Comparative Metabolomics and Molecular Phylogenetics of Melon (Cucumis melo, Cucurbitaceae) Biodiversity

 ,
,  , ,
, ,  , , , ,
, , , ,  , , ,
, , ,  and
and
Abstract
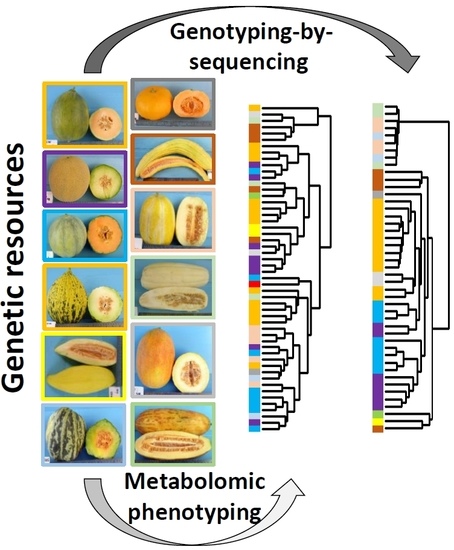
:
1. Introduction
2. Results
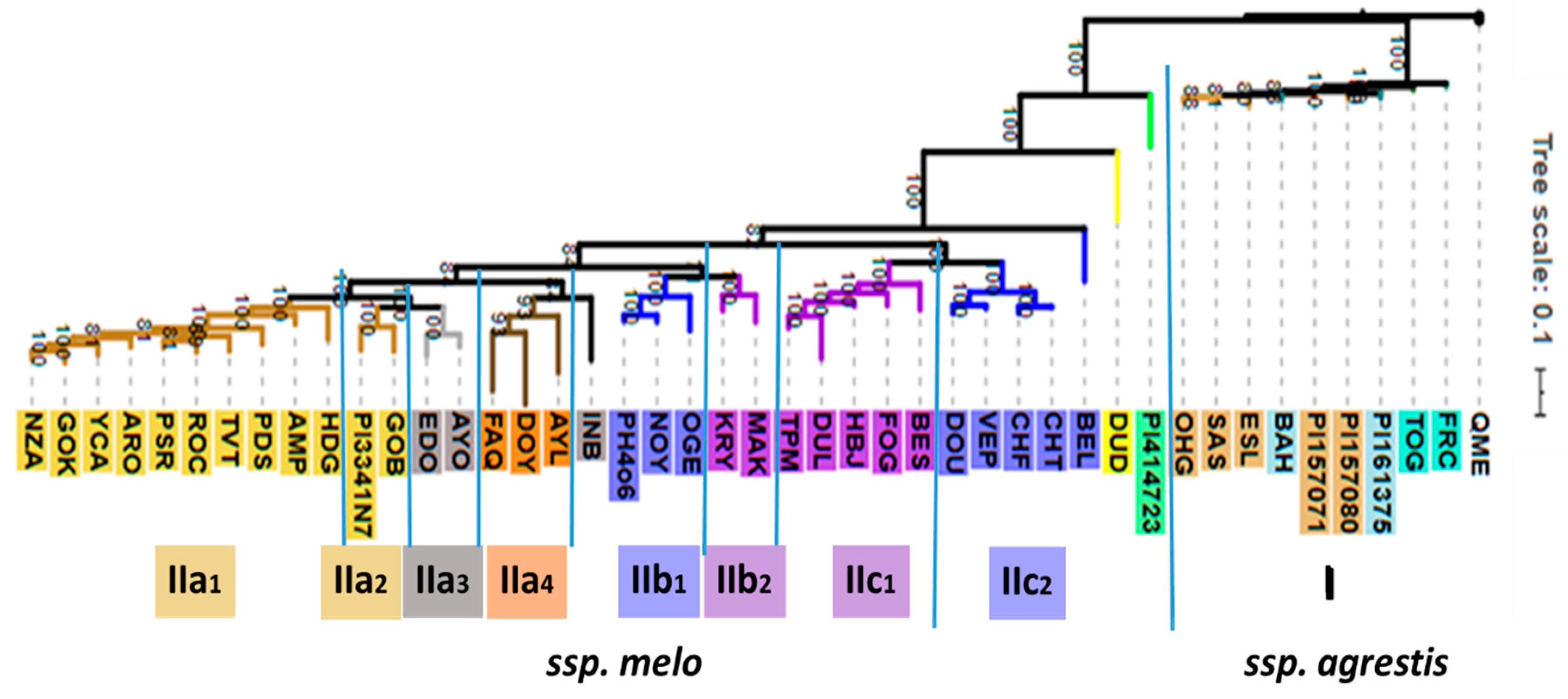
2.1. Infraspecific Structure of Cucumis melo
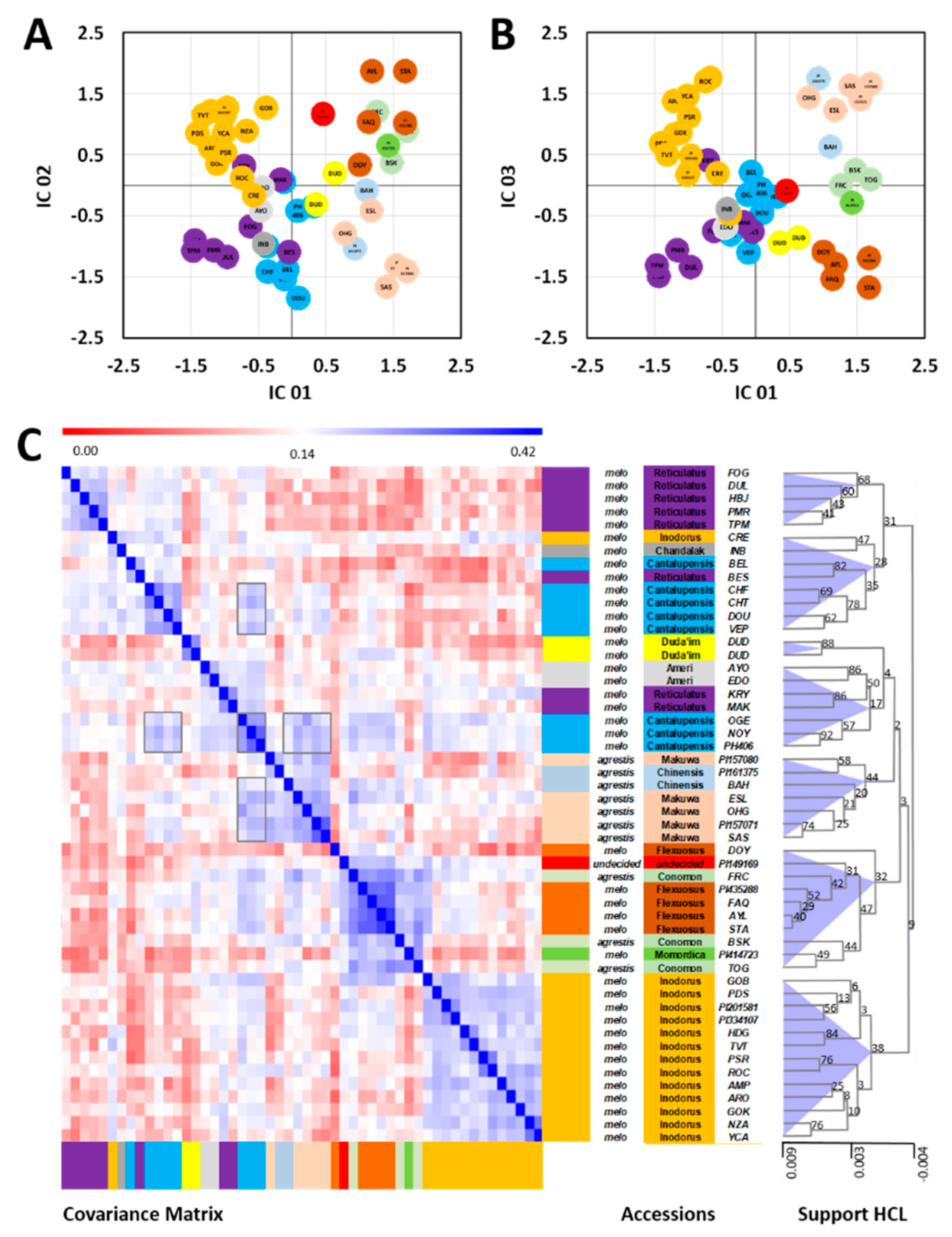
2.2. Combined Analysis of All Metabolomic and Elemental Data
2.3. Platform-Specific Metabolomic Analyses of Melon Flesh or Rind Tissue
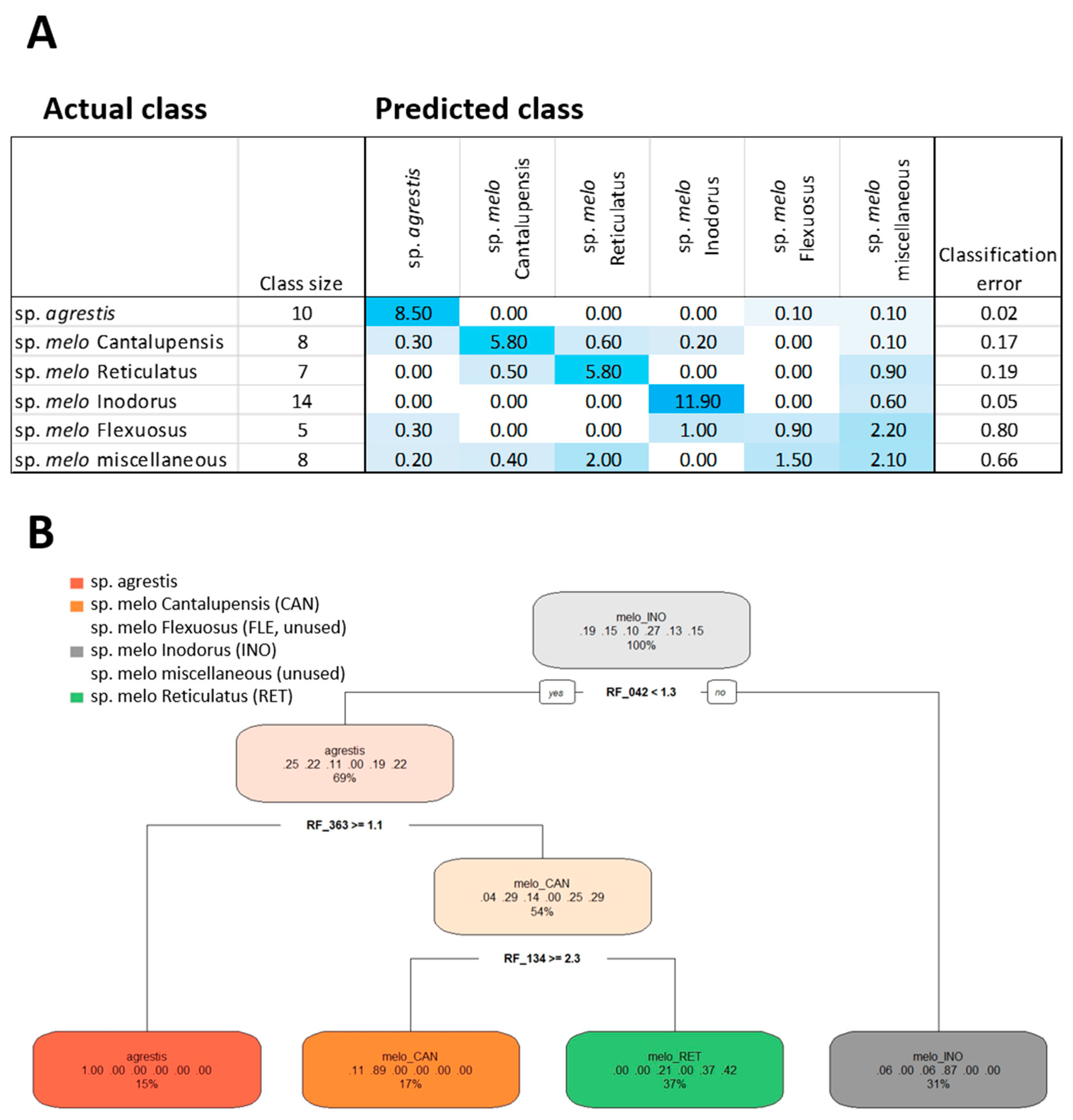
2.4. Feature Selection by Random Forest Technology
3. Discussion
3.1. Phylogenomic and Phytochemical Relationships Partly Coincide in C. melo
3.2. Cantalupensis and Reticulatus Accessions are Separated from Each Other Both Genotypically and Metabolomically
3.3. The Metabolomic-Based Classification May Indicate That Two Independent Evolutionary Events Led to Non-Climacteric Ripening
4. Conclusions
5. Materials and Methods
5.1. Plant Material Description, Cultivation, and Sampling
5.2. Genotype and Phylogenetic Analysis
5.2.1. DNA Isolation for Genotype by Sequencing (GBS)
5.2.2. GBS Analysis
5.2.3. Phylogenetic Analysis
5.3. Global Measurements of Fruit Quality
5.4. Metabolomics and Elementals Analysis
5.4.1. NMR-Based Metabolomic Analyses
5.4.2. GC-MS-Based Metabolomic Analysis of Polar Compounds
5.4.3. GC-MS-Based Metabolomic Analysis of Volatile Compounds
5.4.4. FIE- or LC-MS-Based Metabolomics Analysis
5.4.5. Elemental Analysis
5.4.6. Data Handling and Mining
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pitrat, M. Melon. In Handbook of Plant Breeding: Vegetable; Prohens, J., Nuez, F., Eds.; Springer Science: New York, NY, USA, 2008; Volume I, pp. 283–316. [Google Scholar]
- Burger, Y.; Paris, H.S.; Cohen, R.; Katzir, N.; Tadmor, Y.; Lewinsohn, E.; Schaffer, A.A. Genetic diversity of Cucumis melo. Hortic. Rev. 2010, 36, 165–198. [Google Scholar]
- Pech, J.-C.; Bouzayen, M.; Latche, A. Climacteric fruit ripening: Ethylene-dependent and independent regulation of ripening pathways in melon fruit. Plant Sci. 2008, 175, 114–120. [Google Scholar] [CrossRef] [Green Version]
- Kirkbride, J.H., Jr. Biosystematic Monograph of the Genus Cucumis [Cucurbitaceae]; Parkway Publishers: Boone, NC, USA, 1993. [Google Scholar]
- Robinson, R.W.; Decker-Walters, D.S. Cucurbits; CAB International: Wallingford, Oxon, UK, 1997; pp. 65–70. [Google Scholar]
- Pitrat, M.; Hanelt, P.; Hammer, K. Some comments on infraspecific classification of cultivars of melon. In Proceedings of Cucurbitaceae 2000: The 7th Eucarpia Meeting on Cucurbit Genetics and Breeding, Ma’ale Ha Hamisha, Israel, 19–23 March 2000; Acta Horticulturae No. 510; Katzir, N., Paris, H.S., Eds.; ISHS: Leuven, Belgium, 2000; pp. 29–36. [Google Scholar]
- Decker-Walters, D.S.; Chung, S.M.; Staub, J.E.; Quemada, H.D.; López-Sesé, A.I. The origin and genetic affinities of wild populations of melon [Cucumis melo, Cucurbitaceae] in North America. Plant Syst. Evol. 2002, 233, 183–197. [Google Scholar] [CrossRef]
- Zhao, G.; Lian, Q.; Zhang, Z.; Fu, Q.; He, Y.; Ma, S.; Ruggieri, V.; Monforte, A.J.; Wang, P.; Julca, I.; et al. A comprehensive genome variation map of melon identifies multiple domestication events and loci influencing agronomic traits. Nat. Genet. 2019, 51, 1607–1615. [Google Scholar] [CrossRef]
- Hadfield, K.A.; Rose, J.K.C.; Bennett, A.B. The respiratory climacteric is present in Charentais [Cucumis melo cv. Reticulatus F1 Alpha] melons ripened on or off the plant. J. Exp. Bot. 1995, 46, 1923–1925. [Google Scholar] [CrossRef]
- Aubert, C.; Bourger, N. Investigation of volatiles in Charentais cantaloupe melons [Cucumis melo Var. cantalupensis]. Characterization of aroma constituents in some cultivars. J. Agric. Food Chem. 2004, 52, 4522–4528. [Google Scholar] [CrossRef]
- Renner, S.S.; Schaefer, H.; Kocyan, A. Phylogenetics of Cucumis [Cucurbitaceae]: Cucumber [C. sativus] belongs in an Asian/Australian clade far from melon [C. melo]. BMC Evol. Biol. 2007, 7, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, P.; Schaefer, H.; Telford, I.R.H.; Renner, S.S. Cucumber [Cucumis sativus] and melon [C. melo] have numerous wild relatives in Asia and Australia, and the sister species of melon is from Australia. Proc. Natl. Acad. Sci. USA 2010, 107, 14269–14273. [Google Scholar] [CrossRef] [Green Version]
- Paris, H.S.; Amar, Z.; Lev, E. Medieval emergence of sweet melons, Cucumis melo [Cucurbitaceae]. Ann. Bot. 2012, 110, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Paris, H.S.; Amar, Z.; Lev, E. Medieval history of the duda’im melon [Cucumis melo, Cucurbitaceae]. Econ. Bot. 2012, 66, 276–284. [Google Scholar] [CrossRef]
- Janick, J.; Paris, H.S.; Parrish, D.C. The cucurbits of Mediterranean antiquity: Identification of taxa from ancient images and descriptions. Ann. Bot. 2007, 100, 1441–1457. [Google Scholar] [CrossRef]
- Paris, H.S. Semitic-language records of snake melons [Cucumis melo, Cucurbitaceae] in the medieval period and the “piqqus” of the “faqqous”. Genet. Resour. Crop Evol. 2012, 59, 31–38. [Google Scholar] [CrossRef]
- Deleu, W.; Esteras, C.; Roig, C.; González-To, M.; Fernández-Silva, I.; Gonzalez-Ibeas, D.; Blanca, J.; Aranda, M.A.; Arús, P.; Nuez, F.; et al. A set of EST-SNPs for map saturation and cultivar identification in melon. BMC Plant Biol. 2009, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Blanca, J.M.; Cañizares, J.; Ziarsolo, P.; Esteras, C.; Mir, G.; Nuez, F.; Garcia-Mas, J.; Picó, M.B. Melon transcriptome characterization: Simple sequence repeats and single nucleotide polymorphisms discovery for high throughput genotyping across the species. Plant Genome 2011, 4, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Endl, J.; Achigan-Dako, E.G.; Pandey, A.K.; Monforte, A.J.; Pico, B.; Schaefer, H. Repeated domestication of melon [Cucumis melo] in Africa and Asia and a new close relative from India. Am. J. Bot. 2018, 105, 1662–1671. [Google Scholar] [CrossRef] [Green Version]
- Leida, C.; Moser, C.; Esteras, C.; Sulpice, R.; Lunn, J.E.; de Langen, F.; Monforte, A.J.; Picó, B. Variability of candidate genes, genetic structure and association with sugar accumulation and climacteric behavior in a broad germplasm collection of melon [Cucumis melo L.]. BMC Genet. 2015, 16, 28. [Google Scholar] [CrossRef] [Green Version]
- McNair, J. Angiosperm phylogeny on a Chemical Basis. Bull. Torrey Bot. Club 1935, 62, 515–532. [Google Scholar] [CrossRef]
- Waterman, P.G. The current status of chemical systematics. Phytochemistry 2007, 68, 2896–2903. [Google Scholar] [CrossRef]
- van Treuren, R.; van Eekelen, H.D.; Wehrens, R.; de Vos, R.C. Metabolite variation in the lettuce gene pool: Towards healthier crop varieties and food. Metabolomics 2018, 14, 146. [Google Scholar] [CrossRef] [Green Version]
- Bernillon, S.; Biais, B.; Deborde, C.; Maucourt, M.; Cabasson, C.; Gibon, Y.; Moing, A. Metabolomic and elemental profiling of melon fruit quality as affected by genotype and environment. Metabolomics 2013, 9, 57–77. [Google Scholar] [CrossRef]
- Freilich, S.; Lev, S.; Gonda, I.; Reuveni, E.; Portnoy, V.; Oren, E.; Lohse, M.; Galpaz, N.; Bar, E.; Tzuri, G.; et al. Systems approach for exploring the intricate associations between sweetness, color and aroma in melon fruits. BMC Plant Biol. 2015, 15, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteras, C.; Rambla, J.L.; Sánchez, G.; López-Gresa, M.P.; González-Mas, M.C.; Fernández-Trujillo, J.P.; Bellés, J.M.; Granell, A.; Picó, M.B. Fruit flesh volatile and carotenoid profile analysis within the Cucumis melo L. species reveals unexploited variability for future genetic breeding. J. Sci. Food Agric. 2018, 98, 3915–3925. [Google Scholar] [CrossRef] [PubMed]
- Moing, A.; Aharoni, A.; Biais, B.; Rogachev, I.; Meir, S.; Brodsky, L.; Allwood, J.W.; Erban, A.; Dunn, W.B.; Kay, L.; et al. Extensive metabolic cross talk in melon fruit revealed by spatial and developmental combinatorial metabolomics. New Phytol. 2011, 190, 683–696. [Google Scholar] [CrossRef]
- Daub, C.O.; Kloska, S.; Selbig, J. MetaGeneAlyse: Analysis of integrated transcriptional and metabolite data. Bioinformatics 2003, 19, 2332–2333. [Google Scholar] [CrossRef]
- Erban, A.; Fehrle, I.; Martinez-Seidel, F.; Brigante, F.; Lucini Más, A.; Baroni, V.; Wunderlin, D.; Kopka, J. Discovery of food identity markers by metabolomics and machine learning technology. Sci. Rep. 2019, 9, 9697. [Google Scholar] [CrossRef] [Green Version]
- Wahyuni, Y.; Ballester, A.R.; Tikunov, Y.; de Vos, R.C.; Pelgrom, K.T.; Maharijaya, A.; Sudarmonowati, E.; Bino, R.J.; Bovy, A.G. Metabolomics and molecular marker analysis to explore pepper [Capsicum sp.] biodiversity. Metabolomics 2013, 9, 130–144. [Google Scholar] [CrossRef] [Green Version]
- De Vos, R.C.H.; Schipper, B.; Hall, R.D. High-performance liquid chromatography–mass spectrometry analysis of plant metabolites in Brassicaceae. In Plant Metabolomics; Methods in Molecular Biology [Methods and Protocols]; Hardy, N., Hall, R., Eds.; Humana Press: Totowa, NJ, USA, 2011; Volume 860. [Google Scholar]
- Liu, C.; Jiang, D.; Cheng, Y.; Deng, X.; Chen, F.; Fang, L.; Ma, Z.; Xu, J. Chemotaxonomic study of Citrus, Poncirus and Fortunella genotypes based on peel oil volatile compounds—Deciphering the genetic origin of Mangshanyegan [Citrus nobilis Lauriro]. PLoS ONE 2013, 8, e58411. [Google Scholar] [CrossRef] [Green Version]
- Obando-Ulloa, J.M.; Moreno, E.; García-Mas, J.; Nicolai, B.; Lammertyn, J.; Monforte, A.J.; Fernández-Trujillo, J.P. Climacteric or non-climacteric behavior in melon fruit: 1. Aroma volatiles. Postharvest Biol. Technol. 2008, 49, 27–37. [Google Scholar] [CrossRef]
- Jacquin, H. Monographie Complète Du Melon; Rousselon: Paris, France, 1832. [Google Scholar]
- Paris, H.S. A proposed subspecific classification for Cucurbita pepo. Phytologia 1986, 61, 133–138. [Google Scholar]
- Gong, L.; Paris, H.S.; Nee, M.H.; Stift, G.; Pachner, M.; Vollmann, J.; Lelley, T. Genetic relationships and evolution in Cucurbita pepo [pumpkin, squash, gourd] as revealed by simple sequence repeat polymorphisms. Theor. Appl. Genet. 2012, 124, 875–891. [Google Scholar] [CrossRef] [Green Version]
- Gur, A.; Tzuri, G.; Meir, A.; Sa’ar, U.; Portnoy, V.; Katzir, N.; Schaffer, A.A.; Li, L.; Burger, J.; Tadmor, Y. Genome-wide linkage-disequilibrium mapping to the candidate gene level in melon [Cucumis melo]. Sci. Rep. 2017, 7, 9770. [Google Scholar] [CrossRef]
- Cohen, S.; Itkin, M.; Yeselson, Y.; Tzuri, G.; Portnoy, V.; Harel-Baja, R.; Lev, S.; Sa’ar, U.; Davidovitz-Rikanati, R.; Baranes, N.; et al. The PH gene determines fruit acidity and contributes to the evolution of sweet melons. Nat. Commun. 2014, 5, 4026. [Google Scholar] [CrossRef] [Green Version]
- Harel-Beja, R.; Tzuri, G.; Portnoy, V.; Lotan-Pompan, M.; Lev, S.; Cohen, S.; Dai, N.; Yeselson, L.; Meir, A.; Libhaber, S.E.; et al. A genetic map of melon highly enriched with fruit quality QTLs and EST markers. including sugar and carotenoid metabolism genes. Theor. Appl. Genet. 2010, 121, 511–533. [Google Scholar] [CrossRef]
- Tzuri, G.; Zhou, X.; Chayut, N.; Yuan, H.; Portnoy, V.; Meir, A.; Sa’ar, U.; Baumkoler, F.; Mazourek, M.; Lewinsohn, E.; et al. A ‘golden’ SNP in CmOr governs the fruit flesh color of melon [Cucumis melo]. Plant J. 2015, 82, 267–279. [Google Scholar] [CrossRef]
- Diaz, A.; Fergany, M.; Formisano, G.; Ziarsolo, P.; Blanca, J.; Fei, Z.; Staub, J.E.; Zalapa, J.E.; Cuevas, H.E.; Dace, G.; et al. A consensus linkage map for molecular markers and quantitative trait loci associated with economically important traits in melon [Cucumis melo L.]. BMC Plant Biol. 2011, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Oren, E.; Tzuri, G.; Dafna, A.; Meir, A.; Kumar, R.; Katzir, N.; Elkind, Y.; Freilich, S.; Schaffer, A.A.; Tadmor, Y.; et al. High-density NGS-based map construction and genetic dissection of fruit shape and rind netting in Cucumis melo. Theor. Appl. Genet. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Oren, E.; Tzuri, G.; Vexler, L.; Dafna, A.; Meir, A.; Faigenboim, A.; Kenigswald, M.; Portnoy, V.; Schaffer, A.A.; Levi, A.; et al. The multi-allelic APRR2 gene is associated with fruit pigment accumulation in melon and watermelon. J. Exp. Bot. 2019, 70, 3781–3794. [Google Scholar] [CrossRef] [Green Version]
- Galpaz, N.; Gonda, I.; Shem-Tov, D.; Barad, O.; Tzuri, G.; Lev, S.; Fei, Z.; Xu, Y.; Mao, L.; Jiao, C.; et al. Deciphering genetic factors that determine melon fruit-quality traits using RNA-Seq-based high-resolution QTL and eQTL mapping. Plant J. 2018, 94, 169–191. [Google Scholar] [CrossRef] [Green Version]
- Chayut, N.; Yuan, H.; Ohali, S.; Meir, A.; Sa’ar, U.; Tzuri, G.; Zheng, Y.; Mazourek, M.; Gepstein, S.; Zhou, X.; et al. Distinct mechanisms of the ORANGE protein in controlling carotenoid flux. Plant Physiol. 2017, 173, 376–389. [Google Scholar] [CrossRef]
- Périn, C.; Gomez-Jimenez, M.; Hagen, L.; Dogimont, C.; Pech, J.C.; Latché, A.; Pitrat, M.; Lelièvre, J.M. Molecular and genetic characterization of a non-climacteric phenotype in melon reveals two loci conferring altered ethylene response in fruit. Plant Physiol. 2002, 129, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Ríos, P.; Argyris, J.; Vegas, J.; Leida, C.; Kenigswald, M.; Tzuri, G.; Troadec, C.; Bendahmane, A.; Katzir, N.; Picó, B.; et al. ETHQV6.3 is involved in melon climacteric fruit ripening and is encoded by a NAC domain transcription factor. Plant J. 2017, 91, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Eduardo, I.; Arús, P.; Monforte, A.J. Development of a genomic library of near isogenic lines [NILs] in melon [Cucumis melo L.] from the exotic accession PI161375. Theor. Appl. Genet. 2005, 112, 139–148. [Google Scholar] [CrossRef]
- Vegas, J.; Garcia-Mas, J.; Monforte, A.J. Interaction between QTLs induces an advance in ethylene biosynthesis during melon fruit ripening. Theor. Appl. Genet. 2013, 126, 1531–1544. [Google Scholar] [CrossRef]
- Saladié, M.; Cañizares, J.; Phillips, M.A.; Rodriguez-Concepcion, M.; Larrigaudière, C.; Gibon, Y.; Stitt, M.; Lunn, J.E.; Garcia-Mas, J. Comparative transcriptional profiling analysis of developing melon [Cucumis melo L.] fruit from climacteric and non-climacteric varieties. BMC Genom. 2015, 16, 440. [Google Scholar] [CrossRef]
- Buttery, R.G.; Seifert, R.M.; Ling, L.C.; Soderstrom, E.L.; Ogawa, J.M.; Turnbaugh, J.G. Additional aroma components of honeydew melon. J. Agric. Food Chem. 1982, 30, 1208–1211. [Google Scholar] [CrossRef]
- Gross, K.C.; Sams, C.E. Changes in cell wall neutral sugar composition during fruit ripening: A species survey. Phytochemistry 1984, 23, 2457–2461. [Google Scholar] [CrossRef]
- Jetter, R.; Kunst, L.; Samuels, A.L. Composition of plant cuticular waxes. Biol. Plant Cuticle 2008, 23, 145–181. [Google Scholar]
- Cohen, H.; Dong, Y.; Szymanski, J.; Lashbrooke, J.; Meir, S.; Almekias-Siegl, E.; Zeisler-Diehl, V.V.; Schreiber, L.; Aharoni, A. A multilevel study of melon fruit reticulation provides insight into skin ligno-suberization hallmarks. Plant Physiol. 2019, 179, 1486–1501. [Google Scholar] [CrossRef] [Green Version]
- Dai, N.; Cohen, S.; Portnoy, V.; Tzuri, G.; Harel-Beja, R.; Pompan-Lotan, M.; Carmi, N.; Zhang, G.; Diber, A.; Pollock, S.; et al. Metabolism of soluble sugars in developing melon fruit: A global transcriptional view of the metabolic transition to sucrose accumulation. Plant Mol. Biol. 2011, 76, 1–18. [Google Scholar] [CrossRef]
- Fang, C.; Luo, J. Metabolic GWAS-based dissection of genetic bases underlying the diversity of plant metabolism. Plant J. 2019, 97, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing [GBS] approach for high diversity species. PLoS ONE 2011, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Mas, J.; Benjak, A.; Sanseverino, W.; Bourgeois, M.; Mir, G.; González, V.M.; Hénaff, E.; Câmara, F.; Cozzuto, L.; Lowy, E.; et al. The genome of melon [Cucumis melo L.]. Proc. Natl. Acad. Sci. USA 2012, 109, 11872–11877. [Google Scholar] [CrossRef] [Green Version]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Biais, B.; Allwood, J.W.; Deborde, C.; Xu, Y.; Mickael, M.; Beauvoit, B.; Dunn, W.B.; Jacob, D.; Goodacre, R.; Rolin, D.; et al. 1H NMR, GC−EI-TOFMS, and data set correlation for fruit metabolomics: Application to spatial metabolite analysis in melon. Anal. Chem. 2009, 81, 2884–2894. [Google Scholar] [CrossRef]
- Ward, J.L.; Forcat, S.; Beckmann, M.; Bennett, M.; Miller, S.J.; Baker, J.M.; Hawkins, N.D.; Vermeer, C.P.; Lu, C.; Lin, W.; et al. The metabolic transition during disease following infection of Arabidopsis thaliana by Pseudomonas syringae pv. tomato. Plant J. 2010, 63, 443–457. [Google Scholar] [CrossRef]
- Allwood, J.W.; Erban, A.; de Koning, S.; Dunn, W.B.; Luedemann, A.; Lommen, A.; Kay, L.; Löscher, R.; Kopka, J.; Goodacre, R. Inter-laboratory reproducibility of fast gas chromatography–electron impact–time of flight mass spectrometry [GC–EI–TOF/MS] based plant metabolomics. Metabolomics 2009, 5, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Luedemann, A.; Strassburg, K.; Erban, A.; Kopka, J. TagFinder for the quantitative analysis of gas chromatography-mass spectrometry [GC-MS] based metabolite profiling experiments. Bioinformatics 2008, 24, 732–737. [Google Scholar] [CrossRef]
- Strehmel, N.; Hummel, J.; Erban, A.; Strassburg, K.; Kopka, J. Retention index thresholds for compound matching in GC-MS metabolite profiling. J. Chromatogr. B 2008, 871, 182–190. [Google Scholar] [CrossRef]
- Kopka, J.; Schauer, N.; Krueger, S.; Birkemeyer, C.; Usadel, B.; Bergmueller, E.; Doermann, P.; Weckwerth, W.; Gibon, Y.; Stitt, M.; et al. [email protected]: The Golm Metabolome Database. Bioinformatics 2005, 21, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Verhoeven, H.A.; Jonker, H.; De Vos, R.C.H.; Hall, R.D. Solid Phase Micro-Extraction GC–MS Analysis of Natural Volatile components in Melon and Rice. In Book Humana, Methods for Plant Metabolomics; Hardy, N.G., Hall, R.D., Eds.; Springer–Humana: London, UK, 2012; pp. 85–99. [Google Scholar]
- Lommen, A. MetAlign: Interface-driven, versatile metabolomics tool for hyphenated full-scan mass spectrometry data preprocessing. Anal. Chem. 2009, 81, 3079–3086. [Google Scholar] [CrossRef]
- Tikunov, Y.M.; Laptenok, S.; Hall, R.D.; Bovy, A.; de Vos, R.C.H. MSClust: A tool for unsupervised mass spectra extraction of chromatography-mass spectrometry ion-wise aligned data. Metabolomics 2012, 8, 714–718. [Google Scholar] [CrossRef] [Green Version]
- Moussaieff, A.; Rogachev, I.; Brodsky, L.; Malitsky, S.; Toal, T.W.; Belcher, H.; Yativ, M.; Brady, S.M.; Benfey, P.N.; Aharoni, A. High-resolution metabolic mapping of cell types in plant roots. Proc. Natl. Acad. Sci. USA 2013, 110, E1232–E1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Scholz, M.; Kaplan, F.; Guy, C.L.; Kopka, J.; Selbig, J. Non-linear PCA: A missing data approach. Bioinformatics 2005, 21, 3887–3895. [Google Scholar] [CrossRef] [Green Version]
- Wehrens, R.; Hageman, J.A.; van Eeuwijk, F.; Kooke, R.; Flood, P.J.; Wijnker, E.; Keurentjes, J.J.; Lommen, A.; van Eekelen, H.D.; Hall, R.D.; et al. Improved batch correction in untargeted MS-based metabolomics. Metabolomics 2016, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. BioTechniques 2003, 34, 374–378. [Google Scholar] [CrossRef] [Green Version]
- Probst, P.; Wright, M.; Boulesteix, A.-L. Hyperparameters and tuning strategies for random forest. WIREs Data Min. Knowl. Discov. 2019, 9, e1301. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R. News 2002, 2, 18–22. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Name | Subspecies | Cultivar Group | Accession Abbreviation | Included in GBS | Clade (as in Figure 2) |
|---|---|---|---|---|---|
| Batley’s Hermaphrodite | agrestis | Chinensis | BAH | x | I |
| PI161375 | agrestis | Chinensis | PI161375 | x | I |
| Black Skin | agrestis | Conomon | BSK | NI | |
| Freeman’s Cucumber | agrestis | Conomon | FRC | x | I |
| Tokyo Giant | agrestis | Conomon | TOG | x | I |
| Early Silver Line | agrestis | Makuwa | ESL | x | I |
| Ohgon | agrestis | Makuwa | OHG | x | I |
| PI157071 | agrestis | Makuwa | PI157071 | x | I |
| PI157080 | agrestis | Makuwa | PI157080 | x | I |
| Sakata’s Sweet | agrestis | Makuwa | SAS | x | I |
| Ananas Yoqne‘am | melo | Ameri | AYO | x | IIa2 |
| Ananas ‘En Dor | melo | Ameri | EDO | x | IIa2 |
| Bellegarde | melo | Cantalupensis | BEL | x | IIc2 |
| Charentais Fom 2 Res. | melo | Cantalupensis | CHF | x | IIc2 |
| Charentais | melo | Cantalupensis | CHT | x | IIc2 |
| Doublon | melo | Cantalupensis | DOU | x | IIc2 |
| Noy Yizre‘el | melo | Cantalupensis | NOY | x | IIb1 |
| Ogen | melo | Cantalupensis | OGE | x | IIb1 |
| PH 406 | melo | Cantalupensis | PH406 | x | IIb1 |
| Védrantais | melo | Cantalupensis | VEP | x | IIc2 |
| Indian Best | melo | Khandalak | INB | x | II |
| Duda’im | melo | Duda’im | DUD | x | II |
| PI435288 | melo | Flexuosus | PI435288 | NI | |
| Armenian Yard Long | melo | Flexuosus | AYL | x | IIa3 |
| Doya | melo | Flexuosus | DOY | x | IIa3 |
| Faqqous | melo | Flexuosus | FAQ | x | IIa3 |
| Striped Armenian | melo | Flexuosus | STA | NI | |
| Amarillo Pipa Blanca | melo | Inodorus | AMP | x | IIa1 |
| Amarillo Oro | melo | Inodorus | ARO | x | IIa1 |
| Crenshaw | melo | Inodorus | CRE | NI | |
| Golden Beauty | melo | Inodorus | GOB | x | IIa2 |
| Gold King | melo | Inodorus | GOK | x | IIa1 |
| Honey Dew Green | melo | Inodorus | HDG | x | IIa1 |
| Noy Zahov | melo | Inodorus | NZA | x | IIa1 |
| Piel de Sapo | melo | Inodorus | PDS | x | IIa1 |
| PI 334107, Kirkagac | melo | Inodorus | PI334107 | x | IIa2 |
| PI 201581b, Kirkagac | melo | Inodorus | PI201581 | NI | |
| Piel de Sapo Redon | melo | Inodorus | PSR | x | IIa1 |
| Rochet | melo | Inodorus | ROC | x | IIa1 |
| Tendral Verde Tardio | melo | Inodorus | TVT | x | IIa1 |
| Yellow Canary | melo | Inodorus | YCA | x | IIa1 |
| PI414723 | melo | Momordica | PI414723 | x | II |
| Bender’s Surprise | melo | Reticulatus | BES | x | IIc1 |
| Dulce | melo | Reticulatus | DUL | x | IIc1 |
| Fordhook Gem | melo | Reticulatus | FOG | x | IIc1 |
| Hale’s Best Jumbo | melo | Reticulatus | HBJ | x | IIc1 |
| Krymka | melo | Reticulatus | KRY | x | IIb2 |
| Magyar Kincs | melo | Reticulatus | MAK | x | IIb2 |
| PMR45 | melo | Reticulatus | PMR | NI | |
| Top Mark | melo | Reticulatus | TPM | x | IIc1 |
| PI149169 | undecided | undecided | PI149169 | NI | |
| Qishu Meshullash | (outlier) | (feral) | QME | x | (outlier) |
| Analytical Strategies and Corresponding Examples of Covered Compounds or Compound Families | Number of Molecular Features | |
|---|---|---|
| Flesh | Rind | |
| GC-MS of polar extracts: soluble sugars, sugar-alcohols, organic acids, amino acids, polyamines | 12 397 | 13 200 |
| 1H-NMR fingerprints of polar extracts: major soluble sugars, organic acids, amino acids and other amino compounds | 40 | 28 |
| 1H-NMR quantitative profiles of polar extracts: major soluble sugars, organic acids, amino acids and other amino compounds | 108 | 108 |
| 1H-NMR fingerprints of semi-polar extracts: major soluble sugars, organic acids, amino acids and major semi-polar specialized metabolites | 839 | 819 |
| DI-ESI-MS of semi-polar extracts: positive ionization mode negative ionization mode semi-polar major and specialized metabolites | 931931 | 931931 |
| LC-QTOF-MS of semi-polar extracts: negative ionization mode non-volatile specialized metabolites and their conjugates including the flavonoid- and hydroxycinnamate-families | 20 785 | 30 695 |
| SPME GC-MS of volatiles: volatile specialized metabolites including alcohols, aldehydes, terpenoids | 282 | - |
| ICP-MS of mineral elements: mineral elements including P, K, Fe, Ni, and low-abundant trace elements | 20 | - |
| Analytical Strategy | GC-MS of Polar Extracts | 1H-NMR Fingerprints of Polar Extracts (0.04 ppm VS bucketting) | 1H-NMR Quantitative Profiles of Polar Extracts | 1H-NMR Fingerprints of Semi-Polar Extracts (0.01 ppm bucketting) | FIE-MS of Semi-Polar Extracts | LC-QTOF-MS of Semi-Polar Extracts | SPME GC-MS of Volatiles | ICP-MS of Mineral Elements |
|---|---|---|---|---|---|---|---|---|
| Flesh | ||||||||
| Pearson correlation (r) between the molecular distance matrix and the metabolomic or elemental distance matrix | 0.207 | 0.117 | 0.224 | 0.202 | 0.149 | 0.560 | 0.387 | 0.177 |
| Correlation p-value a | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 |
| Rind | ||||||||
| Pearson correlation (r) between the molecular distance matrix and the metabolomic or elemental distance matrix b | 0.315 | 0.273 | 0.267 | 0.561 | 0.468 | −0.041 | - | - |
| Correlation p-value | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 | 0.189 | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moing, A.; Allwood, J.W.; Aharoni, A.; Baker, J.; Beale, M.H.; Ben-Dor, S.; Biais, B.; Brigante, F.; Burger, Y.; Deborde, C.; et al. Comparative Metabolomics and Molecular Phylogenetics of Melon (Cucumis melo, Cucurbitaceae) Biodiversity. Metabolites 2020, 10, 121. https://doi.org/10.3390/metabo10030121
Moing A, Allwood JW, Aharoni A, Baker J, Beale MH, Ben-Dor S, Biais B, Brigante F, Burger Y, Deborde C, et al. Comparative Metabolomics and Molecular Phylogenetics of Melon (Cucumis melo, Cucurbitaceae) Biodiversity. Metabolites. 2020; 10(3):121. https://doi.org/10.3390/metabo10030121
Chicago/Turabian StyleMoing, Annick, J. William Allwood, Asaph Aharoni, John Baker, Michael H. Beale, Shifra Ben-Dor, Benoît Biais, Federico Brigante, Yosef Burger, Catherine Deborde, and et al. 2020. "Comparative Metabolomics and Molecular Phylogenetics of Melon (Cucumis melo, Cucurbitaceae) Biodiversity" Metabolites 10, no. 3: 121. https://doi.org/10.3390/metabo10030121
APA StyleMoing, A., Allwood, J. W., Aharoni, A., Baker, J., Beale, M. H., Ben-Dor, S., Biais, B., Brigante, F., Burger, Y., Deborde, C., Erban, A., Faigenboim, A., Gur, A., Goodacre, R., Hansen, T. H., Jacob, D., Katzir, N., Kopka, J., Lewinsohn, E., ... Schaffer, A. A. (2020). Comparative Metabolomics and Molecular Phylogenetics of Melon (Cucumis melo, Cucurbitaceae) Biodiversity. Metabolites, 10(3), 121. https://doi.org/10.3390/metabo10030121