccinini, G. Towards a molecular QCA wire: Simulation of write-in and read-out systems. Solid-State Electron. 2012, 77, 101–107. [Google Scholar] [CrossRef]
- Pulimeno, A.; Graziano, M.; Piccinini, G. Molecule Interaction for QCA Computation. In Proceedings of the IEEE-NANO, Birmingham, UK, 20–23 August 2012. [Google Scholar]
- Pulimeno, A.; Graziano, M.; Sanginario, A.; Cauda, V.; Demarchi, D.; Piccinini, G. Bis-ferrocene molecular QCA wire: Ab-initio simulations of fabrication driven fault tolerance. IEEE Trans. Nanotechnol. 2013, 12, 498–507. [Google Scholar] [CrossRef]
- Vankamamidi, V.; Ottavi, M.; Lombardi, F. Clocking and Cell Placement for QCA. In Proceedings of the IEEE-NANO, Cincinnati, OH, USA, 17–20 July 2006; Volume 1, pp. 343–346. [Google Scholar]
- Pulimeno, A.; Graziano, M.; Antidormi, A.; Wang, R.; Zahir, A.; Piccinini, G. Understanding a Bisferrocene Molecular QCA Wire. In Field-Coupled Nanocomputing; Anderson, N., Bhanja, S., Eds.; Lecture Notes in Computer Science; Springer: Berlin, Heidelberg, 2014; pp. 307–338. Volume 8280, ISBN 978-3662437223. [Google Scholar]
- Wang, R.; Chilla, M.; Palucci, A.; Graziano, M.; Piccinini, G. An effective algorithm for clocked field-coupled nanocomputing paradigm. In Proceedings of the IEEE Nanotechnology Materials and Devices Conference (NMDC), Toulouse, France, 9–12 October 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09 Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Ding, F.; Chen, S.; Wang, H. Computational Study of Ferrocene-Based Molecular Frameworks with 2,5-Diethynylpyridine as a Chemical Bridge. Materials 2010, 3, 2668–2683. [Google Scholar] [CrossRef] [Green Version]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Pulimeno, A. Molecular Quantum-Dot Cellular Automata (QCA). Ph.D. Thesis, Politecnico di Torino, Torino, Italy, 2013. [Google Scholar]












© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ardesi, Y.; Pulimeno, A.; Graziano, M.; Riente, F.; Piccinini, G. Effectiveness of Molecules for Quantum Cellular Automata as Computing Devices. J. Low Power Electron. Appl. 2018, 8, 24. https://doi.org/10.3390/jlpea8030024
Ardesi Y, Pulimeno A, Graziano M, Riente F, Piccinini G. Effectiveness of Molecules for Quantum Cellular Automata as Computing Devices. Journal of Low Power Electronics and Applications. 2018; 8(3):24. https://doi.org/10.3390/jlpea8030024
Chicago/Turabian StyleArdesi, Yuri, Azzurra Pulimeno, Mariagrazia Graziano, Fabrizio Riente, and Gianluca Piccinini. 2018. "Effectiveness of Molecules for Quantum Cellular Automata as Computing Devices" Journal of Low Power Electronics and Applications 8, no. 3: 24. https://doi.org/10.3390/jlpea8030024
APA StyleArdesi, Y., Pulimeno, A., Graziano, M., Riente, F., & Piccinini, G. (2018). Effectiveness of Molecules for Quantum Cellular Automata as Computing Devices. Journal of Low Power Electronics and Applications, 8(3), 24. https://doi.org/10.3390/jlpea8030024