Robust Metabolite Quantification from J-Compensated 2D 1H-13C-HSQC Experiments




Abstract
:
1. Introduction
2. Results
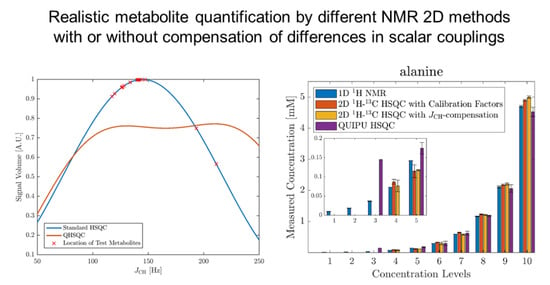
2.1. Efficiency of Scalar Coupling Averaging
2.2. Accuracy and Precision
2.3. Analysis of Matrix Effects
3. Discussion
4. Materials and Methods
4.1. Samples
4.2. Sample Preparation
4.3. NMR Spectroscopy
4.4. Data Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BIRD | Bilinear Rotation Decoupling |
| NMR | Nuclear Magnetic Resonance |
| HSQC | Heteronuclear Single Quantum Coherence |
| LOD | Limit of Detection |
| LLOQ | Lower Limit of Quantification |
| Q-HSQC | Quantitative HSQC |
| QUIPU-HSQC | Quantitative, Perfected and Pure Shifted HSQC |
References
- Lindon, J.C.; Nicholson, J.K.; Holmes, E. The Handbook of Metabonomics and Metabolomics; Elsevier Science & Technology: Oxford, UK, 2007. [Google Scholar]
- Wishart, D.S. Computational Approaches to Metabolomics. Methods Mol. Biol. 2010, 593, 283–313. [Google Scholar]
- Bollard, M.E.; Stanley, E.G.; Lindon, J.C.; Nicholson, J.K.; Holmes, E. NMR-based metabonomic approaches for evaluating physiological influences on biofluid composition. NMR Biomed. 2005, 18, 143–162. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Liebeke, M.; Astle, W.; De Iorio, M.; Bundy, J.G.; Ebbels, T.M.D. Bayesian deconvolution and quantification of metabolites in complex 1D NMR spectra using BATMAN. Nat. Protoc. 2014, 9, 1416–1427. [Google Scholar] [CrossRef]
- Ravanbakhsh, S.; liu, P.; Bjordahl, T.C.; Mandal, R.; Grant, J.R.; Wilson, M.; Eisner, R.; Sinelnikov, I.; Hu, X.; Luchinat, C.; et al. Accurate, Fully-Automated NMR Spectral Profiling for Metabolomics. PLoS ONE 2015, 10, e0124219. [Google Scholar] [CrossRef] [Green Version]
- Hughes, T.S.; Wilson, H.D.; de Vera, I.M.S.; Kojetin, D.J. Deconvolution of Complex 1D NMR Spectra Using Objective Model Selection. PLoS ONE 2015, 10, e0134474. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.; Foxall, P.; Spraul, M.; Farrant, R.; Nicholson, J.; Lindon, J. 750 MHz 1H NMR spectroscopy characterisation of the complex metabolic pattern of urine from patients with inborn errors of metabolism: 2-hydroxyglutaric aciduria and maple syrup urine disease. J. Pharm. Biomed. Anal. 1997, 15, 1647–1659. [Google Scholar] [CrossRef]
- Gronwald, W.; Klein, M.S.; Kaspar, H.; Fagerer, S.R.; Nürnberger, N.; Dettmer, K.; Bertsch, T.; Oefner, P.J. Urinary Metabolite Quantification Employing 2D NMR Spectroscopy. Anal. Chem. 2008, 80, 9288–9297. [Google Scholar] [CrossRef] [PubMed]
- Lewis, I.A.; Schommer, S.C.; Hodis, B.; Robb, K.A.; Tonelli, M.; Westler, W.M.; Sussman, M.R.; Markley, J.L. Method for Determining Molar Concentrations of Metabolites in Complex Solutions from Two-Dimensional 1H-13C NMR Spectra. Anal. Chem. 2007, 79, 9385–9390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, K.; Westler, W.M.; Markley, J.L. Simultaneous Quantification and Identification of Individual Chemicals in Metabolite Mixtures by Two-Dimensional Extrapolated Time-Zero1H-13C HSQC (H0). J. Am. Chem. Soc. 2011, 133, 1662–1665. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, S.; Toikka, M.M.; Karhunen, P.T.; Kilpeläinen, I.A. Quantitative 2D HSQC (Q-HSQC) via Suppression of J-Dependence of Polarization Transfer in NMR Spectroscopy: Application to Wood Lignin. J. Am. Chem. Soc. 2003, 125, 4362–4367. [Google Scholar] [CrossRef]
- Koskela, H.; Kilpeläinen, I.; Heikkinen, S. Some aspects of quantitative 2D NMR. J. Magn. Reson. 2005, 174, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Koskela, H.; Heikkilä, O.; Kilpeläinen, I.; Heikkinen, S. Quantitative two-dimensional HSQC experiment for high magnetic field NMR spectrometers. J. Magn. Reson. 2010, 202, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.J.; Loening, N.M. QQ-HSQC: A quick, quantitative heteronuclear correlation experiment for NMR spectroscopy. Magn. Reson. Chem. 2007, 45, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Mauve, C.; Khlifi, S.; Gilard, F.; Mouille, G.; Farjon, J. Sensitive, highly resolved, and quantitative 1H-13C NMR data in one go for tracking metabolites in vegetal extracts. Chem. Commun. 2016, 52, 6142–6145. [Google Scholar] [CrossRef]
- Farjon, J.; Milande, C.; Martineau, E.; Akoka, S.; Giraudeau, P. The FAQUIRE Approach: FAst, QUantitative, hIghly Resolved and sEnsitivity Enhanced 1H, 13C Data. Anal. Chem. 2018, 90, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Zangger, K. Pure shift NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2015, 86–87, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Friedman, M. The Use of Ranks to Avoid the Assumption of Normality Implicit in the Analysis of Variance. J. Am. Stat. Assoc. 1937, 32, 675–701. [Google Scholar] [CrossRef]
- Nemenyi, P. Distribution-free Multiple Comparisons; Princeton University: Princeton, NJ, USA, 1963. [Google Scholar]
- Garwood, M.; DelaBarre, L. The Return of the Frequency Sweep: Designing Adiabatic Pulses for Contemporary NMR. J. Magn. Reson. 2001, 153, 155–177. [Google Scholar] [CrossRef]
- Mauhart, J.; Glanzer, S.; Sakhaii, P.; Bermel, W.; Zangger, K. Faster and cleaner real-time pure shift NMR experiments. J. Magn. Reson. 2015, 259, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Bhar, B.N. Nuclear spin-lattice relaxation and molecular association in acetic acid. Il Nuovo Cimento B (1965–1970) 1965, 40, 416–423. [Google Scholar] [CrossRef]
- Hrynkiewicz, K.; Krynicki, K.; Waluga, T. Proton spin-lattice relaxation in acetic acid. Arch. Sci. 1961, 14. [Google Scholar]
- Klein, M.S.; Oefner, P.J.; Gronwald, W. MetaboQuant: A tool combining individual peak calibration and outlier detection for accurate metabolite quantification in 1D 1H and 1H-13C HSQC NMR spectra. BioTech 2013, 54, 251–256. [Google Scholar] [CrossRef]
- Food and Drug Administration. Bioanalytical Method Validation; Food and Drug Administration: Washington, DC, USA, 2018. [Google Scholar]
- Von Schlippenbach, T.; Oefner, P.; Gronwald, W. Systematic Evaluation of Non-Uniform Sampling Parameters in the Targeted Analysis of Urine Metabolites by 1H,1H 2D NMR Spectroscopy. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Macura, S. Accelerated Multidimensional NMR Data Acquisition by Varying the Pulse Sequence Repetition Time. J. Am. Chem. Soc. 2009, 131, 9606–9607. [Google Scholar] [CrossRef]
- Foroozandeh, M.; Jeannerat, D. Reconstruction of full high resolution HSQC using signal split in aliased spectra. Magn. Reson. Chem. 2015, 53, 894–900. [Google Scholar] [CrossRef]




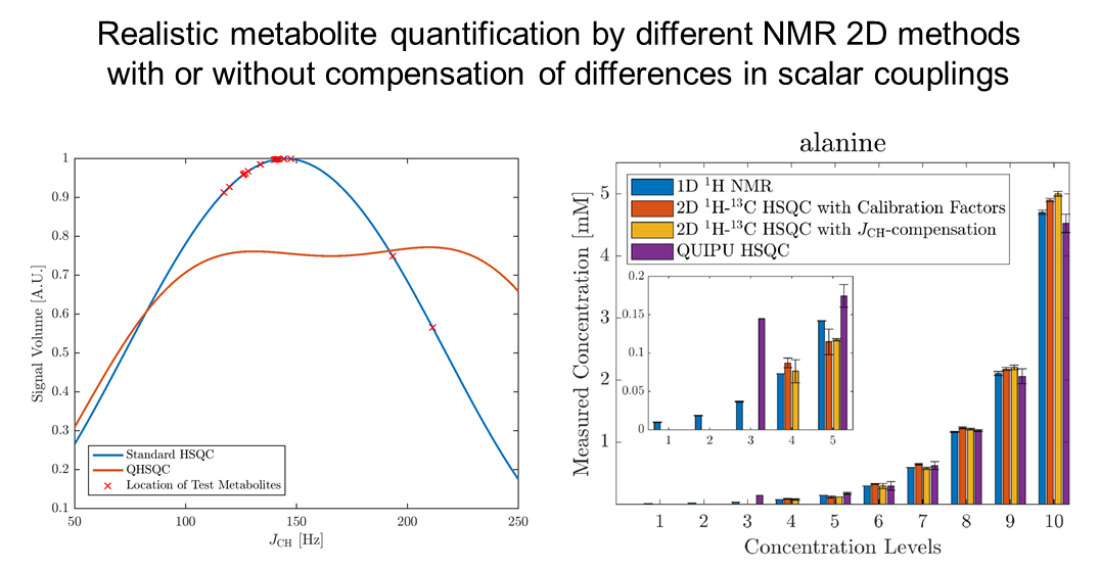{kind=link}
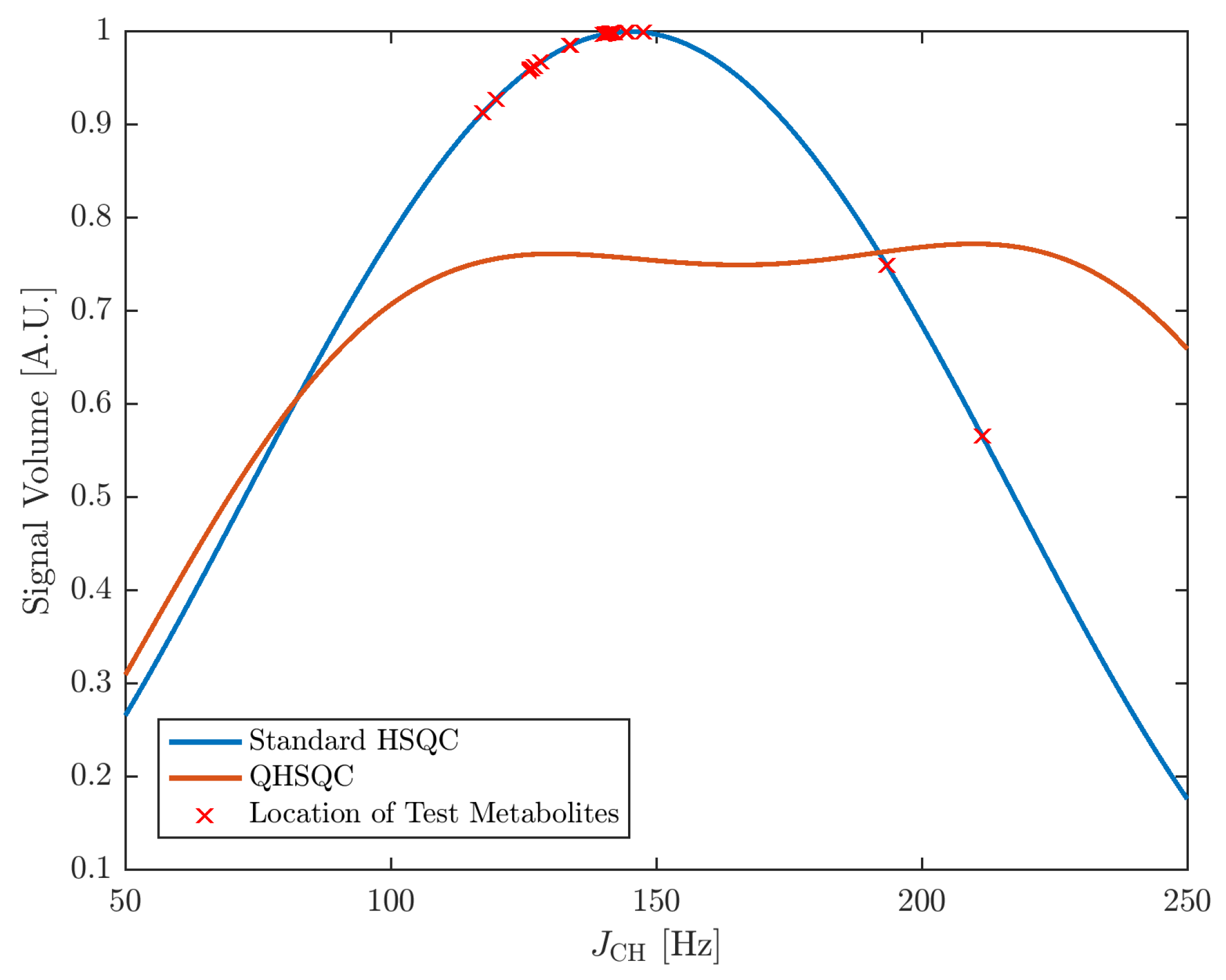{kind=link}
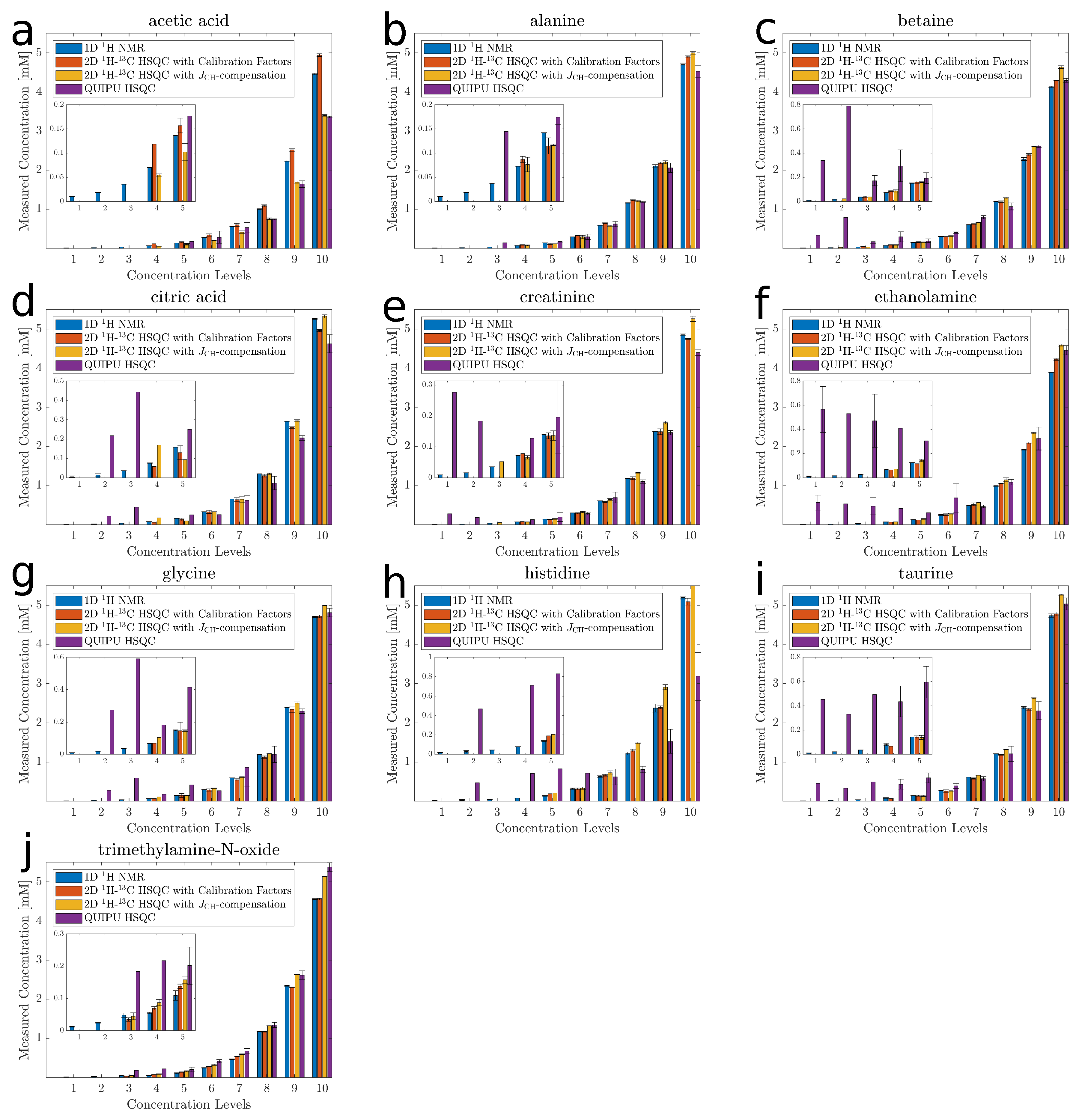{kind=link}
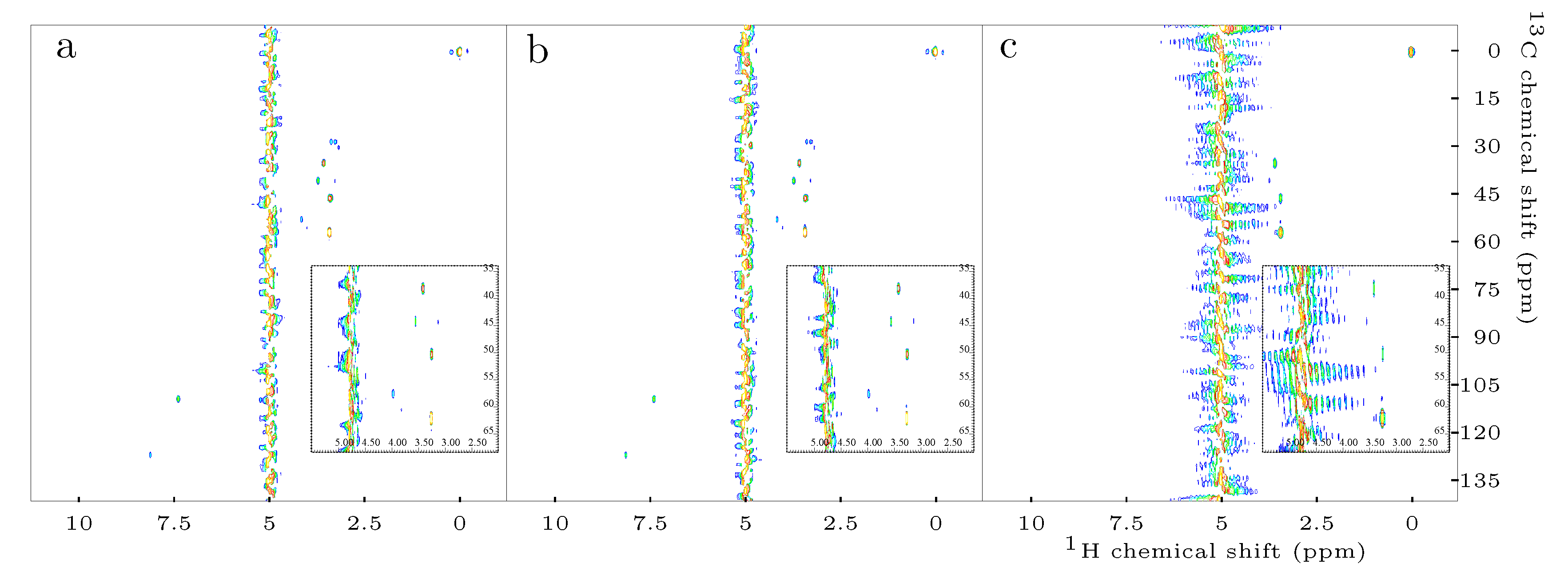{kind=link}
| Coupling Constant [Hz] | Predicted Ratio | Without Averaging | Q-HSQC | QUIPU-HSQC | |
|---|---|---|---|---|---|
| C2 H2 | 140.8 | 1 | 1.00 ± 0.4% | 1.00 ± 1% | 1.00 ± 4% |
| C3 H3A/B | 128.3/117.4 | 0.94 | 0.97 ± 2% | 0.87 ± 2% | 0.83 ± 2% |
| C5 H5 | 193.4 | 0.75 | 0.82 ± 2% | 1.01 ± 0.8% | 0.54 ± 19% |
| C7 H7 | 211.4 | 0.57 | 0.61 ± 5% | 0.93 ± 3.6% | 0.48 ± 6% |
| Standard HSQC | Q-HSQC | QUIPU-HSQC | |
|---|---|---|---|
| mean LOD [M] | 78 | 80 | 71 |
| mean LLOQ [M] | 187 | 148 | 640 |
| Requirement of calibration values | Yes | No | No |
| Sensitivity towards H-C coupling | Yes | No | No |
| Suppression of H-H couplings | No | No | Yes |
| Sensitivity towards residual water signal | Normal | Normal | Increased |
| Measurement time [min] | 55 | 108 | 110 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weitzel, A.; Samol, C.; Oefner, P.J.; Gronwald, W. Robust Metabolite Quantification from J-Compensated 2D 1H-13C-HSQC Experiments. Metabolites 2020, 10, 449. https://doi.org/10.3390/metabo10110449
Weitzel A, Samol C, Oefner PJ, Gronwald W. Robust Metabolite Quantification from J-Compensated 2D 1H-13C-HSQC Experiments. Metabolites. 2020; 10(11):449. https://doi.org/10.3390/metabo10110449
Chicago/Turabian StyleWeitzel, Alexander, Claudia Samol, Peter J. Oefner, and Wolfram Gronwald. 2020. "Robust Metabolite Quantification from J-Compensated 2D 1H-13C-HSQC Experiments" Metabolites 10, no. 11: 449. https://doi.org/10.3390/metabo10110449
APA StyleWeitzel, A., Samol, C., Oefner, P. J., & Gronwald, W. (2020). Robust Metabolite Quantification from J-Compensated 2D 1H-13C-HSQC Experiments. Metabolites, 10(11), 449. https://doi.org/10.3390/metabo10110449