NLRP3 Inflammasome Biomarker—Could Be the New Tool for Improved Cardiometabolic Syndrome Outcome

 ,
,  ,
,  ,
,  and
and 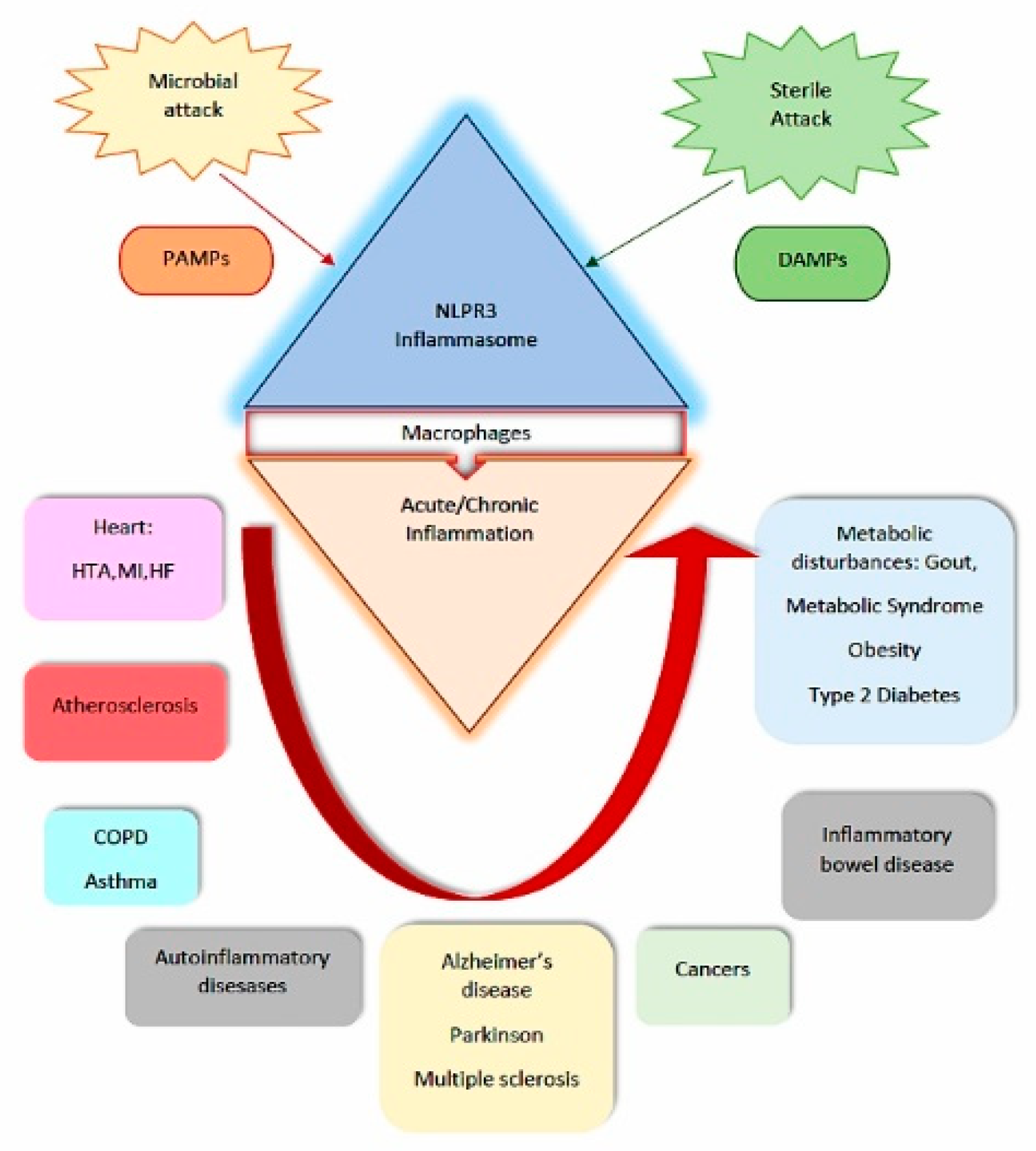{kind=link}
{kind=link}
Abstract
:1. Introduction
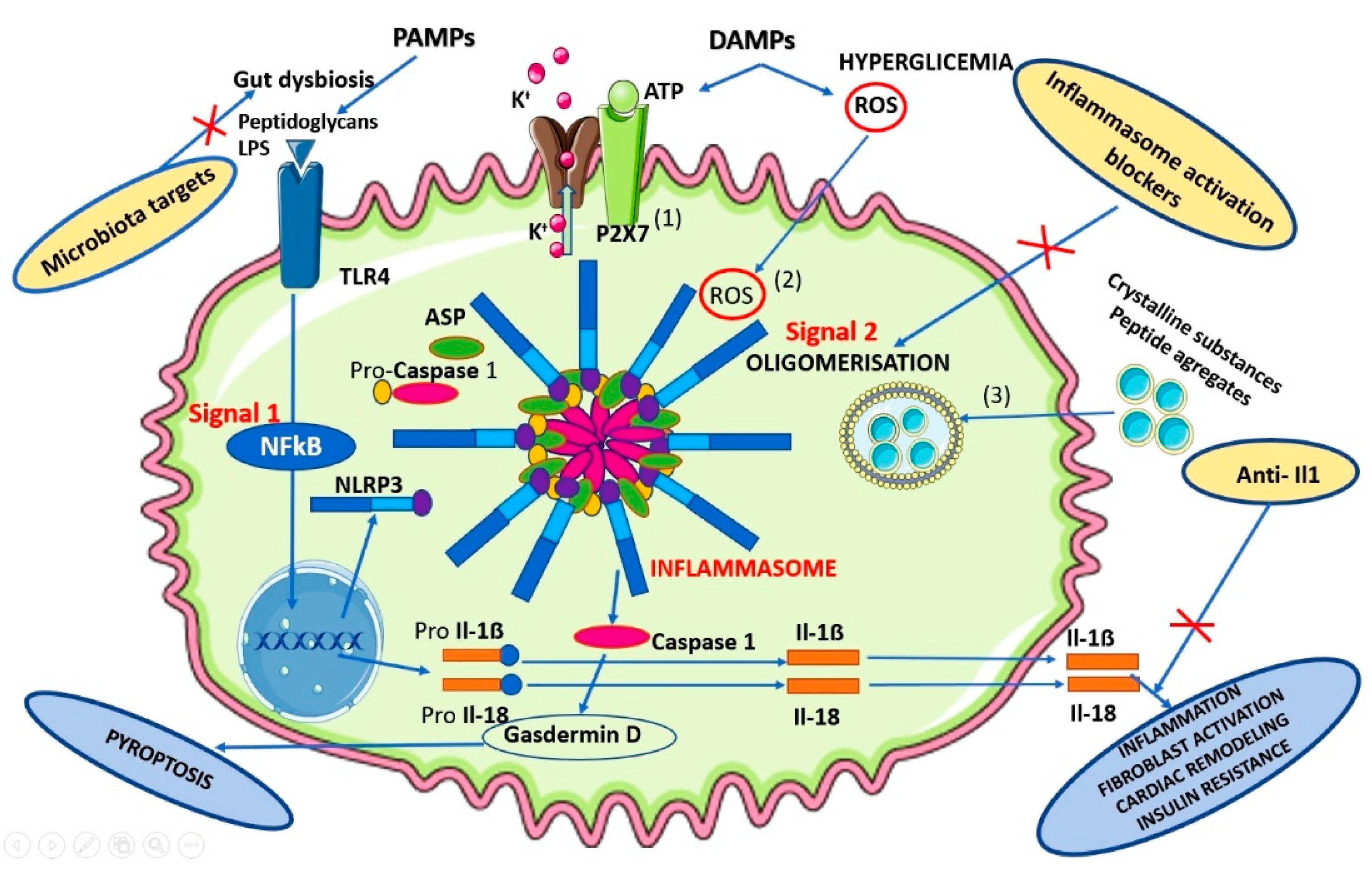
2. Evidence-Based Data on Inflammasome Biomarkers Involvement in CMS Pathogeny
3. The New Paradigm of CMS Therapy
3.1. Targeted Therapies against Specific NLRP3 Inflammasome-Activated Components
3.2. Microbiota Targets
3.3. Diet Targets
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Welty, F.K.; Alfaddagh, A.; Elajami, T.K. Targeting inflammation in metabolic syndrome. Transl. Res. 2016, 167, 257–280. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nardo, D.; Latz, E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011, 32, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, S.P.; Cassel, S.L. Inflammasome-mediated autoinflammatory disorders. Postgrad. Med. 2010, 122, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Rajamäki, K.; Lappalainen, J.; Oörni, K.; Välimäki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 23, e11765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netea, M.G.; Joosten, L.A. Inflammasome Inhibition: Putting Out the Fire. Cell Metab. 2015, 21, P513514. [Google Scholar] [CrossRef] [Green Version]
- Oda, E. Historical perspectives of the metabolic syndrome. Clin. Derm. 2018, 36, 3–8. [Google Scholar] [CrossRef] [PubMed]
- McCracken, E.; Monaghan, M.; Sreenivasan, S. Pathophysiology of the metabolic syndrome. Clin. Derm. 2018, 36, 14–20. [Google Scholar] [CrossRef]
- Kaptoge, S.; Di Angelantonio, E.; Lowe, G.; Pepys, M.B.; Thompson, S.G.; Collins, R.; Danesh, J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. Lancet 2010, 375, 132–140. [Google Scholar]
- Sattar, N.; Murray, H.M.; McConnachie, A.; Blauw, G.J.; Bollen, E.L.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; et al. C-reactive protein and prediction of coronary heart disease and global vascular events in the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER). Circulation 2007, 115, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wensley, F.; Gao, P.; Burgess, S.; Kaptoge, S.; Di Angelantonio, E.; Shah, T.; Engert, J.C.; Clarke, R.; Davey-Smith, G.; Nordestgaard, B.G.; et al. Association between C reactive protein and coronary heart disease: Mendelian randomisation analysis based on individual participant data. BMJ 2011, 342, d548. [Google Scholar] [PubMed] [Green Version]
- Kanhai, D.A.; Kranendonk, M.E.; Uiterwaal, C.S.; van der Graaf, Y.; Kappelle, L.J.; Visseren, F.L. Adiponectin and incident coronary heart disease and stroke. A systematic review and meta-analysis of prospective studies. Obes. Rev. 2013, 14, 555–567. [Google Scholar] [CrossRef]
- Sook Lee, E.; Park, S.S.; Kim, E.; Sook Yoon, Y.; Ahn, H.Y.; Park, C.Y.; Ho Yun, Y.; Woo Oh, S. Association between adiponectin levels and coronary heart disease and mortality: A systematic review and meta-analysis. Int. J. Epidemiol. 2013, 42, 1029–1039. [Google Scholar] [CrossRef] [Green Version]
- Borges, M.C.; Lawlor, D.A.; de Oliveira, C.; White, J.; Horta, B.L.; Barros, A.J. Role of Adiponectin in coronary heart disease risk: A Mendelian randomization study. Circ. Res. 2016, 22, 119491–119499. [Google Scholar] [CrossRef] [Green Version]
- Roujeau, C.; Jockers, R.; Dam, J. New pharmacological perspectives for the leptin receptor in the treatment of obesity. Front. Endocrinol. 2014, 5, 167. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Xu, C.H.; Xu, Y.N.; Wang, Y.L.; Wang, M. Association of leptin levels with pathogenetic risk of coronary heart disease and stroke: A meta-analysis. Arq. Bras. Endocrinol. Metab. 2014, 58, 817–823. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Guo, W.; Li, J.; Cao, S.; Zhang, J.; Pan, J.; Wang, Z.; Wen, P.; Shi, X.; Zhang, S. Leptin concentration and risk of coronary heart disease and stroke: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0166360. [Google Scholar] [CrossRef] [Green Version]
- Chai, S.B.; Sun, F.; Nie, X.L.; Wang, J. Leptin and coronary heart disease: A systematic review and meta-analysis. Atherosclerosis 2014, 233, 3–10. [Google Scholar] [CrossRef]
- Wu, L.; Sun, D. Leptin receptor gene polymorphism and the risk of cardiovascular disease: A systemic review and meta-analysis. Int. J. Environ. Res. Public Health 2017, 14, 375. [Google Scholar] [CrossRef] [Green Version]
- Janket, S.J.; Javaheri, H.; Ackerson, L.K.; Ayilavarapu, S.; Meurman, J.H. Oral infections, metabolic inflammation, genetics, and cardiometabolic diseases. J. Dent. Res. 2015, 94 (Suppl. 9), 119S–127S. [Google Scholar] [CrossRef]
- Bansal, A.; Henao-Mejia, J.; Simmons, R.A. Immune system: An emerging player in mediating effects of endocrine disruptors on metabolic health. Endocrinology 2018, 159, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Chowdhury, N.; Govindarajan, G.; Karuparthi, P.R.; Habibi, J.; Sowers, J.R. Myocardial myocyte remodeling and fibrosis in the cardiometabolic syndrome. J. Cardiometab. Syndr. 2006, 1, 326–333. [Google Scholar] [CrossRef]
- Guo, R.; Rogers, O.; Nair, S. Targeting apelinergic system in cardiometabolic disease. Curr. Drug Targets 2017, 18, 1785–1791. [Google Scholar] [CrossRef]
- Newgard, C.B. Metabolomics and metabolic diseases: Where do we stand? Cell Metab. 2017, 25, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Njock, M.S.; Fish, J.E. Endothelial miRNAs as cellular messengers in cardiometabolic diseases. Trends Endocrinol. Metab. 2017, 28, 237–246. [Google Scholar] [CrossRef]
- Rotllan, N.; Price, N.; Pati, P.; Goedeke, L.; Fernández-Hernando, C. microRNAs in lipoprotein metabolism and cardiometabolic disorders. Atherosclerosis 2016, 246, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Assmann, T.S.; Milagro, F.I.; Martínez, J.A. Crosstalk between microRNAs, the putative target genes and the lncRNA network in metabolic diseases. Mol. Med. Rep. 2019, 20, 3543–3554. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.; Ting, J. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.Z.; Zu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Man, S.; Kanneganti, T. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Coll, R.; Robertson, A.; Chae, J.; Higgins., S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 4–14. [Google Scholar] [CrossRef]
- Westermeier, F.; Navarro-Marquez, M.; Lopez-Crisosto, C.; Bravo-Sagua, R.; Quiroga, C.; Bustamante, M.; Verdejo, H.E.; Zalaquett, R.; Ibacache, M.; Parra, V.; et al. Defective insulin signaling and mitochondrial dynamics in diabetic cardiomyopathy. Biochim. Biophys. Acta 2015, 1853, 1113–1118. [Google Scholar] [CrossRef] [Green Version]
- Kuethe, F.; Sigusch, H.H.; Bornstein, S.R.; Hilbig, K.; Kamvissi, V.; Figulla, H.R. Apoptosis in patients with dilated cardiomyopathy and diabetes: A feature of diabetic cardiomyopathy? Horm. Metab. Res. 2007, 39, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ding, W.Y.; Zhao, J.; Wang, Z.H.; Zhong, M.; Zhang, W.; Chen, Y.; Zhang, Y.; Li, L.; Tang, M. Activin receptor-like kinase 7 mediates high glucose-induced H9c2 cardiomyoblast apoptosis through activation of Smad2/3. Int. J. Biochem. Cell Biol. 2013, 45, 2027–2035. [Google Scholar] [CrossRef]
- Luo, B.; Huang, F.; Liu, Y.; Liang, Y.; Wei, Z.; Ke, H.; Zeng, Z.; Huang, W.; He, Y. NLRP3 Inflammasome as a Molecular Marker in Diabetic Cardiomyopathy. Front. Physiol. 2017, 8, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunwald, E. Biomarkers in heart failure. N. Engl. J. Med. 2008, 358, 2148–2159. [Google Scholar] [CrossRef] [Green Version]
- Aiyar, N.; Disa, J.; Ao, Z.; Ju, H.; Nerurkar, S.; Willette, R.N.; Macphee, C.H.; Johns, D.G.; Douglas, S.A. Lysophosphatidylcholine induces inflammatory activation of human coronary artery smooth muscle cells. Mol. Cell Biochem. 2007, 295, 113–120. [Google Scholar] [CrossRef]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Investig. 2002, 110, 851–860. [Google Scholar] [CrossRef]
- Scola, L.; Giarratana, R.M.; Torre, S.; Argano, V.; Lio, D.; Balistreri, C.R. On the Road to Accurate Biomarkers for Cardiometabolic Diseases by Integrating Precision and Gender Medicine Approaches. Int. J. Mol. Sci. 2019, 20, 6015. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Frangogiannis, N.G. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim. Biophys Acta 2013, 1833, 945–953. [Google Scholar] [CrossRef] [Green Version]
- Bracey, N.A.; Gershkovich, B.; Chun, J.; Vilaysane, A.; Meijndert, H.C.; Wright, J.R., Jr.; Fedak, P.W.; Beck, P.L.; Muruve, D.A.; Duff, J.H. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 2014, 11, 19571–19584. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I.; Glass, C.K. Anti-inflammatory therapy in chronic disease: Challenges and opportunities. Science 2013, 336, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Volund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.L.; Nidorf, S.M. Anti-inflammatory therapy with canakinumab for atherosclerotic disease: Lessons from the CANTOS trial. J. Thorac. Dis. 2018, 10, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, Y.G.; Kim, D.J.; Park, S.H.; Jeong, K.-H.; Lee, Y.H.; Lim, S.J.; Lee, S.; Moon, J. Inflammasome-Independent Role of NLRP3 Mediates Mitochondrial Regulation in Renal Injury. Front. Immunol. 2018, 9, 2563. [Google Scholar] [CrossRef] [Green Version]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Mastrocola, R.; Penna, C.; Tullio, F.; Femmino, S.; Nigro, D.; Chiazza, F.; Serpe, L.; Collotta, D.; Alloatti, G.; Cocco, M.; et al. Pharmacological inhibition of NLRP3 inflammasome attenuates myocardial ischemia/reperfusion injury by activation of RISK and mitochondrial pathways. Oxid. Med. Cell. Longev. 2016, 5271251. [Google Scholar] [CrossRef]
- Butts, B.; Gary, R.A.; Dunbar, S.B.; Butler, J. The importance of NLRP3 inflammasome in heart failure. J. Card. Fail. 2015, 21, 586–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, L.P. The NLRP3 inflammasome and diabetic cardiomyopathy: Editorial to: “Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model” by Beibei Luo et al. Cardiovasc. Drugs 2014, 28, 5–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, S.; Dunne, A.; Subramanian, S.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Iulia-Suceveanu, A.; Micu, S.I.; Voinea, C.; Manea, M.E.; Catrinoiu, D.; Mazilu, L.; Stoian, A.P.; Parepa, I.; Stoica, R.A.; Suceveanu, A.-P. Metformin and Its Benefits in Improving Gut Microbiota Disturbances in Diabetes Patients. Metformin 2019. [Google Scholar] [CrossRef] [Green Version]
- Suceveanu, A.I.; Stoian, A.P.; Parepa, I.R.; Voinea, C.; Hainarosie, R.; Manuc, D.; Nitipir, C.; Mazilu, L.; Suceveanu, A.P. Gut Microbiota Patterns in Obese and Type 2 Diabetes (T2D) Patients from Romanian Black Sea Coast. Rev.Chim. 2018, 69, 2260–2267. [Google Scholar] [CrossRef]
- Macia, L.; Tan, J.; Vieira, A.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; McKenzie, C.I.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wang, Y.; Wang, P.; Huang, Y.; Wang, F. Function through the Inhibition of NLRP3 Inflammasome and Autophagy. Cell. Physiol. Biochem. 2018, 49, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Traba, J.; Kwarteng-Siaw, M.; Okoli, T.C.; Li, J.; Huffstutler, R.D.; Bray, A.; Waclawiw, M.A.; Han, K.; Pelletier, M.; Sauve, A.A.; et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J. Clin. Investig. 2015, 125, 4592–4600. [Google Scholar] [CrossRef] [Green Version]
- Dror, E.; Dalmas, E.; Meier, D.; Wueest, S.; Thévenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Hajmrle, C.; Smith, N.; Spigelman, A.F.; Dai, X.; Senior, L.; Bautista, A.; Ferdaoussi, M.; MacDonald, P.E. Interleukin-1 signaling contributes to acute islet compensation. JCI Insight 2016, 1, e86055. [Google Scholar] [CrossRef]
- Donath, M.Y.; Meier, D.T.; Böni-Schnetzler, M. Inflammation in the Pathophysiology and Therapy of Cardiometabolic Disease. Endocr. Rev. 2019, 40, 1080–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenorio-Jiménez, C.; Martínez-Ramírez, M.J.; Gil, Á.; Gómez-Llorente, C. Effects of Probiotics on Metabolic Syndrome: A Systematic Review of Randomized Clinical Trials. Nutrients 2020, 12, 124. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suceveanu, A.-I.; Mazilu, L.; Katsiki, N.; Parepa, I.; Voinea, F.; Pantea-Stoian, A.; Rizzo, M.; Botea, F.; Herlea, V.; Serban, D.; et al. NLRP3 Inflammasome Biomarker—Could Be the New Tool for Improved Cardiometabolic Syndrome Outcome. Metabolites 2020, 10, 448. https://doi.org/10.3390/metabo10110448
Suceveanu A-I, Mazilu L, Katsiki N, Parepa I, Voinea F, Pantea-Stoian A, Rizzo M, Botea F, Herlea V, Serban D, et al. NLRP3 Inflammasome Biomarker—Could Be the New Tool for Improved Cardiometabolic Syndrome Outcome. Metabolites. 2020; 10(11):448. https://doi.org/10.3390/metabo10110448
Chicago/Turabian StyleSuceveanu, Andra-Iulia, Laura Mazilu, Niki Katsiki, Irinel Parepa, Felix Voinea, Anca Pantea-Stoian, Manfredi Rizzo, Florin Botea, Vlad Herlea, Dragos Serban, and et al. 2020. "NLRP3 Inflammasome Biomarker—Could Be the New Tool for Improved Cardiometabolic Syndrome Outcome" Metabolites 10, no. 11: 448. https://doi.org/10.3390/metabo10110448
APA StyleSuceveanu, A.-I., Mazilu, L., Katsiki, N., Parepa, I., Voinea, F., Pantea-Stoian, A., Rizzo, M., Botea, F., Herlea, V., Serban, D., & Suceveanu, A.-P. (2020). NLRP3 Inflammasome Biomarker—Could Be the New Tool for Improved Cardiometabolic Syndrome Outcome. Metabolites, 10(11), 448. https://doi.org/10.3390/metabo10110448