Absolute Quantification of the Central Carbon Metabolome in Eight Commonly Applied Prokaryotic and Eukaryotic Model Systems

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
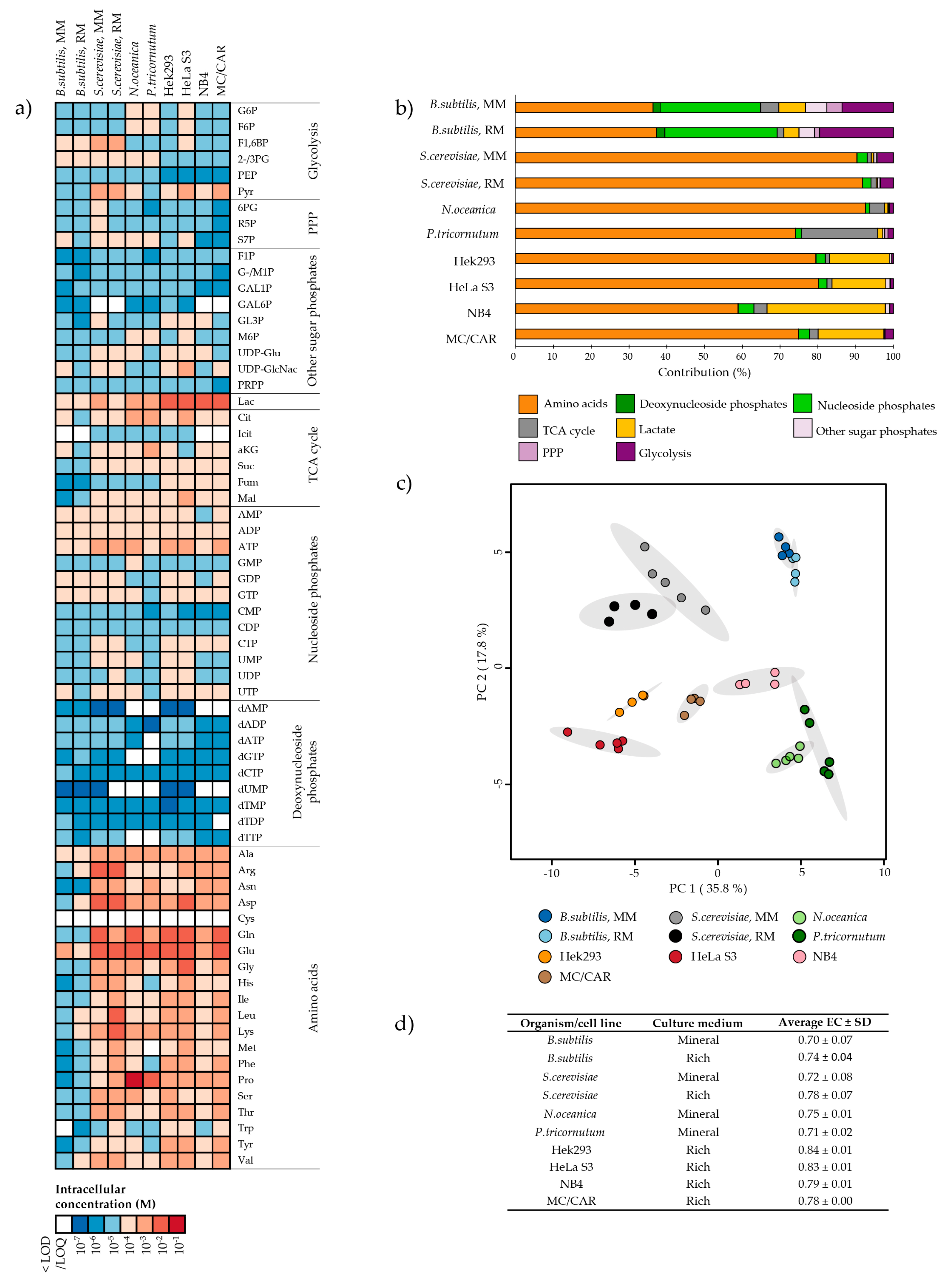
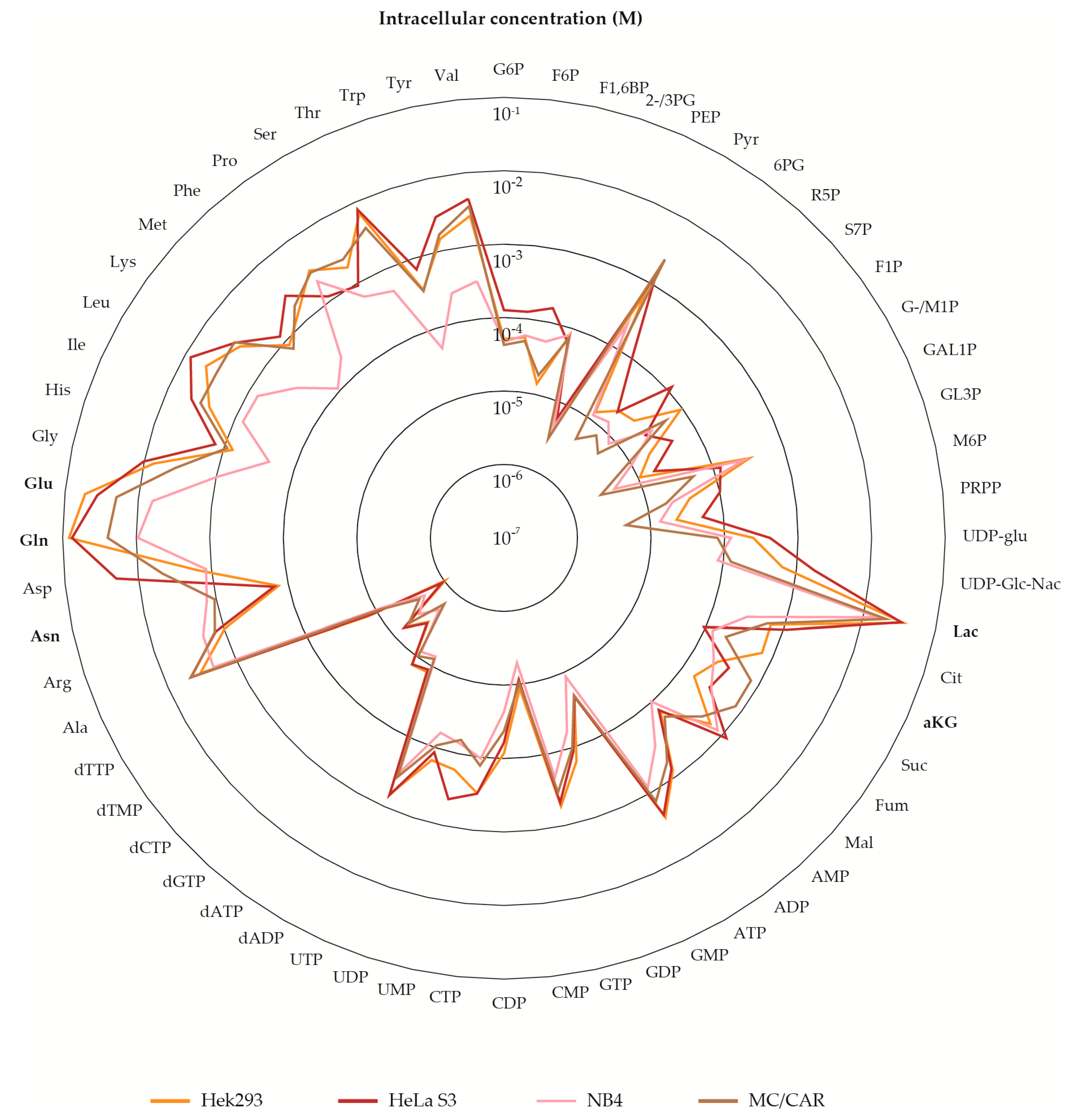
2.1. Metabolite Pools of Central Carbon Metabolism Vary Over at Least Three Orders of Magnitude
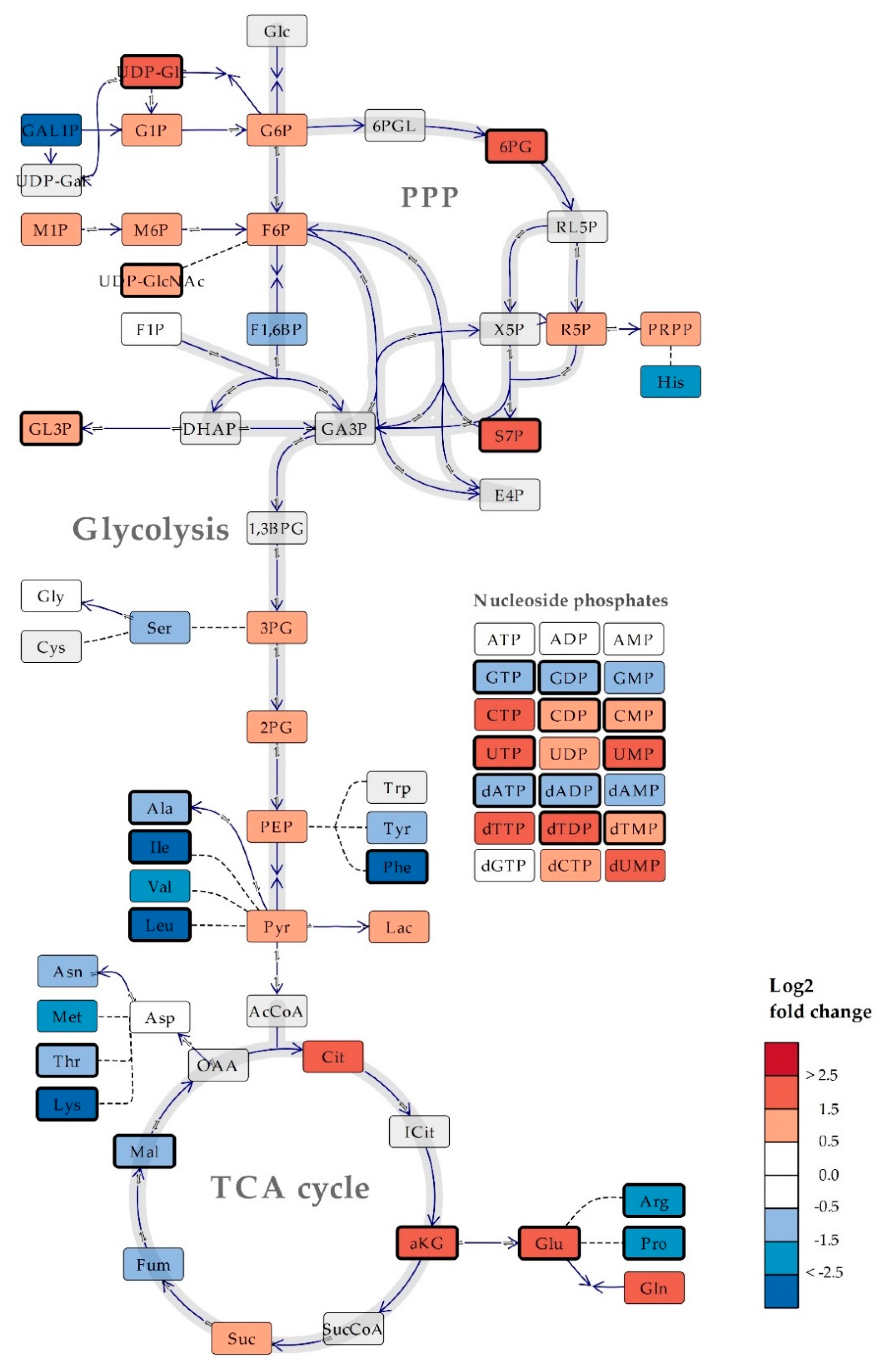
2.2. Intracellular Metabolite Levels in Bacillus subtilis are Highly Dependent on Culture Medium Composition
2.3. Several Metabolic Features are Conserved Across Saccharomyces cerevisiae and Bacillus subtilis Cultured in Mineral Compared to Rich Media
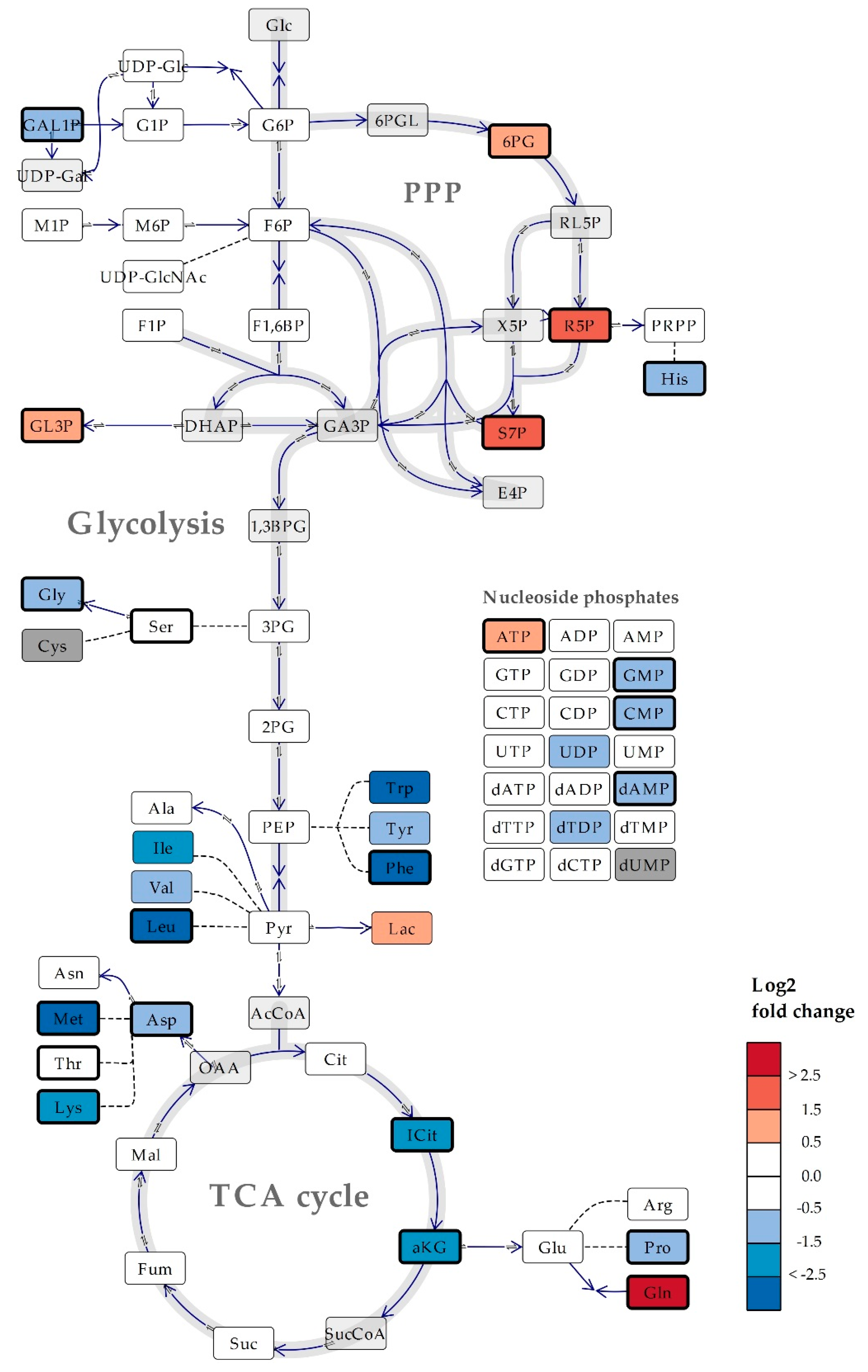
2.4. The Microalgae Nannochloropsis oceanica and Phaeodactylum tricornutum are Both High in Proline
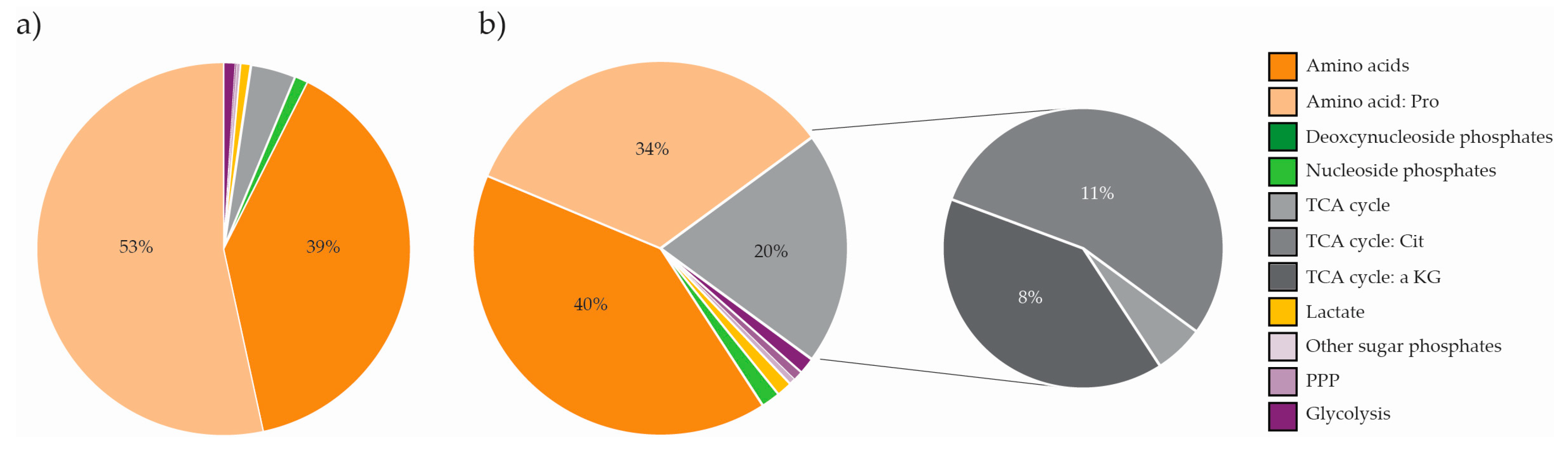
2.5. The Metabolite Profile of Human Cell Lines Varies with Tissue of Origin
2.6. Concluding Remarks
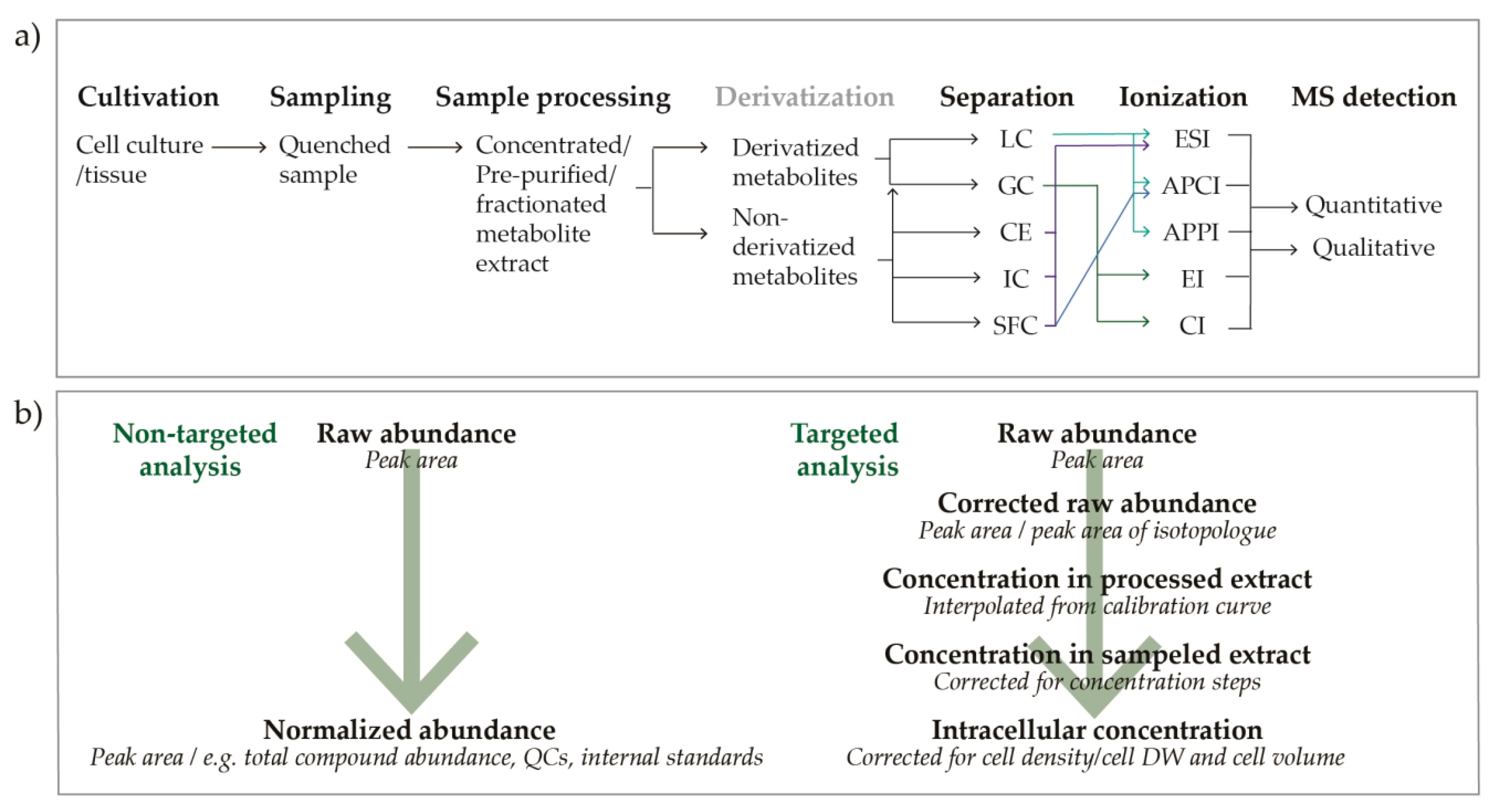
3. Materials and Methods
3.1. Cultivation
3.1.1. Cell Lines
3.1.2. Bacillus subtilis
3.1.3. Saccharomyces cerevisiae
3.1.4. Nannocloropsis oceanica and Phaeodactylum tricornutum
3.2. Sampling
3.2.1. Suspension Cell Lines
3.2.2. Adherent Cell Lines
3.2.3. Microorganisms
3.3. Preparation of Metabolite Extracts
3.4. Targeted Mass Spectrometric Metabolite Profiling
3.4.1. CapIC-MS/MS Analysis of Phosphorylated Metabolites and TCA Cycle Intermediates
3.4.2. LC-MS/MS Analysis of Organic Acids
3.4.3. LC-MS/MS Analysis of Amino Acids
3.5. Data Analysis
3.5.1. Data Processing
3.5.2. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Becker, J.; Wittmann, C. From systems biology to metabolically engineered cells — An omics perspective on the development of industrial microbes. Curr. Opin. Microbiol. 2018, 45, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, H.; Yoshihara, K.; Hara, K.Y.; Hasunuma, T.; Ogino, C.; Kondo, A. Metabolome analysis-based design and engineering of a metabolic pathway in Corynebacterium glutamicum to match rates of simultaneous utilization of D-glucose and L-arabinose. Microb. Cell Fact. 2018, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.H.; Sun, H.; Yan, G.L.; Wang, P.; Wang, X.J. Mass spectrometry-based metabolomics: Applications to biomarker and metabolic pathway research. Biomed. Chromatogr. 2016, 30, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Nakahigashi, K.; Baba, T.; Robert, M.; Soga, T.; Kanai, A.; Hirasawa, T.; Naba, M.; Hirai, K.; Hoque, A.; et al. Multiple high-throughput analyses monitor the response of E-coli to perturbations. Science 2007, 316, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chu, J.; Zhuang, Y.P.; van Gulik, W.; Noorman, H. A dynamic model-based preparation of uniformly-C-13-labeled internal standards facilitates quantitative metabolomics analysis of Penicillium chrysogenum. J. Biotechnol. 2019, 299, 21–31. [Google Scholar] [CrossRef]
- Misra, B.B.; Mohapatra, S. Tools and resources for metabolomics research community: A 2017–2018 update. Electrophoresis 2019, 40, 227–246. [Google Scholar] [CrossRef]
- Salek, R.M.; Arita, M.; Dayalan, S.; Ebbels, T.; Jones, A.R.; Neumann, S.; Rocca-Serra, P.; Viant, M.R.; Vizcaino, J.A. Embedding standards in metabolomics: The Metabolomics Society data standards task group. Metabolomics 2015, 11, 782–783. [Google Scholar] [CrossRef] [Green Version]
- Vuckovic, D. Improving metabolome coverage and data quality: Advancing metabolomics and lipidomics for biomarker discovery. Chem. Commun. 2018, 54, 6728–6749. [Google Scholar] [CrossRef]
- Lu, W.; Su, X.; Klein, M.S.; Lewis, I.A.; Fiehn, O.; Rabinowitz, J.D. Metabolite Measurement: Pitfalls to Avoid and Practices to Follow. Annu. Rev. Biochem. 2017, 86, 277–304. [Google Scholar] [CrossRef]
- Edison, A.S.; Hall, R.D.; Junot, C.; Karp, P.D.; Kurland, I.J.; Mistrik, R.; Reed, L.K.; Saito, K.; Salek, R.M.; Steinbeck, C.; et al. The Time Is Right to Focus on Model Organism Metabolomes. Metabolites 2016, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Nishino, S.; Okahashi, N.; Matsuda, F.; Shimizu, H. Absolute quantitation of glycolytic intermediates reveals thermodynamic shifts in Saccharomyces cerevisiae strains lacking PFK1 or ZWF1 genes. J. Biosci. Bioeng. 2015, 120, 280–286. [Google Scholar] [CrossRef] [Green Version]
- Ji, F.F.; Shen, Y.; Tang, L.H.; Cai, Z.W. Determination of intracellular metabolites concentrations in Escherichia coli under nutrition stress using liquid chromatography-tandem mass spectrometry. Talanta 2018, 189, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Lee, Y.J.; Lee, S.Y.; Oh, H.B.; Lee, J. Determination of the Intracellular Concentrations of Metabolites in Escherichia coli Collected during the Exponential and Stationary Growth Phases using Liquid Chromatography-Mass Spectrometry. Bull. Korean Chem. Soc. 2011, 32, 524–530. [Google Scholar] [CrossRef] [Green Version]
- Bennett, B.D.; Kimball, E.H.; Gao, M.; Osterhout, R.; Van Dien, S.J.; Rabinowitz, J.D. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5, 593–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hameri, T.; Fengos, G.; Ataman, M.; Miskovic, L.; Hatzimanikatis, V. Kinetic models of metabolism that consider alternative steady-state solutions of intracellular fluxes and concentrations. Metab. Eng. 2019, 52, 29–41. [Google Scholar] [CrossRef]
- Willemsen, A.M.; Hendrickx, D.M.; Hoefsloot, H.C.J.; Hendriks, M.; Wahl, S.A.; Teusink, B.; Smilde, A.K.; van Kampen, A.H.C. MetDFBA: Incorporating time-resolved metabolomics measurements into dynamic flux balance analysis. Mol. Biosyst. 2015, 11, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Bordbar, A.; Yurkovich, J.T.; Paglia, G.; Rolfsson, O.; Sigurjonsson, O.E.; Palsson, B.O. Elucidating dynamic metabolic physiology through network integration of quantitative time-course metabolomics. Sci. Rep. 2017, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Martinez, V.S.; Kromer, J.O. Quantification of Microbial Phenotypes. Metabolites 2016, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Guo, A.C.; Jewison, T.; Wilson, M.; Liu, Y.F.; Knox, C.; Djoumbou, Y.; Lo, P.; Mandal, R.; Krishnamurthy, R.; Wishart, D.S. ECMDB: The E-coli Metabolome Database. Nucleic Acids Res. 2013, 41, D625–D630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Gaona, M.; Marcu, A.; Pon, A.; Guo, A.C.; Sajed, T.; Wishart, N.A.; Karu, N.; Feunang, Y.D.; Arndt, D.; Wishart, D.S. YMDB 2.0: A significantly expanded version of the yeast metabolome database. Nucleic Acids Res. 2017, 45, D440–D445. [Google Scholar] [CrossRef] [PubMed]
- Schaub, J.; Reuss, M. In Vivo Dynamics of Glycolysis in Escherichia coli Shows Need for Growth-Rate Dependent Metabolome Analysis. Biotechnol. Prog. 2008, 24, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.L.B.; Hebly, M.; Duong, G.H.; Wahl, S.A.; Pronk, J.T.; Heijnen, J.J.; Daran-Lapujade, P.; van Gulik, W.M. Similar temperature dependencies of glycolytic enzymes: An evolutionary adaptation to temperature dynamics? BMC Syst. Biol. 2012, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaechter, M.; Maaloe, O.; Kjeldgaard, N.O. Dependency on medium and temperature of cell size and chemical composition during balances growth of Salmonella typhimurium. J. Gen. Microbiol. 1958, 19, 592–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patejko, M.; Jacyna, J.; Markuszewski, M.J. Sample preparation procedures utilized in microbial metabolomics: An overview. J. Chromatogr. B-Anal. Technol. Biomed. Life Sci. 2017, 1043, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Wordofa, G.G.; Kristensen, M.; Schrubbers, L.; McCloskey, D.; Forster, J.; Schneider, K. Quantifying the Metabolome of Pseudomonas taiwanensis VLB120: Evaluation of Hot and Cold Combined Quenching/Extraction Approaches. Anal. Chem. 2017, 89, 8738–8747. [Google Scholar] [CrossRef]
- Kvitvang, H.F.N.; Bruheim, P. Fast filtration sampling protocol for mammalian suspension cells tailored for phosphometabolome profiling by capillary ion chromatography—Tandem mass spectrometry. J. Chromatogr. B-Anal. Technol. Biomed. Life Sci. 2015, 998, 45–49. [Google Scholar] [CrossRef]
- Kapoore, R.V.; Vaidyanathan, S. Towards quantitative mass spectrometry-based metabolomics in microbial and mammalian systems. Philos. Trans. R. Soc. A-Math. Phys. Eng. Sci. 2016, 374, 14. [Google Scholar] [CrossRef]
- Zakhartsev, M.; Vielhauer, O.; Horn, T.; Yang, X.L.; Reuss, M. Fast sampling for quantitative microbial metabolomics: New aspects on cold methanol quenching: Metabolite co-precipitation. Metabolomics 2015, 11, 286–301. [Google Scholar] [CrossRef]
- Siegel, D.; Permentier, H.; Reijngoud, D.J.; Bischoff, R. Chemical and technical challenges in the analysis of central carbon metabolites by liquid-chromatography mass spectrometry. J. Chromatogr. B-Anal. Technol. Biomed. Life Sci. 2014, 966, 21–33. [Google Scholar] [CrossRef]
- Chang, J.W.; Lee, G.; Coukos, J.S.; Moellering, R.E. Profiling Reactive Metabolites via Chemical Trapping and Targeted Mass Spectrometry. Anal. Chem. 2016, 88, 6658–6661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, A.; Siegel, D.; Permentier, H.; Reijngoud, D.J.; Dekker, F.; Bischoff, R. Stability of energy metabolitesAn often overlooked issue in metabolomics studies: A review. Electrophoresis 2015, 36, 2156–2169. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Fatemeh, T.; Tang, L.H.; Cai, Z.W. Quantitative metabolic network profiling of Escherichia coli: An overview of analytical methods for measurement of intracellular metabolites. TrAC-Trends Anal. Chem. 2016, 75, 141–150. [Google Scholar] [CrossRef]
- Teleki, A.; Sanchez-Kopper, A.; Takors, R. Alkaline conditions in hydrophilic interaction liquid chromatography for intracellular metabolite quantification using tandem mass spectrometry. Anal. Biochem. 2015, 475, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Bajad, S.U.; Lu, W.Y.; Kimball, E.H.; Yuan, J.; Peterson, C.; Rabinowitz, J.D. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J. Chromatogr. A 2006, 1125, 76–88. [Google Scholar] [CrossRef]
- Jandera, P. Stationary and mobile phases in hydrophilic interaction chromatography: A review. Anal. Chim. Acta 2011, 692, 1–25. [Google Scholar] [CrossRef]
- Kvitvang, H.F.N.; Andreassen, T.; Adam, T.; Villas-Boas, S.G.; Bruheim, P. Highly Sensitive GC/MS/MS Method for Quantitation of Amino and Nonamino Organic Acids. Anal. Chem. 2011, 83, 2705–2711. [Google Scholar] [CrossRef]
- Maruyama, A.; Kami, K.; Sasaki, K.; Sato, H.; Sato, Y.; Tsuchihara, K.; Makinoshima, H. Extraction of Aqueous Metabolites from Cultured Adherent Cells for Metabolomic Analysis by Capillary Electrophoresis-Mass Spectrometry. J. Vis. Exp. 2019, e59551. [Google Scholar] [CrossRef] [Green Version]
- Kvitvang, H.F.N.; Kristiansen, K.A.; Bruheim, P. Assessment of capillary anion exchange ion chromatography tandem mass spectrometry for the quantitative profiling of the phosphometabolome and organic acids in biological extracts. J. Chromatogr. A 2014, 1370, 70–79. [Google Scholar] [CrossRef]
- Stafsnes, M.H.; Rost, L.M.; Bruheim, P. Improved phosphometabolome profiling applying isotope dilution strategy and capillary ion chromatography-tandem mass spectrometry. J. Chromatogr. B-Anal. Technol. Biomed. Life Sci. 2018, 1083, 278–283. [Google Scholar] [CrossRef]
- Patacq, C.; Chaudet, N.; Letisse, F. Absolute Quantification of ppGpp and pppGpp by Double-Spike Isotope Dilution Ion Chromatography-High-Resolution Mass Spectrometry. Anal. Chem. 2018, 90, 10715–10723. [Google Scholar] [CrossRef]
- Reyes-Garces, N.; Gionfriddo, E. Recent developments and applications of solid phase microextraction as a sample preparation approach for mass-spectrometry-based metabolomics and lipidomics. TrAC-Trends Anal. Chem. 2019, 113, 172–181. [Google Scholar] [CrossRef]
- Wu, L.; Mashego, M.R.; van Dam, J.C.; Proell, A.M.; Vinke, J.L.; Ras, C.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly C-13-labeled cell extracts as internal standards. Anal. Biochem. 2005, 336, 164–171. [Google Scholar] [CrossRef]
- Weiner, M.; Trondle, J.; Schmideder, A.; Albermann, C.; Binder, K.; Sprenger, G.A.; Weuster-Botz, D. Parallelized small-scale production of uniformly C-13-labeled cell extract for quantitative metabolome analysis. Anal. Biochem. 2015, 478, 134–140. [Google Scholar] [CrossRef]
- Schatschneider, S.; Abdelrazig, S.; Safo, L.; Henstra, A.M.; Millat, T.; Kim, D.H.; Winzer, K.; Minton, N.P.; Barrett, D.A. Quantitative Isotope-Dilution High-Resolution-Mass-Spectrometry Analysis of Multiple Intracellular Metabolites in Clostridium autoethanogenum with Uniformly C-13-Labeled Standards Derived from Spirulina. Anal. Chem. 2018, 90, 4470–4477. [Google Scholar] [CrossRef]
- Atkinson, D.E.; Walton, G.M. Adenosone triphosphate conservation in metabolic regulation — Rat liver citrate cleavage enzyme. J. Biol. Chem. 1967, 242, 3239–3241. [Google Scholar]
- De la Fuente, I.M.; Cortes, J.M.; Valero, E.; Desroches, M.; Rodrigues, S.; Malaina, I.; Martinez, L. On the Dynamics of the Adenylate Energy System: Homeorhesis vs Homeostasis. PLoS ONE 2014, 9, e108676. [Google Scholar] [CrossRef] [Green Version]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- van Tilburg, A.Y.; Cao, H.J.; van der Meulen, S.B.; Solopova, A.; Kuipers, O.P. Metabolic engineering and synthetic biology employing Lactococcus lactis and Bacillus subtilis cell factories. Curr. Opin. Biotechnol. 2019, 59, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kunst, F.; Ogasawara, N.; Moszer, I.; Albertini, A.M.; Alloni, G.; Azevedo, V.; Bertero, M.G.; Bessieres, P.; Bolotin, A.; Borchert, S.; et al. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 1997, 390, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Rauhut, R.; Klug, G. mRNA degradation in bacteria. FEMS Microbiol. Rev. 1999, 23, 353–370. [Google Scholar] [CrossRef]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. Engl. 2005, 44, 7342–7372. [Google Scholar] [CrossRef]
- Huergo, L.F.; Dixon, R. The Emergence of 2-Oxoglutarate as a Master Regulator Metabolite. Microbiol. Mol. Biol. Rev. 2015, 79, 419–435. [Google Scholar] [CrossRef] [Green Version]
- Lagunas, R.; Gancedo, C. Role of phosphate in the regulation of the Pasteur effect in Saccharomyces cerevisiae. Eur. J. Biochem. 1983, 137, 479–483. [Google Scholar] [CrossRef]
- Guo, L.; Liang, S.; Zhang, Z.; Liu, H.; Wang, S.; Pan, K.; Xu, J.; Ren, X.; Pei, S.; Yang, G. Genome assembly of Nannochloropsis oceanica provides evidence of host nucleus overthrow by the symbiont nucleus during speciation. Commun. Biol. 2019, 2, 249. [Google Scholar] [CrossRef] [Green Version]
- Bowler, C.; Allen, A.E.; Badger, J.H.; Grimwood, J.; Jabbari, K.; Kuo, A.; Maheswari, U.; Martens, C.; Maumus, F.; Otillar, R.P.; et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 2008, 456, 239–244. [Google Scholar] [CrossRef]
- Alipanah, L.; Winge, P.; Rohloff, J.; Najafi, J.; Brembu, T.; Bones, A.M. Molecular adaptations to phosphorus deprivation and comparison with nitrogen deprivation responses in the diatom Phaeodactylum tricornutum. PLoS ONE 2018, 13, e0193335. [Google Scholar] [CrossRef]
- Adarme-Vega, T.C.; Lim, D.K.Y.; Timmins, M.; Vernen, F.; Li, Y.; Schenk, P.M. Microalgal biofactories: A promising approach towards sustainable omega-3 fatty acid production. Microb. Cell Fact. 2012, 11. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Cho, J.M.; Chang, Y.K.; Oh, Y.K. Cell disruption and lipid extraction for microalgal biorefineries: A review. Bioresour. Technol. 2017, 244, 1317–1328. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, J.; Cui, J.; Feng, Y.; Cui, Q. Metabolic profiles of Nannochloropsis oceanica IMET1 under nitrogen-deficiency stress. Bioresour. Technol. 2013, 130, 731–738. [Google Scholar] [CrossRef]
- Wegmann, K. Osmoregulation in eukaryotic algae. FEMS Microbiol. Lett. 1986, 39, 37–43. [Google Scholar] [CrossRef]
- Matthijs, M.; Fabris, M.; Obata, T.; Foubert, I.; Franco-Zorrilla, J.M.; Solano, R.; Fernie, A.R.; Vyverman, W.; Goossens, A. The transcription factor bZIP14 regulates the TCA cycle in the diatom Phaeodactylum tricornutum. EMBO J. 2017, 36, 1559–1576. [Google Scholar] [CrossRef] [Green Version]
- Scherer, W.F.; Syverton, J.T.; Gey, G.O. Studies on the propagation of invitro of poliomyelitis viruses. 4. Viral multiplication in a stable strain of human malignantephithelial cells (strain HeLa derived from an epidermoid carcinoma of the cervix. J. Exp. Med. 1953, 97, 695–710. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Posener, K.; Negelein, E. Über den stoffwechsel der tumoren. Biochem. Z. 1924, 152, 319–344. [Google Scholar]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, Y.; Kinoshita, A.; Tomita, M. Dynamic simulation of red blood cell metabolism and its application to the analysis of a pathological condition. Theor. Biol. Med. Model. 2005, 2, 18. [Google Scholar] [CrossRef] [Green Version]
- Gausdal, G.; Gjertsen, B.T.; McCormack, E.; Van Damme, P.; Hovland, R.; Krakstad, C.; Bruserud, O.; Gevaert, K.; Vandekerckhove, J.; Doskeland, S.O. Abolition of stress-induced protein synthesis sensitizes leukemia cells to anthracycline-induced death. Blood 2008, 111, 2866–2877. [Google Scholar] [CrossRef] [Green Version]
- Guillard, R.R.; Ryther, J.H. Studies of marine planktonic diatoms. I. Cyclotella nana Hustedt, and Detonula confervacea (cleve) Gran. Can. J. Microbiol. 1962, 8, 229–239. [Google Scholar] [CrossRef]
- Tan, B.; Lu, Z.; Dong, S.; Zhao, G.; Kuo, M.S. Derivatization of the tricarboxylic acid intermediates with O-benzylhydroxylamine for liquid chromatography-tandem mass spectrometry detection. Anal. Biochem. 2014, 465, 134–147. [Google Scholar] [CrossRef]
- Jeong, J.W.; Snay, J.; Ataai, M.M. A mathematical model for examining growth and sporulation processes of Bacillus subtilis. Biotechnol. Bioeng. 1990, 35, 160–184. [Google Scholar] [CrossRef]
- Klis, F.M.; de Koster, C.G.; Brul, S. Cell wall-related bionumbers and bioestimates of Saccharomyces cerevisiae and Candida albicans. Eukaryot. Cell 2014, 13, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.P.; Williams, E.; Wang, D.Z.; Xie, Z.X.; Hsia, R.C.; Jenck, A.; Halden, R.; Li, J.; Chen, F.; Place, A.R. Responses of Nannochloropsis oceanica IMET1 to Long-Term Nitrogen Starvation and Recovery. Plant Physiol. 2013, 162, 1110–1126. [Google Scholar] [CrossRef] [Green Version]
- Ashour, M.; Kamel, A. Enhance growth and biochemical composition of Nannochloropsis oceanica cultured under nutrient limitation using commercial agricultural fertilizers. J. Mar. Sci. Res. Dev 2017, 7, 233. [Google Scholar] [CrossRef] [Green Version]
- Nordic microalgea. Phaeodactylum Tricornutum Bohlin. 1897. Available online: http://nordicmicroalgae.org/taxon/Phaeodactylum%20tricornutum?media_id=Phaeodactylum%20tricornutum_5.jpg (accessed on 15 November 2019).
- Fidalgo Paredes, P.; Cid, Á.; Abalde, J.; Herrero, C. Culture of the marine diatom Phaeodactylum tricornutum with different nitrogen sources: Growth, nutrient conversion and biochemical composition. Cah. Biol. Mar. 1995, 36, 165–173. [Google Scholar]
- Dean, R.B.; Dixon, W.J. Simplified Statistics for Small Numbers of Observations. Anal. Chem. 1951, 23, 636–638. [Google Scholar] [CrossRef]
- Droste, P.; Noh, K.; Wiechert, W. Omix—A Visualization Tool for Metabolic Networks with Highest Usability and Customizability in Focus. Chem. Ing. Tech. 2013, 85, 849–862. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Vacuum Pressure (Below ambient, mbar) | Sampling Volume (mL) |
|---|---|---|
| B. subtilis | 700 | 10 |
| S. cerevisiae | 400 | 10 |
| N. oceanica | 600 | 15 |
| P. tricornutum | 600 | 15 |
| Organism | Cell DW (g/cell) | Cell Volume (L/cell) | Specific Cell Volume (L/g) | Reference |
|---|---|---|---|---|
| B. subtilis | 2.2 × 10−13 | 9 × 10−16 | 4.09 × 10−3 | [71] |
| S. cerevisiae | 1.65 × 10−11 | 4.4 × 10−14 | 2.66 × 10−3 | [72] |
| N. oceanica | 1.19 × 10−11 | 1.4 × 10−14 | 1.18 × 10−3 | * Cell volume [73], cell DW [74] |
| P. tricornutum | 4.88 × 10−11 | 1.22 × 10−13 | 2.51 × 10−3 | Cell volume [75], cell DW [76] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Røst, L.M.; Brekke Thorfinnsdottir, L.; Kumar, K.; Fuchino, K.; Eide Langørgen, I.; Bartosova, Z.; Kristiansen, K.A.; Bruheim, P. Absolute Quantification of the Central Carbon Metabolome in Eight Commonly Applied Prokaryotic and Eukaryotic Model Systems. Metabolites 2020, 10, 74. https://doi.org/10.3390/metabo10020074
Røst LM, Brekke Thorfinnsdottir L, Kumar K, Fuchino K, Eide Langørgen I, Bartosova Z, Kristiansen KA, Bruheim P. Absolute Quantification of the Central Carbon Metabolome in Eight Commonly Applied Prokaryotic and Eukaryotic Model Systems. Metabolites. 2020; 10(2):74. https://doi.org/10.3390/metabo10020074
Chicago/Turabian StyleRøst, Lisa M., Lilja Brekke Thorfinnsdottir, Kanhaiya Kumar, Katsuya Fuchino, Ida Eide Langørgen, Zdenka Bartosova, Kåre Andre Kristiansen, and Per Bruheim. 2020. "Absolute Quantification of the Central Carbon Metabolome in Eight Commonly Applied Prokaryotic and Eukaryotic Model Systems" Metabolites 10, no. 2: 74. https://doi.org/10.3390/metabo10020074
APA StyleRøst, L. M., Brekke Thorfinnsdottir, L., Kumar, K., Fuchino, K., Eide Langørgen, I., Bartosova, Z., Kristiansen, K. A., & Bruheim, P. (2020). Absolute Quantification of the Central Carbon Metabolome in Eight Commonly Applied Prokaryotic and Eukaryotic Model Systems. Metabolites, 10(2), 74. https://doi.org/10.3390/metabo10020074