Chaperones and Proteostasis: Role in Parkinson’s Disease

Abstract
:1. Introduction
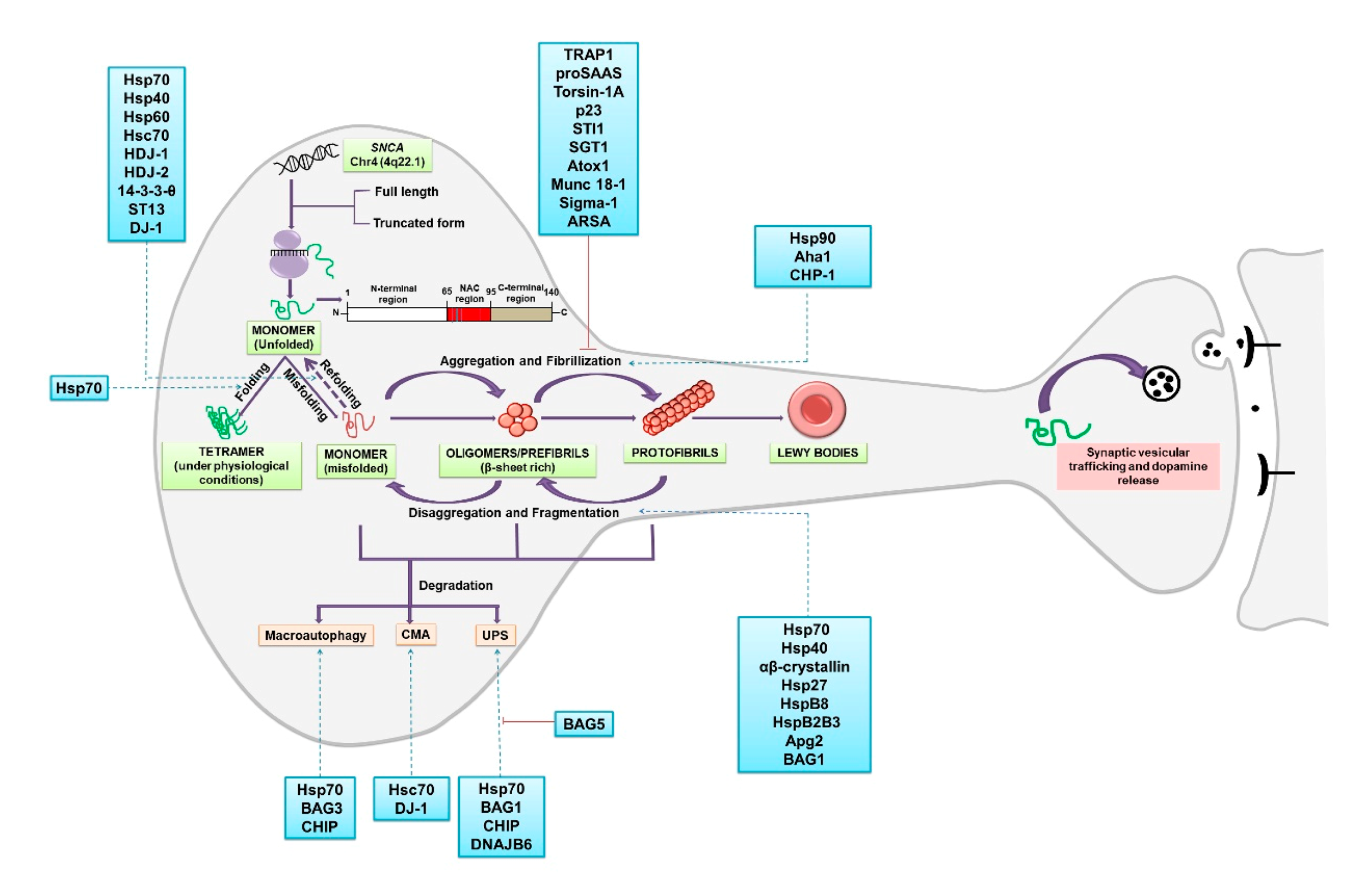
2. α-Synuclein: Folding, Higher-Order Structures and Degradation
2.1. α-Synuclein: Chaperones Involved in Misfolding, Aggregation and Disaggregation
2.2. α-Synuclein: Chaperones Involved in Refolding
2.3. α-Synuclein: Chaperones Involved in Degradation
2.3.1. Degradation via Ubiquitin Proteasome System (UPS)
2.3.2. Degradation via Chaperone Mediated Autophagy (CMA)
2.3.3. Degradation via Macroautophagy
2.4. Extracellular α-Synuclein
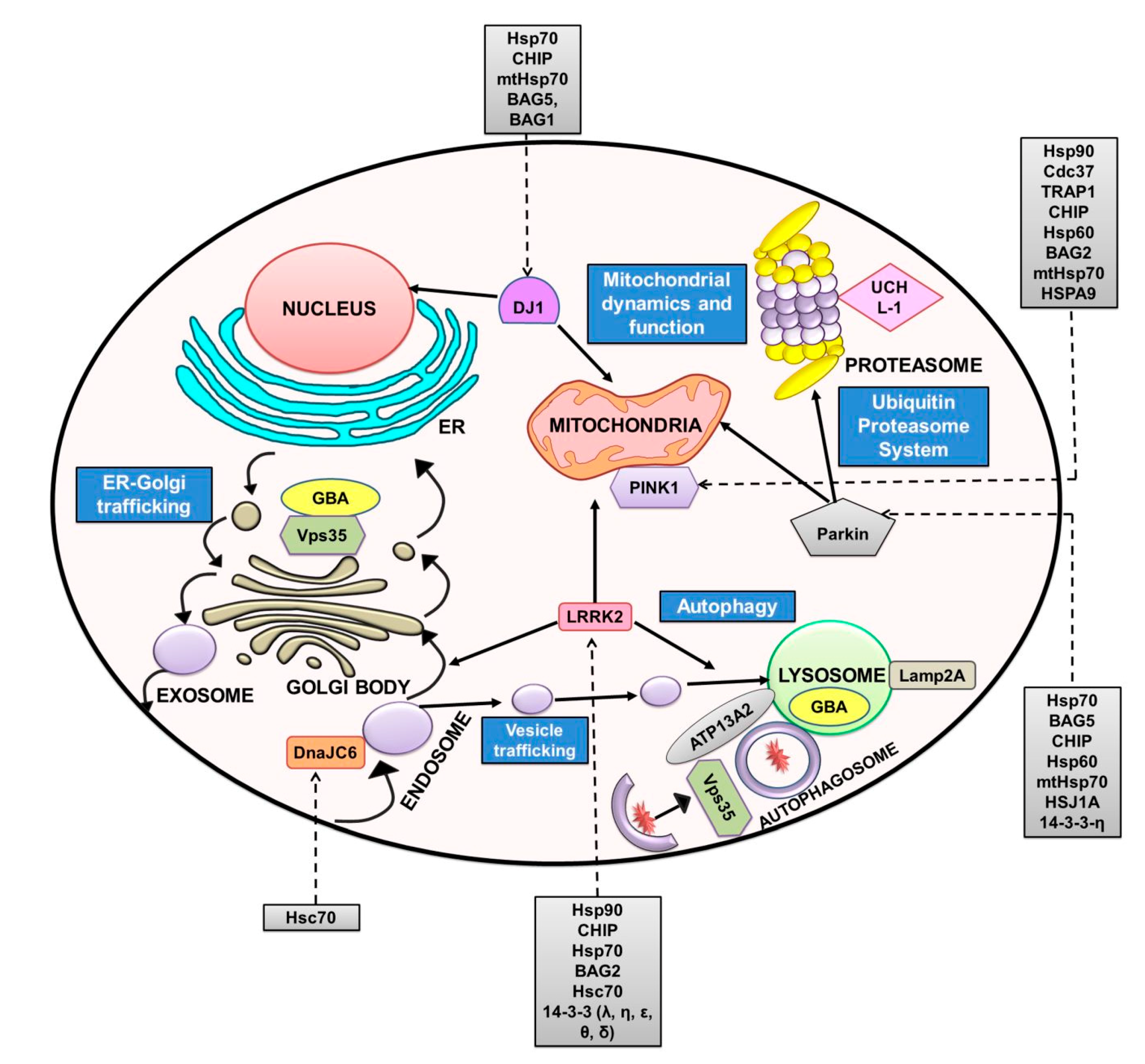
3. Other PD-Related Proteins and Chaperones Regulating Them
3.1. Parkin
3.2. PINK1
3.3. DJ-1
3.4. LRRK2
3.5. DNAJ
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandopadhyay, R.; de Belleroche, J. Pathogenesis of Parkinson’s disease: Emerging role of molecular chaperones. Trends. Mol. Med. 2010, 16, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Dimant, H.; Ebrahimi-Fakhari, D.; McLean, P.J. Molecular chaperones and co-chaperones in Parkinson disease. Neuroscientist 2012, 18, 589–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [Green Version]
- Miller, I.N.; Cronin-Golomb, A. Gender differences in Parkinson’s disease: Clinical characteristics and cognition. Mov. Disord. 2010, 25, 2695–2703. [Google Scholar] [CrossRef] [Green Version]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Stoker, T.; Greenland, J. Pathological Mechanisms and Clinical Aspects of GBA1 Mutation-Associated Parkinson’s Disease. Parkinson’s Disease: Pathogenesis and Clinical Aspects; Codon Publications: Brisbane, Australia, 2018. [Google Scholar]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 1–21. [Google Scholar] [CrossRef]
- Sironi, L.; Restelli, L.M.; Tolnay, M.; Neutzner, A.; Frank, S. Dysregulated interorganellar crosstalk of mitochondria in the pathogenesis of Parkinson’s disease. Cells 2020, 9, 233. [Google Scholar] [CrossRef] [Green Version]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Pickart, C.M.; Cohen, R.E. Proteasomes and their kin: Proteases in the machine age. Nat. Rev. Mol. Cell Biol. 2004, 5, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, J.A.; Guarné, A.; Ortega, J. ClpP: A structurally dynamic protease regulated by AAA+ proteins. J. Struct. Biol. 2012, 179, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Lucius, A.L. Examination of the polypeptide substrate specificity for Escherichia coli ClpA. Biochemistry 2013, 52, 4941–4954. [Google Scholar] [CrossRef]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Gene. Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of native brain α-synuclein. Nature 2013, 498, E4–E6. [Google Scholar] [CrossRef]
- Lopes da Fonseca, T.; Villar-Piqué, A.; Outeiro, T.F. The interplay between alpha-synuclein clearance and spreading. Biomolecules 2015, 5, 435–471. [Google Scholar] [CrossRef] [Green Version]
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4976. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
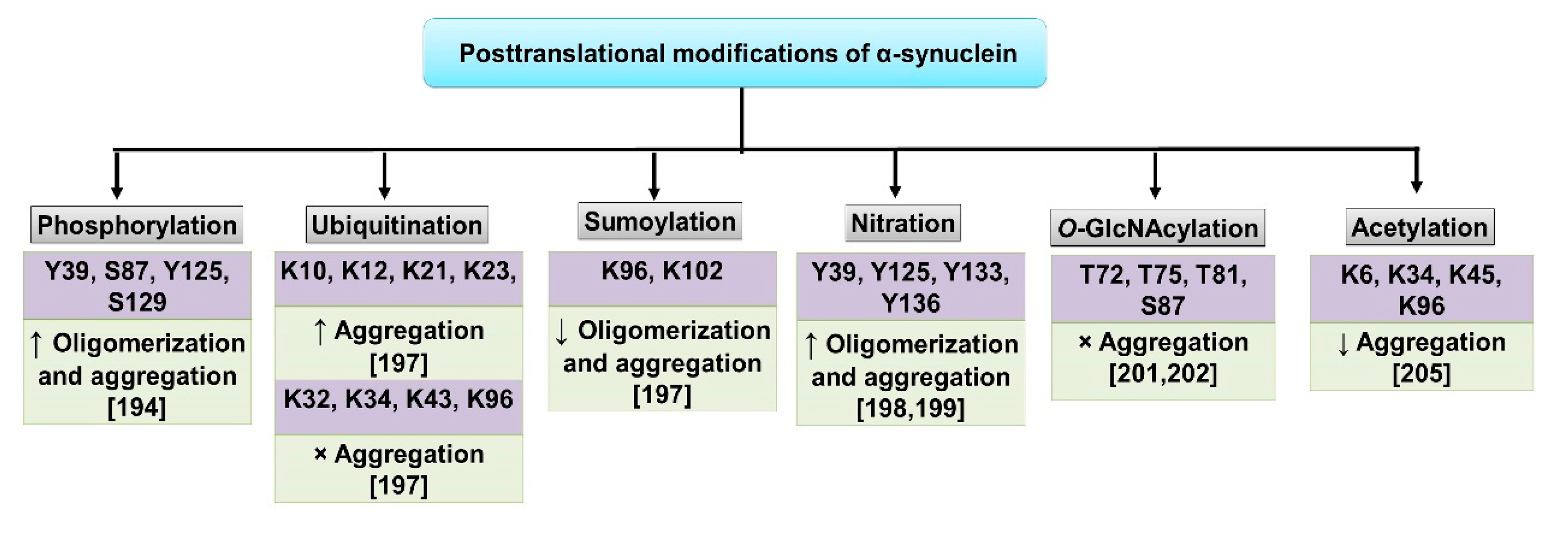
- Li, J.; Zhang, J.; Li, X. The Roles of Post-translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease–lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narkiewicz, J.; Giachin, G.; Legname, G. In vitro aggregation assays for the characterization of α-synuclein prion-like properties. Prion 2014, 8, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falsone, S.F.; Kungl, A.J.; Rek, A.; Cappai, R.; Zangger, K. The molecular chaperone Hsp90 modulates intermediate steps of amyloid assembly of the Parkinson-related protein α-synuclein. J. Biol. Chem. 2009, 284, 31190–31199. [Google Scholar] [CrossRef] [Green Version]
- Uryu, K.; Richter-Landsberg, C.; Welch, W.; Sun, E.; Goldbaum, O.; Norris, E.H.; Pham, C.-T.; Yazawa, I.; Hilburger, K.; Micsenyi, M. Convergence of heat shock protein 90 with ubiquitin in filamentous α-synuclein inclusions of α-synucleinopathies. Am. J. Clin. Pathol. 2006, 168, 947–961. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Clark-Dixon, C.; Wang, S.; Flower, T.R.; Williams-Hart, T.; Zweig, R.; Robinson, L.C.; Tatchell, K.; Witt, S.N. Novel suppressors of α-synuclein toxicity identified using yeast. Hum. Mol. Genet. 2008, 17, 3784–3795. [Google Scholar] [CrossRef] [Green Version]
- Putcha, P.; Danzer, K.M.; Kranich, L.R.; Scott, A.; Silinski, M.; Mabbett, S.; Hicks, C.D.; Veal, J.M.; Steed, P.M.; Hyman, B.T. Brain-permeable small-molecule inhibitors of Hsp90 prevent α-synuclein oligomer formation and rescue α-synuclein-induced toxicity. J. Pharmacol. Exp. Ther. 2010, 332, 849–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, E.K.; Voigt, A.; Lutz, A.K.; Toegel, J.P.; Gerhardt, E.; Karsten, P.; Falkenburger, B.; Reinartz, A.; Winklhofer, K.F.; Schulz, J.B. The mitochondrial chaperone protein TRAP1 mitigates α-Synuclein toxicity. PLoS Genet. 2012, 8, e1002488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rane, A.; Rajagopalan, S.; Ahuja, M.; Thomas, B.; Chinta, S.J.; Andersen, J.K. Hsp90 Co-chaperone p23 contributes to dopaminergic mitochondrial stress via stabilization of PHD2: Implications for Parkinson’s disease. Neurotoxicology 2018, 65, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Carver, J.A.; Ecroyd, H. Preventing α-synuclein aggregation: The role of the small heat-shock molecular chaperone proteins. Biochim. Biophys. Acta 2014, 1842, 1830–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waudby, C.A.; Knowles, T.P.; Devlin, G.L.; Skepper, J.N.; Ecroyd, H.; Carver, J.A.; Welland, M.E.; Christodoulou, J.; Dobson, C.M.; Meehan, S. The interaction of αB-crystallin with mature α-synuclein amyloid fibrils inhibits their elongation. Biophys. J. 2010, 98, 843–851. [Google Scholar] [CrossRef]
- Cox, D.; Whiten, D.R.; Brown, J.W.; Horrocks, M.H.; San Gil, R.; Dobson, C.M.; Klenerman, D.; van Oijen, A.M.; Ecroyd, H. The small heat shock protein Hsp27 binds α-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 2018, 293, 4486–4497. [Google Scholar] [CrossRef] [Green Version]
- Bruinsma, I.B.; Bruggink, K.A.; Kinast, K.; Versleijen, A.A.; Segers-Nolten, I.M.; Subramaniam, V.; Bea Kuiperij, H.; Boelens, W.; de Waal, R.M.; Verbeek, M.M. Inhibition of α-synuclein aggregation by small heat shock proteins. Proteins 2011, 79, 2956–2967. [Google Scholar] [CrossRef]
- Sharma, S.K.; Priya, S. Expanding role of molecular chaperones in regulating α-synuclein misfolding; implications in Parkinson’s disease. Cell. Mol. Life Sci. 2017, 74, 617–629. [Google Scholar] [CrossRef]
- Gao, X.; Carroni, M.; Nussbaum-Krammer, C.; Mogk, A.; Nillegoda, N.B.; Szlachcic, A.; Guilbride, D.L.; Saibil, H.R.; Mayer, M.P.; Bukau, B. Human Hsp70 disaggregase reverses Parkinson’s-linked α-synuclein amyloid fibrils. Mol. Cell 2015, 59, 781–793. [Google Scholar] [CrossRef] [Green Version]
- Bianco, C.L.; Shorter, J.; Régulier, E.; Lashuel, H.; Iwatsubo, T.; Lindquist, S.; Aebischer, P. Hsp104 antagonizes α-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J. Clin. Investig. 2008, 118, 3087–3097. [Google Scholar] [CrossRef] [Green Version]
- McLean, P.J.; Kawamata, H.; Shariff, S.; Hewett, J.; Sharma, N.; Ueda, K.; Breakefield, X.O.; Hyman, B.T. TorsinA and heat shock proteins act as molecular chaperones: Suppression of α-synuclein aggregation. J. Neurochem. 2002, 83, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Jarvela, T.S.; Lam, H.A.; Helwig, M.; Lorenzen, N.; Otzen, D.E.; McLean, P.J.; Maidment, N.T.; Lindberg, I. The neural chaperone proSAAS blocks α-synuclein fibrillation and neurotoxicity. Proc. Natl. Acad. Sci. USA 2016, 113, E4708–E4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Jiang, T.; Han, G.A.; Malintan, N.T.; Xie, L.; Wang, L.; Tse, F.W.; Gaisano, H.Y.; Collins, B.M.; Meunier, F.A. Rescue of Munc18-1 and-2 double knockdown reveals the essential functions of interaction between Munc18 and closed syntaxin in PC12 cells. Mol. Biol. Cell 2009, 20, 4962–4975. [Google Scholar] [CrossRef] [Green Version]
- Chai, Y.J.; Sierecki, E.; Tomatis, V.M.; Gormal, R.S.; Giles, N.; Morrow, I.C.; Xia, D.; Götz, J.; Parton, R.G.; Collins, B.M. Munc18-1 is a molecular chaperone for α-synuclein, controlling its self-replicating aggregation. J. Cell Biol. 2016, 214, 705–718. [Google Scholar] [CrossRef]
- Mishina, M.; Ishiwata, K.; Ishii, K.; Kitamura, S.; Kimura, Y.; Kawamura, K.; Oda, K.; Sasaki, T.; Sakayori, O.; Hamamoto, M. Function of sigma1 receptors in Parkinson’s disease. Acta Neurol. Scand. 2005, 112, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Wang, L.; Zhang, T.; Zhang, B.; Chen, L. Sigma-1 receptor knockout increases α-synuclein aggregation and phosphorylation with loss of dopaminergic neurons in substantia nigra. Neurobiol. Aging 2017, 59, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Bubacco, L. Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant? Biomolecules 2020, 10, 195. [Google Scholar] [CrossRef] [Green Version]
- Carboni, E.; Lingor, P. Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson’s disease. Metallomics 2015, 7, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Horvath, I.; Blockhuys, S.; Sulskis, D.; Holgersson, S.; Kumar, R.; Burmann, B.r.M.; Wittung-Stafshede, P. Interaction between Copper Chaperone Atox1 and Parkinson’s Disease Protein α-Synuclein Includes Metal-Binding Sites and Occurs in Living Cells. ACS Chem. Neurosci. 2019, 10, 4659–4668. [Google Scholar] [CrossRef]
- Horvath, I.; Werner, T.; Kumar, R.; Wittung-Stafshede, P. Copper chaperone blocks amyloid formation via ternary complex. Q. Rev. Biophys. 2018, 51, e6. [Google Scholar] [CrossRef] [Green Version]
- Okita, Y.; Rcom-H’cheo-Gauthier, A.N.; Goulding, M.; Chung, R.S.; Faller, P.; Pountney, D.L. Metallothionein, copper and alpha-synuclein in alpha-synucleinopathies. Front. Neurosci. 2017, 11, 114. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kanai, K.; Suzuki, M.; Kim, W.S.; Yoo, H.S.; Fu, Y.; Kim, D.-K.; Jung, B.C.; Choi, M.; Oh, K.W. Arylsulfatase A, a genetic modifier of Parkinson’s disease, is an α-synuclein chaperone. Brain 2019, 142, 2845–2859. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Rohde, M.; Jäättelä, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, W.L. The J-domain family and the recruitment of chaperone power. Trends Biochem. Sci. 1998, 23, 222–227. [Google Scholar] [CrossRef]
- Sharma, S.K.; De Los Rios, P.; Christen, P.; Lustig, A.; Goloubinoff, P. The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat. Chem. Biol. 2010, 6, 914. [Google Scholar] [CrossRef] [PubMed]
- Höfeld, J.; Minami, Y.; Hartl, F.-U. Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell 1995, 83, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Nollen, E.A.; Brunsting, J.F.; Song, J.; Kampinga, H.H.; Morimoto, R.I. Bag1 functions in vivo as a negative regulator of Hsp70 chaperone activity. Mol. Cellular Biol. 2000, 20, 1083–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherzer, C.R.; Eklund, A.C.; Morse, L.J.; Liao, Z.; Locascio, J.J.; Fefer, D.; Schwarzschild, M.A.; Schlossmacher, M.G.; Hauser, M.A.; Vance, J.M. Molecular markers of early Parkinson’s disease based on gene expression in blood. Proc. Natl. Acad. Sci. USA 2007, 104, 955–960. [Google Scholar] [CrossRef] [Green Version]
- Klucken, J.; Shin, Y.; Masliah, E.; Hyman, B.T.; McLean, P.J. Hsp70 reduces α-synuclein aggregation and toxicity. J. Biol. Chem. 2004, 279, 25497–25502. [Google Scholar] [CrossRef] [Green Version]
- Auluck, P.K.; Chan, H.E.; Trojanowski, J.Q.; Lee, V.M.-Y.; Bonini, N.M. Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef]
- Pemberton, S.; Madiona, K.; Pieri, L.; Kabani, M.; Bousset, L.; Melki, R. Hsc70 protein interaction with soluble and fibrillar α-synuclein. J. Biol. Chem. 2011, 286, 34690–34699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priya, S.; Sharma, S.K.; Sood, V.; Mattoo, R.U.; Finka, A.; Azem, A.; De Los Rios, P.; Goloubinoff, P. GroEL and CCT are catalytic unfoldases mediating out-of-cage polypeptide refolding without ATP. Proc. Natl. Acad. Sci. USA 2013, 110, 7199–7204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, H.; Fukui, N.; Adachi, M.; Saiki, E.; Yamasaki, A.; Matsumura, R.; Kuroyanagi, D.; Hongo, K.; Mizobata, T.; Kawata, Y. Human Molecular Chaperone Hsp60 and Its Apical Domain Suppress Amyloid Fibril Formation of α-Synuclein. Int. J. Mol. Sci. 2020, 21, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sluchanko, N.N.; Gusev, N.B. Moonlighting chaperone-like activity of the universal regulatory 14-3-3 proteins. FEBS J. 2017, 284, 1279–1295. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Underwood, R.; Kamath, A.; Britain, C.; McFerrin, M.B.; McLean, P.J.; Volpicelli-Daley, L.A.; Whitaker, R.H.; Placzek, W.J.; Becker, K. 14-3-3 Proteins Reduce Cell-to-Cell Transfer and Propagation of Pathogenic α-Synuclein. J. Neurosci. 2018, 38, 8211–8232. [Google Scholar] [CrossRef] [Green Version]
- Martinat, C.; Shendelman, S.; Jonason, A.; Leete, T.; Beal, M.F.; Yang, L.; Floss, T.; Abeliovich, A. Sensitivity to oxidative stress in DJ-1-deficient dopamine neurons: An ES-derived cell model of primary Parkinsonism. PLoS Biol. 2004, 2, e327. [Google Scholar] [CrossRef]
- Zondler, L.; Miller-Fleming, L.; Repici, M.; Gonçalves, S.; Tenreiro, S.; Rosado-Ramos, R.; Betzer, C.; Straatman, K.; Jensen, P.H.; Giorgini, F. DJ-1 interactions with α-synuclein attenuate aggregation and cellular toxicity in models of Parkinson’s disease. Cell Death Dis. 2014, 5, e1350. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. α-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A.; Kwon, Y.T. Protein quality control by molecular chaperones in neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi-Fakhari, D.; Wahlster, L.; McLean, P.J. Molecular chaperones in Parkinson’s disease–present and future. J. Parkinsons Dis. 2011, 1, 299–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.C.; Fishman, P.S.; Thakor, N.V.; Oyler, G.A. Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J. Biol. Chem. 2003, 278, 22044–22055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesen, E.L.; De Snoo, M.L.; Rajendran, L.; Kalia, L.V.; Kalia, S.K. Chaperone-based therapies for disease modification in Parkinson’s disease. Parkinson’s Dis. 2017, 22017, 5015307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connell, P.; Ballinger, C.A.; Jiang, J.; Wu, Y.; Thompson, L.J.; Höhfeld, J.; Patterson, C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001, 3, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Kalia, S.K.; Chau, H.; Lozano, A.M.; Hyman, B.T.; McLean, P.J. Ubiquitinylation of α-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5). PLoS ONE 2011, 6, e14695. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates α-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Ballinger, C.A.; Wu, Y.; Dai, Q.; Cyr, D.M.; Höhfeld, J.; Patterson, C. CHIP is a U-box-dependent E3 ubiquitin ligase identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem. 2001, 276, 42938–42944. [Google Scholar] [CrossRef] [Green Version]
- Tetzlaff, J.E.; Putcha, P.; Outeiro, T.F.; Ivanov, A.; Berezovska, O.; Hyman, B.T.; McLean, P.J. CHIP targets toxic α-synuclein oligomers for degradation. J. Biol. Chem. 2008, 283, 17962–17968. [Google Scholar] [CrossRef] [Green Version]
- Takayama, S.; Bimston, D.N.; Matsuzawa, S.I.; Freeman, B.C.; Aime-Sempe, C.; Xie, Z.; Morimoto, R.I.; Reed, J.C. BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J. 1997, 16, 4887–4896. [Google Scholar] [CrossRef] [Green Version]
- Lüders, J.; Demand, J.; Höhfeld, J. The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J. Biol. Chem. 2000, 275, 4613–4617. [Google Scholar] [CrossRef] [Green Version]
- Demand, J.; Alberti, S.; Patterson, C.; Höhfeld, J. Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 2001, 11, 1569–1577. [Google Scholar] [CrossRef] [Green Version]
- Deshayes, N.; Arkan, S.; Hansen, C. The Molecular Chaperone DNAJB6, but Not DNAJB1, Suppresses the Seeded Aggregation of Alpha-Synuclein in Cells. Int. J. Mol. Sci. 2019, 20, 4495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelender, S. Ubiquitination of α-synuclein and autophagy in Parkinson’s disease. Autophagy 2008, 4, 372–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rott, R.; Szargel, R.; Haskin, J.; Shani, V.; Shainskaya, A.; Manov, I.; Liani, E.; Avraham, E.; Engelender, S. Monoubiquitylation of α-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 2008, 283, 3316–3328. [Google Scholar] [CrossRef] [Green Version]
- Rott, R.; Szargel, R.; Haskin, J.; Bandopadhyay, R.; Lees, A.J.; Shani, V.; Engelender, S. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 18666–18671. [Google Scholar] [CrossRef] [Green Version]
- Machiya, Y.; Hara, S.; Arawaka, S.; Fukushima, S.; Sato, H.; Sakamoto, M.; Koyama, S.; Kato, T. Phosphorylated α-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J. Biol. Chem. 2010, 285, 40732–40744. [Google Scholar] [CrossRef] [Green Version]
- Bentea, E.; Verbruggen, L.; Massie, A. The proteasome inhibition model of Parkinson’s disease. J. Parkinsons Dis. 2017, 7, 31–63. [Google Scholar] [CrossRef] [Green Version]
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [CrossRef] [Green Version]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced α-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef]
- Zondler, L.; Kostka, M.; Garidel, P.; Heinzelmann, U.; Hengerer, B.; Mayer, B.; Weishaupt, J.H.; Gillardon, F.; Danzer, K.M. Proteasome impairment by α-synuclein. PLoS ONE 2017, 12, e0184040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghee, M.; Fournier, A.; Mallet, J. Rat α-synuclein interacts with Tat binding protein 1, a component of the 26S proteasomal complex. J. Neurochem. 2000, 75, 2221–2224. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.K.; Rajput, C.; Mishra, S.; ur Rasheed, M.S.; Singh, M.P. Malfunctioning of chaperone-mediated autophagy in Parkinson’s disease: Feats, constraints, and flaws of modulators. Neurotox. Res. 2019, 35, 260–270. [Google Scholar] [CrossRef]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, S.K.; McCormack, A.L.; Manning-Boğ, A.B.; Cuervo, A.M.; Di Monte, D.A. Lysosomal degradation of α-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.-C.; Follenzi, A. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Rodriguez-Oroz, M.C.; Obeso, J.A.; Cooper, J. Influence of microRNA deregulation on chaperone-mediated autophagy and α-synuclein pathology in Parkinson’s disease. Cell Death Dis. 2013, 4, e545. [Google Scholar] [CrossRef]
- Li, G.; Yang, H.; Zhu, D.; Huang, H.; Liu, G.; Lun, P. Targeted suppression of chaperone-mediated autophagy by miR-320a promotes α-synuclein aggregation. Int. J. Mol. Sci. 2014, 15, 15845–15857. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Polissidis, A.; Chrysanthou-Piterou, M.; Kloukina, I.; Stefanis, L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016, 12, 2230–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Lian, Y.; Ding, J.; Meng, Y.; Li, C.; Chen, L.; Qiu, P. The role of chaperone-mediated autophagy in neurotoxicity induced by alpha-synuclein after methamphetamine exposure. Brain Behav. 2019, 9, e01352. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-Y.; Kang, W.-Y.; Chen, Y.-M.; Jiang, T.-F.; Zhang, J.; Zhang, L.-N.; Ding, J.-Q.; Liu, J.; Chen, S.-D. DJ-1 inhibits α-synuclein aggregation by regulating chaperone-mediated autophagy. Front. Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, T.; Suzuki, K.; Ohsumi, Y. Yeast autophagosomes: De novo formation of a membrane structure. Trends Cell Biol. 2002, 12, 231–235. [Google Scholar] [CrossRef]
- Kraft, C.; Martens, S. Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 2012, 24, 496–501. [Google Scholar] [CrossRef]
- Reggiori, F.; Tucker, K.A.; Stromhaug, P.E.; Klionsky, D.J. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev. Cell 2004, 6, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Young, A.R.; Chan, E.Y.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef] [Green Version]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schüchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5–Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef]
- Tanida, I.; Sou, Y.-s.; Ezaki, J.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3-and GABAA receptor-associated protein-phospholipid conjugates. J. Biol. Chem. 2004, 279, 36268–36276. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Chegini, F.; Chua, G.; Murphy, K.; Gai, W.; Halliday, G.M. Macroautophagy in sporadic and the genetic form of Parkinson’s disease with the A53T α-synuclein mutation. Transl. Neurodegener. 2012, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.-S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkin, V.; Lamark, T.; Sou, Y.-S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.-A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.M.; Wong, E.S.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.-P.; Ho, M.W.; Troncoso, J.; Gygi, S.P.; Lee, M.K. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Gen. 2008, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Mori, F.; Tanji, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Accumulation of histone deacetylase 6, an aggresome-related protein, is specific to Lewy bodies and glial cytoplasmic inclusions. Neuropathology 2011, 31, 561–568. [Google Scholar] [CrossRef]
- Du, Y.; Wang, F.; Zou, J.; Le, W.; Dong, Q.; Wang, Z.; Shen, F.; Yu, L.; Li, Y. Histone deacetylase 6 regulates cytotoxic α-synuclein accumulation through induction of the heat shock response. Neurobiol. Aging 2014, 35, 2316–2328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qian, S.-B. Chaperone-mediated hierarchical control in targeting misfolded proteins to aggresomes. Mol. Biol. Cell 2011, 22, 3277–3288. [Google Scholar] [CrossRef]
- Cao, Y.-L.; Yang, Y.-P.; Mao, C.-J.; Zhang, X.-Q.; Wang, C.-T.; Yang, J.; Lv, D.-J.; Wang, F.; Hu, L.-F.; Liu, C.-F. A role of BAG3 in regulating SNCA/α-synuclein clearance via selective macroautophagy. Neurobiol. Aging 2017, 60, 104–115. [Google Scholar] [CrossRef]
- McGlinchey, R.P.; Lee, J.C. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc. Natl. Acad. Sci. USA 2015, 112, 9322–9327. [Google Scholar] [CrossRef] [Green Version]
- Vidoni, C.; Follo, C.; Savino, M.; Melone, M.A.; Isidoro, C. The role of cathepsin D in the pathogenesis of human neurodegenerative disorders. Med. Res. Rev. 2016, 36, 845–870. [Google Scholar] [CrossRef]
- Cullen, V.; Sardi, S.P.; Ng, J.; Xu, Y.H.; Sun, Y.; Tomlinson, J.J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J. Acid β-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α-synuclein processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef]
- Abdelmotilib, H.; Maltbie, T.; Delic, V.; Liu, Z.; Hu, X.; Fraser, K.B.; Moehle, M.S.; Stoyka, L.; Anabtawi, N.; Krendelchtchikova, V. α-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic neurodegeneration. Neurobiol. Dis. 2017, 105, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Bae, E.-J.; Lee, S.-J. Extracellular α-synuclein—A novel and crucial factor in Lewy body diseases. Nat. Rev. Neurol. 2014, 10, 92. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Ruf, W.P.; Putcha, P.; Joyner, D.; Hashimoto, T.; Glabe, C.; Hyman, B.T.; McLean, P.J. Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 2011, 25, 326–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehay, B.; Vila, M.; Bezard, E.; Brundin, P.; Kordower, J.H. Alpha-synuclein propagation: New insights from animal models. Mov. Disord. 2016, 31, 161–168. [Google Scholar] [CrossRef]
- Fontaine, S.N.; Zheng, D.; Sabbagh, J.J.; Martin, M.D.; Chaput, D.; Darling, A.; Trotter, J.H.; Stothert, A.R.; Nordhues, B.A.; Lussier, A. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016, 35, 1537–1549. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, J.-P.; Shi, M.; Quinn, T.; Bradner, J.; Beyer, R.; Chen, S.; Zhang, J. Rab11a and HSP90 regulate recycling of extracellular α-synuclein. J. Neurosci. 2009, 29, 1480–1485. [Google Scholar] [CrossRef] [Green Version]
- Rey, N.; George, S.; Brundin, P. Spreading the word: Precise animal models and validated methods are vital when evaluating prion-like behaviour of alpha-synuclein. Neuropathol. Appl. Neurobiol. 2016, 42, 51–76. [Google Scholar] [CrossRef]
- Tyson, T.; Steiner, J.A.; Brundin, P. Sorting out release, uptake and processing of alpha-synuclein during prion-like spread of pathology. J. Neurochem. 2016, 139, 275–289. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. Elife 2018, 7, e36402. [Google Scholar] [CrossRef]
- Surguchev, A.A.; Emamzadeh, F.N.; Surguchov, A. Cell responses to extracellular α-synuclein. Molecules 2019, 24, 305. [Google Scholar] [CrossRef] [Green Version]
- Diógenes, M.J.; Dias, R.B.; Rombo, D.M.; Miranda, H.V.; Maiolino, F.; Guerreiro, P.; Näsström, T.; Franquelim, H.G.; Oliveira, L.M.; Castanho, M.A. Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J. Neurosci. 2012, 32, 11750–11762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, A.N.; Redeker, V.; Fritz, N.; Pieri, L.; Almeida, L.G.; Spolidoro, M.; Liebmann, T.; Bousset, L.; Renner, M.; Léna, C. α-synuclein assemblies sequester neuronal α3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 2015, 34, 2408–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiten, D.R.; Cox, D.; Horrocks, M.H.; Taylor, C.G.; De, S.; Flagmeier, P.; Tosatto, L.; Kumita, J.R.; Ecroyd, H.; Dobson, C.M.; et al. Single-Molecule Characterization of the Interactions between Extracellular Chaperones and Toxic α-Synuclein Oligomers. Cell Rep. 2018, 23, 3492–3500. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Trempe, J.-F.; Fon, E.A. Structure and function of Parkin, PINK1, and DJ-1, the three musketeers of neuroprotection. Front. Neurol. 2013, 4, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahabi, K.; Perwez, A.; Rizvi, M.A. Parkin in Parkinson’s disease and Cancer: A double-edged sword. Mol. Neurobiol. 2018, 55, 6788–6800. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Schlossmacher, M.G.; Frosch, M.P.; Gai, W.P.; Medina, M.; Sharma, N.; Forno, L.; Ochiishi, T.; Shimura, H.; Sharon, R.; Hattori, N. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am. J. Clin. Pathol. 2002, 160, 1655–1667. [Google Scholar] [CrossRef] [Green Version]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25, S32–S39. [Google Scholar] [CrossRef]
- Imai, Y.; Soda, M.; Hatakeyama, S.; Akagi, T.; Hashikawa, T.; Nakayama, K.-I.; Takahashi, R. CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol. Cell 2002, 10, 55–67. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Henn, I.H.; Kay-Jackson, P.C.; Heller, U.; Tatzelt, J. Inactivation of Parkin by oxidative stress and C-terminal truncations a protective role of molecular chaperones. J. Biol. Chem. 2003, 278, 47199–47208. [Google Scholar] [CrossRef] [Green Version]
- Kalia, S.K.; Lee, S.; Smith, P.D.; Liu, L.; Crocker, S.J.; Thorarinsdottir, T.E.; Glover, J.R.; Fon, E.A.; Park, D.S.; Lozano, A.M. BAG5 inhibits parkin and enhances dopaminergic neuron degeneration. Neuron 2004, 44, 931–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, X.; Liu, X.; Yang, L.; Chen, Q.; Zhao, D.; Zuo, J.; Liu, W. Mitochondrial dysfunction induced by knockdown of mortalin is rescued by Parkin. Biochem. Biophys. Res. Commun. 2011, 410, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.M.; Novoselov, S.S.; Robinson, P.A.; Cheetham, M.E. Molecular chaperone-mediated rescue of mitophagy by a Parkin RING1 domain mutant. Hum. Mol. Gen. 2011, 20, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakkar, V.; Kuiper, E.E.; Pandey, A.; Braakman, I.; Kampinga, H.H. Versatile members of the DNAJ family show Hsp70 dependent anti-aggregation activity on RING1 mutant parkin C289G. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Davison, E.J.; Pennington, K.; Hung, C.C.; Peng, J.; Rafiq, R.; Ostareck-Lederer, A.; Ostareck, D.H.; Ardley, H.C.; Banks, R.E.; Robinson, P.A. Proteomic analysis of increased Parkin expression and its interactants provides evidence for a role in modulation of mitochondrial function. Proteomics 2009, 9, 4284–4297. [Google Scholar] [CrossRef]
- Rakovic, A.; Grünewald, A.; Voges, L.; Hofmann, S.; Orolicki, S.; Lohmann, K.; Klein, C. PINK1-interacting proteins: Proteomic analysis of overexpressed PINK1. Parkinsons Dis. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Loh, S.; Martins, L.M. Drosophila Trap1 protects against mitochondrial dysfunction in a PINK1/parkin model of Parkinson’s disease. Cell Death Dis. 2013, 4, e467. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Chiba, T.; Tanaka, K.; Mizuno, Y.; Hattori, N. 14-3-3eta is a novel regulator of parkin ubiquitin-ligase. Mov. Disord. 2006, 25, 211–221. [Google Scholar]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.S.; Miljan, E.A.; Keen, G.; Stanyer, L.; Hargreaves, I.; Klupsch, K.; Deas, E. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 2008, 3, e2455. [Google Scholar] [CrossRef]
- Weihofen, A.; Ostaszewski, B.; Minami, Y.; Selkoe, D.J. Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum. Mol. Gen. 2008, 17, 602–616. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, Y.; Kim, Y.-J.; Ido, Y.; Misawa, H.; Kawashima, K.; Endo, S.; Takahashi, R. L347P PINK1 mutant that fails to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a proteasome-dependent manner. Neurosci. Res. 2008, 61, 43–48. [Google Scholar] [CrossRef]
- Lin, W.; Kang, U.J. Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 2008, 106, 464–474. [Google Scholar] [CrossRef] [Green Version]
- Qu, D.; Hage, A.; Don-Carolis, K.; Huang, E.; Joselin, A.; Safarpour, F.; Marcogliese, P.C.; Rousseaux, M.W.; Hewitt, S.J.; Huang, T. BAG2 gene-mediated regulation of PINK1 protein is critical for mitochondrial translocation of PARKIN and neuronal survival. J. Biol. Chem. 2015, 290, 30441–30452. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Karsten, P.; Hamm, S.; Pogson, J.H.; Müller-Rischart, A.K.; Exner, N.; Haass, C.; Whitworth, A.J.; Winklhofer, K.F.; Schulz, J.B. TRAP1 rescues PINK1 loss-of-function phenotypes. Hum. Mol. Gen. 2013, 22, 2829–2841. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-H.; Song, K.; Yoon, S.-H.; Shehzad, O.; Kim, Y.-S.; Son, J.H. Rescue of PINK1 protein null-specific mitochondrial complex IV deficits by ginsenoside Re activation of nitric oxide signaling. J. Biol. Chem. 2012, 287, 44109–44120. [Google Scholar] [CrossRef] [Green Version]
- Honbou, K.; Suzuki, N.N.; Horiuchi, M.; Niki, T.; Taira, T.; Ariga, H.; Inagaki, F. The crystal structure of DJ-1, a protein related to male fertility and Parkinson’s disease. J. Biol. Chem. 2003, 278, 31380–31384. [Google Scholar] [CrossRef] [Green Version]
- Malgieri, G.; Eliezer, D. Structural effects of Parkinson’s disease linked DJ-1 mutations. Protein Sci. 2008, 17, 855–868. [Google Scholar] [CrossRef] [Green Version]
- Ariga, H.; Takahashi-Niki, K.; Kato, I.; Maita, H.; Niki, T.; Iguchi-Ariga, S.M. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 683920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagakubo, D.; Taira, T.; Kitaura, H.; Ikeda, M.; Tamai, K.; Iguchi-Ariga, S.M.; Ariga, H. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation withras. Biochem. Biophys. Res. Commun. 1997, 231, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Miyasaka, T.; Hatsuta, H.; Takahashi-Niki, K.; Hayashi, K.; Mita, Y.; Kusano-Arai, O.; Iwanari, H.; Ariga, H.; Hamakubo, T. Immunostaining of oxidized DJ-1 in human and mouse brains. J. Neuropathol. Exp. Neurol. 2014, 73, 714–728. [Google Scholar] [CrossRef]
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 2004, 127, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Li, H.M.; Niki, T.; Taira, T.; Iguchi-Ariga, S.M.; Ariga, H. Association of DJ-1 with chaperones and enhanced association and colocalization with mitochondrial Hsp70 by oxidative stress. Free Radic. Res. 2005, 39, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Maita, C.; Maita, H.; Iguchi-Ariga, S.M.; Ariga, H. Monomer DJ-1 and its N-terminal sequence are necessary for mitochondrial localization of DJ-1 mutants. PLoS ONE 2013, 8, e540878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.-X.; Tan, J.-Q.; Zhang, H.-N.; Rizwana, K.; Lu, J.-H.; Tang, J.-G.; Jiang, B.; Shen, X.-M.; Guo, J.-F.; Tang, B.-S. BAG5 Interacts with DJ-1 and inhibits the neuroprotective effects of DJ-1 to combat mitochondrial oxidative damage. Oxid. Med. Cell. Longev. 2017, 2017, 5094934. [Google Scholar] [CrossRef]
- Deeg, S.; Gralle, M.; Sroka, K.; Bähr, M.; Wouters, F.S.; Kermer, P. BAG1 restores formation of functional DJ-1 L166P dimers and DJ-1 chaperone activity. J. Cell Biol. 2010, 188, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.J.; Zhang, L.; Troncoso, J.; Lee, M.K.; Hattori, N.; Mizuno, Y.; Dawson, T.M.; Dawson, V.L. Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum. Mol. Gen. 2005, 14, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Bosgraaf, L.; Van Haastert, P.J. Roc, a Ras/GTPase domain in complex proteins. Mol. Cell. Res. 2003, 1643, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Tsika, E.; Moore, D.J. Contribution of GTPase activity to LRRK2-associated Parkinson disease. Small GTPases 2013, 4, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The role of LRRK2 in neurodegeneration of parkinson disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Berwick, D.C.; Heaton, G.R.; Azeggagh, S.; Harvey, K. LRRK2 Biology from structure to dysfunction: Research progresses, but the themes remain the same. Mol. Neurodegener. 2019, 14, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xie, C.; Greggio, E.; Parisiadou, L.; Shim, H.; Sun, L.; Chandran, J.; Lin, X.; Lai, C.; Yang, W.-J. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J. Neurosci. 2008, 28, 3384–3391. [Google Scholar] [CrossRef]
- Ko, H.S.; Bailey, R.; Smith, W.W.; Liu, Z.; Shin, J.-H.; Lee, Y.-I.; Zhang, Y.-J.; Jiang, H.; Ross, C.A.; Moore, D.J. CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 2897–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Goldberg, M.S. Regulation of LRRK2 stability by the E3 ubiquitin ligase CHIP. PLoS ONE 2009, 4, e5949. [Google Scholar] [CrossRef]
- Fukuzono, T.; Pastuhov, S.I.; Fukushima, O.; Li, C.; Hattori, A.; Iemura, S.i.; Natsume, T.; Shibuya, H.; Hanafusa, H.; Matsumoto, K. Chaperone complex BAG 2–HSC 70 regulates localization of Caenorhabditis elegans leucine-rich repeat kinase LRK-1 to the Golgi. Genes Cells 2016, 21, 311–324. [Google Scholar] [CrossRef] [Green Version]
- Orenstein, S.J.; Kuo, S.-H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394. [Google Scholar] [CrossRef] [Green Version]
- Sala, G.; Marinig, D.; Arosio, A.; Ferrarese, C. Role of chaperone-mediated autophagy dysfunctions in the pathogenesis of Parkinson’s disease. Front. Mol. Neurosci. 2016, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.J.; Dzamko, N.; Morrice, N.A.; Campbell, D.G.; Deak, M.; Ordureau, A.; Macartney, T.; Tong, Y.; Shen, J.; Prescott, A.R.; et al. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem. J. 2010, 430, 393–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, T.; Yoshida, S.; Sugeno, N.; Kobayashi, J.; Aoki, M. DnaJ/Hsp40 family and Parkinson’s disease. Front. Neurosci. 2018, 11, 743. [Google Scholar] [CrossRef]
- Zarouchlioti, C.; Parfitt, D.A.; Li, W.; Gittings, L.M.; Cheetham, M.E. DNAJ Proteins in neurodegeneration: Essential and protective factors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160534. [Google Scholar] [CrossRef] [Green Version]
- Olgiati, S.; Quadri, M.; Fang, M.; Rood, J.P.; Saute, J.A.; Chien, H.F.; Bouwkamp, C.G.; Graafland, J.; Minneboo, M.; Breedveld, G.J. DNAJC6 Mutations Associated with Early-Onset Parkinson’s Disease. Ann. Neurol. 2016, 79, 244–256. [Google Scholar] [CrossRef]
- Ungewickell, E.; Ungewickell, H.; Holstein, S.E.; Lindner, R.; Prasad, K.; Barouch, W.; Martini, B.; Greene, L.E.; Eisenberg, E. Role of auxilin in uncoating clathrin-coated vesicles. Nature 1995, 378, 632–635. [Google Scholar] [CrossRef]
- Morgan, J.R.; Prasad, K.; Jin, S.; Augustine, G.J.; Lafer, E.M. Uncoating of clathrin-coated vesicles in presynaptic terminals: Roles for Hsc70 and auxilin. Neuron 2001, 32, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Roosen, D.A.; Blauwendraat, C.; Cookson, M.R.; Lewis, P.A. DNAJC proteins and pathways to parkinsonism. FEBS J. 2019, 286, 3080–3094. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.; Krainc, D. LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 5576–5581. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.R.; Moussaud, S.; McLean, P. Targeting heat shock proteins to modulate α-synuclein toxicity. Ther. Adv. Neurol. Disord. 2014, 7, 33–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varkey, J.; Isas, J.M.; Mizuno, N.; Jensen, M.B.; Bhatia, V.K.; Jao, C.C.; Petrlova, J.; Voss, J.C.; Stamou, D.G.; Steven, A.C. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J. Biol. Chem. 2010, 285, 32486–32493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K. A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Chen, M.; Mi, N.; Yang, W.; Li, X.; Wang, P.; Yin, N.; Li, Y.; Yue, F.; Chan, P. Increased oligomerization and phosphorylation of α-synuclein are associated with decreased activity of glucocerebrosidase and protein phosphatase 2A in aging monkey brains. Neurobiol. Aging 2015, 36, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.-Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits α-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef]
- Oueslati, A. Implication of Alpha-Synuclein Phosphorylation at S129 in Synucleinopathies: What Have We Learned in the Last Decade? J. Parkinsons Dis. 2016, 6, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Meier, F.; Abeywardana, T.; Dhall, A.; Marotta, N.P.; Varkey, J.; Langen, R.; Chatterjee, C.; Pratt, M.R. Semisynthetic, site-specific ubiquitin modification of α-synuclein reveals differential effects on aggregation. J. Am. Chem. Soc. 2012, 134, 5468–5471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rott, R.; Szargel, R.; Shani, V.; Hamza, H.; Savyon, M.; Elghani, F.A.; Bandopadhyay, R.; Engelender, S. SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 13176–13181. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M.-Y. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Liu, Y.; Qiang, M.; Wei, Y.; He, R. A novel molecular mechanism for nitrated α-synuclein-induced cell death. J. Mol. Cell Biol. 2011, 3, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Danielson, S.R.; Held, J.M.; Schilling, B.; Oo, M.; Gibson, B.W.; Andersen, J.K. Preferentially increased nitration of α-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson’s disease. Anal. Chem. 2009, 81, 7823–7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, Y.E.; Galesic, A.; Levine, P.M.; De Leon, C.A.; Lamiri, N.; Brennan, C.K.; Pratt, M.R. O-GlcNAcylation of α-synuclein at serine 87 reduces aggregation without affecting membrane binding. ACS Chem. Biol. 2017, 12, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Levine, P.M.; Galesic, A.; Balana, A.T.; Mahul-Mellier, A.-L.; Navarro, M.X.; De Leon, C.A.; Lashuel, H.A.; Pratt, M.R. α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duce, J.A.; Wong, B.X.; Durham, H.; Devedjian, J.-C.; Smith, D.P.; Devos, D. Post translational changes to α-synuclein control iron and dopamine trafficking; a concept for neuron vulnerability in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 45. [Google Scholar] [CrossRef] [Green Version]
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V.; Lang, A.E. Parkinson disease in 2015: Evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 2016, 12, 65. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| PD Associated Protein | Associated Chaperones and Co-Chaperones | Function in Proteostasis | Localization | Reference | |
|---|---|---|---|---|---|
| 1. | α-synuclein (PARK1) | Hsp70 (HspA) | Refolding of misfolded α-synuclein and aids in degradation | Mitochondria, Cytosol, ER, Nucleus | [39,61] |
| Hsp40 (DNAJ) | Refolding of misfolded α-synuclein | Cytosol, Nucleus | [61] | ||
| DNAJB6 | Prevents formation of α-synuclein fibrils and is involved in its degradation via UPS | Cytosol, Nucleus | [83] | ||
| ST13 (Hip) | Co-chaperone of Hsp70 | Cytosol | [57] | ||
| BAG1 (Hap, Rap46) | Co-chaperone of Hsp70 and Hsc70; prevents aggregation | Cytosol, Nucleus | [58] | ||
| BAG5 | Co-chaperone of Hsp70 and Hsc70; role in of α-synuclein degradation | Cytosol, Nucleus, Mitochondria | [76] | ||
| HSPA8 (Hsc70) | Prevents formation of α-synuclein fibrils and is involved in its degradation via CMA | Cytosol, Nucleus, Lysosomes, Plasma membrane | [62] | ||
| HSPA9 (mtHsp70/GRP78/Mortalin) | Prevents formation of α-synuclein fibrils | Mitochondria | [74] | ||
| BAG3 | Co-chaperone of Hsp70; involved in the degradation of α-synuclein via macroautophagy | Cytosol, Nucleus, Plasma membrane | [118] | ||
| Hsp90 (HspC) | Modulates assembly of α-synuclein to form fibrils | Cytosol, ER, Nucleus, Lysosome | [29] | ||
| Aha1 (p38) | Co-chaperone of Hsp90; involved in α-synuclein fibrillation | Cytosol, ER, | [21] | ||
| TRAP1 (HspC5) | Prevents formation of α-synuclein fibrils | Mitochondria | [33] | ||
| p23 | Co-chaperone of Hsp90; prevents formation of α-synuclein fibrils | Cytosol, Nucleus | [21] | ||
| STI1 (Hop) | Co-chaperone of Hsp90; prevents formation of α-synuclein fibrils | Cytosol, Nucleus | [21] | ||
| CHIP (Stub1) | Co-chaperone of Hsp90 and Hsp70; involved in degradation of α-synuclein | Cytosol, Nucleus | [21,117] | ||
| Sgt1 | Co-chaperone of Hsp90; prevents aggregation of α-synuclein | Cytosol, Nucleus | [21] | ||
| CHP-1 | Co-chaperone of Hsp90; involved in aggregation of α-synuclein | Cytosol | [21] | ||
| Hsp60 (HspD1) | Prevents α-synuclein aggregation | Mitochondria, Cytosol | [64] | ||
| DJ-1 (PARK7) | Inhibits α-synuclein glycation and aggregation | Mitochondria, Nucleus Cytosol | [67] | ||
| Hsp27 (HspB) | Helps in disaggregation of α-synuclein fibrils and prevents aggregation | Cytosol, Nucleus, Cytoskeleton | [37] | ||
| αB-crystallin (HSPB) | Helps in disaggregation of α-synuclein fibrils prevents aggregation | Nucleus, Cytosol | [36] | ||
| HspB8 | Helps in disaggregation of α-synuclein fibrils prevents aggregation | Cytosol, Nucleus | [38] | ||
| HspB2B3 | Helps in disaggregation of α-synuclein fibrils prevents aggregation | Cytosol, Nucleus | [38] | ||
| Apg2 (Hsp110) | Helps in disaggregation of α-synuclein fibrils prevents aggregation | Nucleus, Cytosol | [40] | ||
| 14-3-3-θ | Involved in protein trafficking and refolding of α-synuclein | Nucleus, Cytosol | [66] | ||
| proSAAS/7B2 | Prevents formation of α-synuclein fibrils | [43] | |||
| Torsin-1A (Tor1A) | Prevents formation of α-synuclein fibrils | Cytoskeleton, Nucleus, ER | [42] | ||
| HDJ1 | Prevents formation of α-synuclein fibrils | Cytosol, Nucleus | [42] | ||
| HDJ2 | Prevents formation of α-synuclein fibrils | Cytosol, Nucleus | [42] | ||
| Atox1 | Prevents formation of α-synuclein fibrils | Cytosol | [50] | ||
| Sigma-1 | Prevents formation of α-synuclein fibrils | Nucleus, ER | [47] | ||
| Munc 18-1 | Prevents formation of α-synuclein fibrils | Plasma membrane, Nucleus, Cytosol | [45] | ||
| Arylsulfatase A (ARSA) | Prevents formation of α-synuclein fibrils | Lysosome, Cytosol | [53] | ||
| Clusterin (CLU) | Prevents formation of α-synuclein fibrils | Extracellular, Mitochondria, Nucleus | [134] | ||
| α2-macroglobulin (α2M) | Prevents formation of α-synuclein fibrils | Extracellular, Cytosol | [134] | ||
| 2. | Parkin ( PARK2) | Hsp70 (HspA) | Refolding of misfolded Parkin | Mitochondria, Cytosol, ER, Nucleus | [142,169] |
| BAG5 | Co-chaperone of Hsp70 and Hsc70; inhibits E3 ubiquitin activity of Parkin | Cytosol, Mitochondria, Nucleus | [143] | ||
| CHIP | Co-chaperone of Hsp70; enhances the E3 activity of Parkin through dissociation of Hsp70 from Parkin-Pael-R complexes | Cytosol, Nucleus | [141,169] | ||
| mtHsp70 (Mortalin/HspA9) | Helps in maintaining mitochondrial homeostasis through its interaction with Parkin | Mitochondria | [146] | ||
| HSJ1a | Co-chaperone of Hsp70; reduces misfolding and aggregation of mutant Parkin | Nucleus, Cytosol, ER | [146] | ||
| 14-3-3-η | Negatively regulated E3 ubiquitin ligase activity of Parkin | Mitochondria | [151] | ||
| Hsp60 (HspD1) | Involved in mitochondrial protein folding | Mitochondria, Cytosol | [148] | ||
| 3. | DJ-1 (PARK7) | Hsp70 (HspA) | Stabilizes DJ-1 and facilitates its protease activity | Mitochondria, Cytosol, ER, Nucleus | [166] |
| CHIP | Facilitates protease activity of DJ-1 | Cytosol, Nucleus | [166] | ||
| mtHsp70 | Facilitates protease activity of DJ-1 | Mitochondria | [166] | ||
| BAG1 | Co-chaperone of Hsp70; stabilizes DJ-1 and facilitates activity | Cytosol, Nucleus | [169] | ||
| BAG5 | Reduces DJ-1 dimerization and attenuates its stability | Cytosol, Mitochondria, Nucleus | [168] | ||
| 4. | PINK1 (PARK6) | Hsp90 (HspC) | Stabilization of cleaved forms of PINK1 | Cytosol, ER, Nucleus, Lysosome | [156] |
| Cdc37 | Co-chaperone of Hsp90; stabilizes Pink1 | Cytosol | [156] | ||
| TRAP1 (HSPC5) | Restores Pink1 loss-of-function phenotypes | Mitochondrion, Nucleus | [150] | ||
| CHIP | Decreases ubiquitinated Pink1 | Cytosol, Nucleus | [157] | ||
| Hsp60 (HspD1) | Involved in mitochondrial protein folding | Mitochondria, Cytosol | [149] | ||
| BAG2 | Regulates the level of Pink1 | Cytosol, Nucleus | [157] | ||
| mtHsp70 (HspA9 GRP75/Mortalin) | Regulates Pink1-Parkin dependent homeostasis of mitochondrial proteins | Mitochondria, Nucleus | [149] | ||
| LRPPRC | Regulates Pink1-Parkin dependent homeostasis of mitochondrial proteins | ER, Nucleus, Mitochondria, Cytoskeleton | [149] | ||
| 5. | LRRK2 (PARK8) | Hsp90 (HspC) | Stabilization of LRRK2 | Cytosol, ER, Nucleus, Lysosome | [177] |
| Cdc37 | Co-chaperone of Hsp90; stabilizes LRRK2 | Cytosol | [177] | ||
| CHIP | CHIP mediated proteasomal degradation of LRRK2 | Cytosol, Nucleus | [178] | ||
| Hsp70 | Proteasomal degradation of LRRK2 | Mitochondria, Cytosol, ER, Nucleus | [177] | ||
| Hsc70 | Proteasomal degradation of LRRK2 | Plasma membrane, Cytosol, Nucleus, and Lysosome | [177] | ||
| 14-3-3 | Interacts with LRRK2 and regulates its kinase activity | Mitochondria, Cytosol, Plasma membrane, Nucleus | [183] | ||
| BAG2 | Interacts with LRRK2 | Cytosol, Nucleus | [180] | ||
| 6. |
DnaJC6/Auxilin (PARK9) | Hsc70 | Clathrin-mediated endocytosis | Plasma membrane, Cytosol, Nucleus, and Lysosome | [183] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, N.; Raveendran, A.; Nagotu, S. Chaperones and Proteostasis: Role in Parkinson’s Disease. Diseases 2020, 8, 24. https://doi.org/10.3390/diseases8020024
Joshi N, Raveendran A, Nagotu S. Chaperones and Proteostasis: Role in Parkinson’s Disease. Diseases. 2020; 8(2):24. https://doi.org/10.3390/diseases8020024
Chicago/Turabian StyleJoshi, Neha, Atchaya Raveendran, and Shirisha Nagotu. 2020. "Chaperones and Proteostasis: Role in Parkinson’s Disease" Diseases 8, no. 2: 24. https://doi.org/10.3390/diseases8020024
APA StyleJoshi, N., Raveendran, A., & Nagotu, S. (2020). Chaperones and Proteostasis: Role in Parkinson’s Disease. Diseases, 8(2), 24. https://doi.org/10.3390/diseases8020024