Identification and Validation of Metabolic Markers for Adulteration Detection of Edible Oils Using Metabolic Networks


 ,
, 
 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Sample Preparation
2.3. UPLC-MS/MS Analysis of the Target Compounds
2.4. Data Processing and Statistical Analysis of Metabolites
3. Results and Discussion
3.1. Separation Parameters of Liquid Chromatography
3.2. Optimization of the Mass Spectrometry Parameters for Standard Solutions
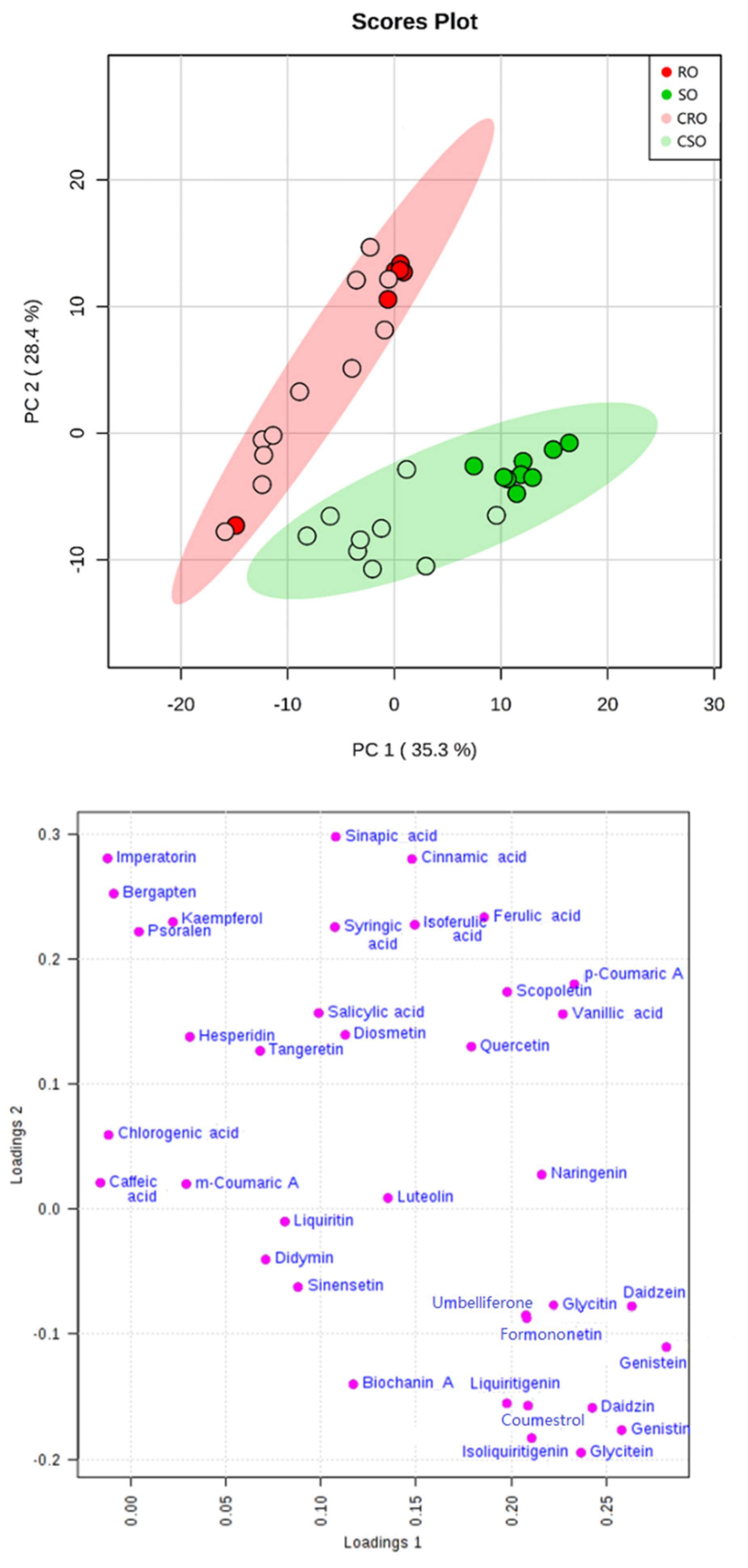
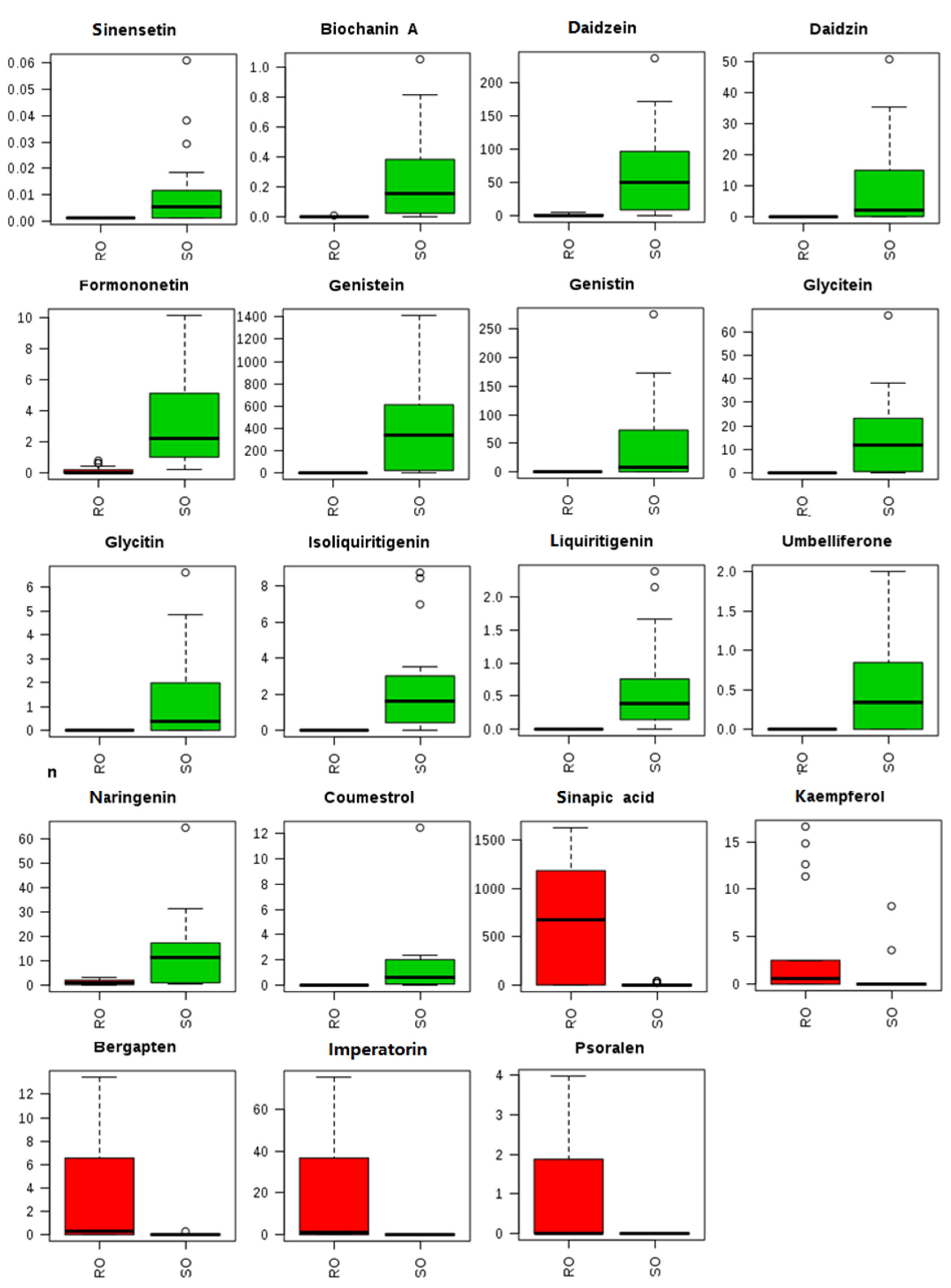
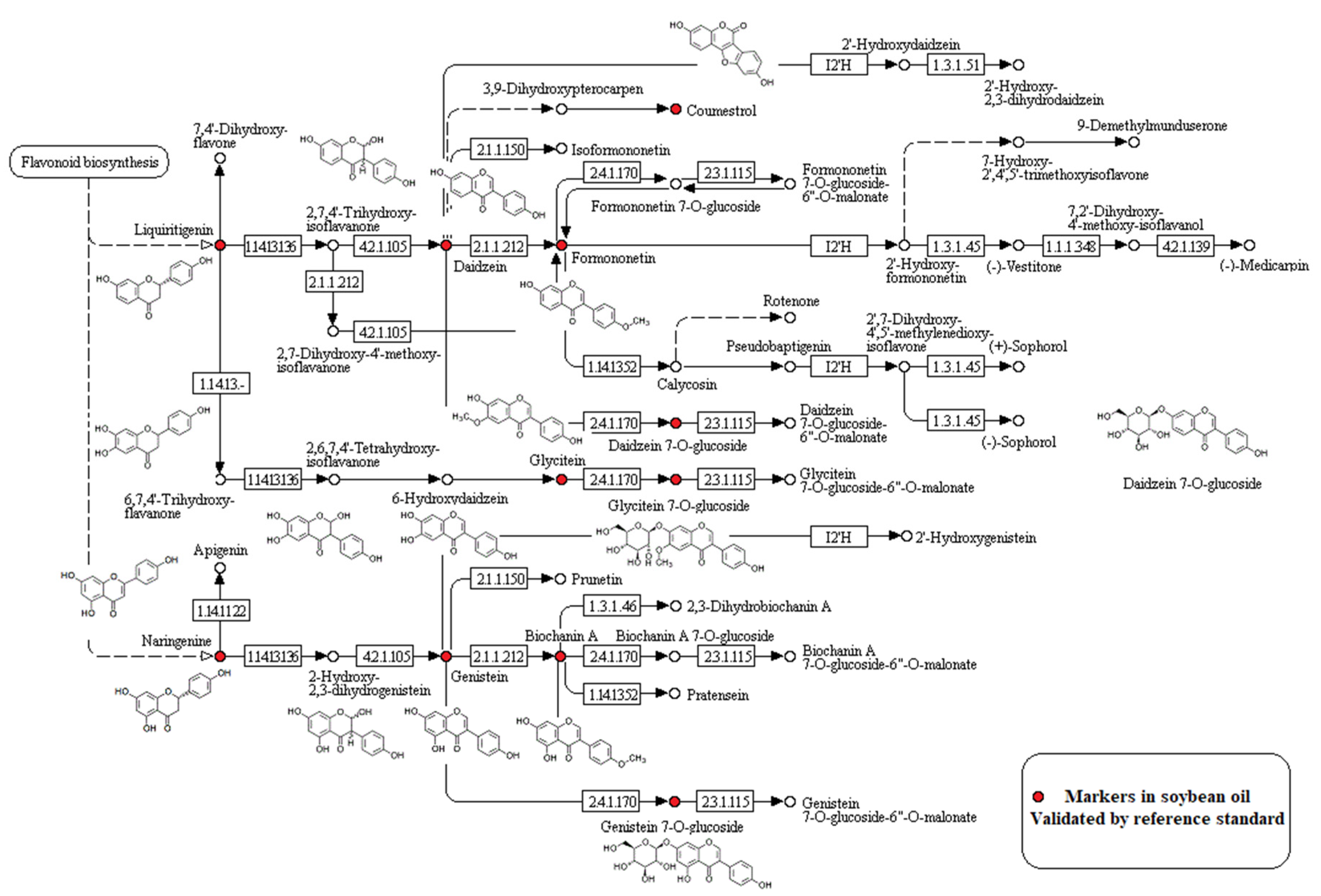
3.3. Oil Sample Analysis and Marker Screening
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moore:, J.C.; Spink, J.; Lipp, M. Development and application of a database of food ingredient fraud and economically motivated adulteration from 1980 to 2010. J. Food Sci. 2012, 77, R118–R126. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.D.; Gunning, Y.; Rigby, N.M.; Philo, M.; Kemsley, E.K. Meat Authentication via Multiple Reaction Monitoring Mass Spectrometry of Myoglobin Peptides. Anal. Chem. 2015, 87, 10315–10322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, R.S.; Morrissey, M.T. DNA-Based methods for the identification of commercial fish and seafood species. Compr. Rev. Food Sci. Food Saf. 2008, 7, 280–295. [Google Scholar] [CrossRef]
- Zhang, L.X.; Li, P.W.; Sun, X.M.; Wang, X.F.; Xu, B.C.; Wang, X.P.; Ma, F.; Zhang, Q.; Ding, X.X. Classification and Adulteration Detection of Vegetable Oils Based on Fatty Acid Profiles. J. Agric. Food Chem. 2014, 62, 8745–8751. [Google Scholar] [CrossRef] [PubMed]
- Karoui, R.; Baerdemaeker, J.D. A review of the analytical methods coupled with chemometric tools for the determination of the quality and identity of dairy products. Food Chem. 2007, 102, 621–640. [Google Scholar] [CrossRef]
- Gerhardt, N.; Birkenmeier, M.; Schwolow, S.; Rohn, S.; Weller, P. Volatile-Compound Fingerprinting by Headspace-Gas-Chromatography Ion-Mobility Spectrometry (HS-GC-IMS) as a Benchtop Alternative to 1H NMR Profiling for Assessment of the Authenticity of Honey. Anal. Chem. 2018, 90, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, R.; Morales, M.T.; Alonso, V. Authentication of European Virgin Olive Oils by Their Chemical Compounds, Sensory Attributes, and Consumers’ Attitudes. J. Agric. Food Chem. 1997, 45, 1076–1083. [Google Scholar] [CrossRef]
- Lockley, A.K.; Bardsley, R.G. DNA-based methods for food authentication. Trends Food Sci. Technol. 2000, 11, 67–77. [Google Scholar] [CrossRef]
- Chan, T.W.D.; But, P.P.H.; Cheng, S.W.; Kwok, I.M.Y.; Lau, F.W.; Xu, H.X. Differentiation and authentication of Panax ginseng, Panax quinquefolius, and ginseng products by using HPLC/MS. Anal. Chem. 2000, 72, 1281–1287. [Google Scholar] [CrossRef]
- Aparicio, R.; Aparicio-Ruíz, R. Authentication of vegetable oils by chromatographic techniques. J. Chromatogr. A 2000, 881, 93–104. [Google Scholar] [CrossRef]
- Lerma-García, M.J.; Ramis-Ramos, G.; Herrero-Martínez, J.M.; Simó-Alfonso, E.F. Authentication of extra virgin olive oils by Fourier-transform infrared spectroscopy. Food Chem. 2010, 118, 78–83. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, P.; Yu, L.L.; Harrington, P.B. Authentication of Organically and Conventionally Grown Basils by Gas Chromatography/Mass Spectrometry Chemical Profiles. Anal. Chem. 2013, 85, 2945–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodcock, T.; Downey, G.; Donnell, C.P.; O’Donnell, C.P. Confirmation of Declared Provenance of European Extra Virgin Olive Oil Samples by NIR Spectroscopy. J. Agric. Food Chem. 2008, 56, 11520–11525. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, D.; Smyth, H.E.; Gishen, M. Feasibility Study on the Use of Visible and Near-Infrared Spectroscopy Together with Chemometrics To Discriminate between Commercial White Wines of Different Varietal Origins. J. Agric. Food Chem. 2003, 51, 7703–7708. [Google Scholar] [CrossRef]
- Sikorska, E.; Górecki, T.; Khmelinskii, I.V.; Sikorski, M.; Kozioł, J. Classification of edible oils using synchronous scanning fluorescence spectroscopy. Food Chem. 2005, 89, 217–225. [Google Scholar] [CrossRef]
- Eliasson, C.; Matousek, P. Noninvasive Authentication of Pharmaceutical Products through Packaging Using Spatially Offset Raman Spectroscopy. Anal. Chem. 2007, 79, 1696–1701. [Google Scholar] [CrossRef]
- Sacchi, R.; Mannina, L.; Fiordiponti, P.; Barone, P.; Paolillo, L.; Patumi, M.; Segre, A. Characterization of Italian Extra Virgin Olive Oils Using 1H-NMR Spectroscopy. J. Agric. Food Chem. 1998, 46, 3947–3951. [Google Scholar] [CrossRef]
- Zhang, L.X.; Shuai, Q.; Li, P.W.; Zhang, Q.; Ma, F.; Zhang, W.; Ding, X.X. Ion mobility spectrometry fingerprints: A rapid detection technology for adulteration of sesame oil. Food Chem. 2016, 192, 60–66. [Google Scholar] [CrossRef]
- Shuai, Q.; Zhang, L.X.; Li, P.W.; Zhang, Q.; Wang, X.P.; Ding, X.X.; Zhang, W. Rapid adulteration detection for flaxseed oil using ion mobility spectrometry and chemometric methods. Anal. Methods 2014, 6, 9575–9580. [Google Scholar] [CrossRef]
- Cosio, M.S.; Ballabio, D.; Benedetti, S.; Gigliotti, C. Geographical origin and authentication of extra virgin olive oils by an electronic nose in combination with artificial neural networks. Anal. Chim. Acta 2006, 567, 202–210. [Google Scholar] [CrossRef]
- Fasciotti, M.; Netto, A.D.P. Optimization and application of methods of triacylglycerol evaluation for characterization of olive oil adulteration by soybean oil with HPLC-APCI-MS-MS. Talanta 2010, 81, 1116–1125. [Google Scholar] [CrossRef]
- Sun, X.M.; Zhang, L.X.; Li, P.W.; Xu, B.C.; Ma, F.; Zhang, Q.; Zhang, W. Fatty acid profiles based adulteration detection for flaxseed oil by gas chromatography mass spectrometry. LWT-Food Sci. Technol. 2015, 63, 430–436. [Google Scholar] [CrossRef]
- Zhang, L.X.; Huang, X.R.; Li, P.W.; Na, W.; Jiang, J.; Mao, J.; Ding, X.X.; Zhang, Q. Multivariate adulteration detection for sesame oil. Chemometr. Intell. Lab. 2017, 161, 147–150. [Google Scholar] [CrossRef]
- Zhang, L.X.; Yuan, Z.; Li, P.W.; Wang, X.F.; Mao, J.; Zhang, Q.; Hu, C.D. Targeted multivariate adulteration detection based on fatty acid profiles and Monte Carlo one-class partial least squares. Chemometr. Intell. Lab. 2017, 169, 94–99. [Google Scholar] [CrossRef]
- Xu, B.C.; Zhang, L.X.; Ma, F.; Zhang, W.; Wang, X.P.; Zhang, Q.; Luo, D.L.; Ma, H.Y.; Li, P.W. Determination of free steroidal compounds in vegetable oils by comprehensive two-dimensional gas chromatography coupled to time-of-flight mass spectrometry. Food Chem. 2018, 245, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, L.X.; Li, P.W.; Wang, X.P.; Zhang, Q.; Xu, B.C.; Sun, X.M.; Ma, F.; Ding, X.X. Characterization of volatile components in four vegetable oils by headspace two-dimensional comprehensive chromatography time-of-flight mass spectrometry. Talanta 2014, 129, 629–635. [Google Scholar] [CrossRef]
- Rahmati, S.; Julkapli, N.M.; Yehye, W.A.; Basirun, W.J. Identification of meat origin in food products—A review. Food Control 2016, 68, 379–390. [Google Scholar] [CrossRef]
- Gryson, N.; Ronsse, F.; Messens, K.; Loose, M.D.; Verleyen, T.; Dewettinck, K. Detection of DNA during the refining of soybean oil. J. Am. Oil Chem. Soc. 2002, 79, 171–174. [Google Scholar] [CrossRef]
- Damirchi, S.A.; Savage, G.P.; Dutta, P.C. Sterol fractions in hazelnut and virgin olive oils and 4,4′-dimethylsterols as possible markers for detection of adulteration of virgin olive oil. J. Am. Oil Chem. Soc. 2005, 82, 717–725. [Google Scholar] [CrossRef]
- Flores, G.; del Castillo, M.L.R.; Blanch, G.P.; Herraiz, M. Detection of the adulteration of olive oils by solid phase microextraction and multidimensional gas chromatography. Food Chem. 2006, 97, 336–342. [Google Scholar] [CrossRef]
- Jabeur, H.; Zribi, A.; Makni, J.; Rebai, A.; Abdelhedi, R.; Bouaziz, M. Detection of Chemlali Extra-Virgin Olive Oil Adulteration Mixed with Soybean Oil, Corn Oil, and Sunflower Oil by Using GC and HPLC. J. Agric. Food Chem. 2014, 62, 4893–4904. [Google Scholar] [CrossRef] [PubMed]
- Dulf, F.V.; Bele, C.; Unguresan, M.L.; Socaciu, C. Qualitative and quantitative markers to identify the quality and adulteration of olive oil with rapeseed oil. Bull. Univ. Agric. Sci. Vet. Med. Cluj-Napoca Agric. 2007, 63, 575–580. [Google Scholar]
- Bueno, M.J.M.; Díaz-Galiano, F.J.; Rajski, Ł.; Cutillas, V.; Fernández-Alba, A.R. A non-targeted metabolomic approach to identify food markers to support discrimination between organic and conventional tomato crops. J. Chromatogr. A 2018, 1546, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Gil-Solsona, R.; Raro, M.; Sales, C.; Lacalle, L.; Díaz, R.; Ibáñez, M.; Beltran, J.; Sancho, J.V.; Hernández, F.J. Metabolomic approach for Extra virgin olive oil origin discrimination making use of ultra-high performance liquid chromatography-Quadrupole time-of-flight mass spectrometry. Food Control 2016, 70, 350–359. [Google Scholar] [CrossRef]
- Wang, H.J.; Murphy, P.A. Isoflavone content in commercial soybean foods. J. Agric. Food Chem. 1994, 42, 1666–1673. [Google Scholar] [CrossRef]
- Zhao, X.; Ma, F.; Li, P.W.; Li, G.M.; Zhang, L.X.; Zhang, Q.; Zhang, W.; Wang, X.P. Simultaneous determination of isoflavones and resveratrols for adulteration detection of soybean and peanut oils by mixed-mode SPE LC-MS/MS. Food Chem. 2015, 176, 465–471. [Google Scholar] [CrossRef]
- Pizarro, M.L.; Becerra, M.; Sayago, A.; Beltrán, M.; Beltrán, R. Comparison of Different Extraction Methods to Determine Phenolic Compounds in Virgin Olive Oil. Food Anal. Methods 2013, 6, 123–132. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffi, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Lendoiro, E.; Quintela, O.; de Castro, A.; Cruz, A.; Lopez-Rivadulla, M.; Concheiro, M. Target screening and confirmation of 35 licit and illicit drugs and metabolites in hair by LC-MSMS. Forensic Sci. Int. 2012, 217, 207–215. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular datasets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, R.R.; Dorrestein, P.C.; Quinn, R.A. Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. USA 2015, 112, 12549–12550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI: FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dou, X.; Zhang, L.; Wang, X.; Yang, R.; Wang, X.; Ma, F.; Yu, L.; Mao, J.; Li, H.; Wang, X.; et al. Identification and Validation of Metabolic Markers for Adulteration Detection of Edible Oils Using Metabolic Networks. Metabolites 2020, 10, 85. https://doi.org/10.3390/metabo10030085
Dou X, Zhang L, Wang X, Yang R, Wang X, Ma F, Yu L, Mao J, Li H, Wang X, et al. Identification and Validation of Metabolic Markers for Adulteration Detection of Edible Oils Using Metabolic Networks. Metabolites. 2020; 10(3):85. https://doi.org/10.3390/metabo10030085
Chicago/Turabian StyleDou, Xinjing, Liangxiao Zhang, Xiao Wang, Ruinan Yang, Xuefang Wang, Fei Ma, Li Yu, Jin Mao, Hui Li, Xiupin Wang, and et al. 2020. "Identification and Validation of Metabolic Markers for Adulteration Detection of Edible Oils Using Metabolic Networks" Metabolites 10, no. 3: 85. https://doi.org/10.3390/metabo10030085
APA StyleDou, X., Zhang, L., Wang, X., Yang, R., Wang, X., Ma, F., Yu, L., Mao, J., Li, H., Wang, X., & Li, P. (2020). Identification and Validation of Metabolic Markers for Adulteration Detection of Edible Oils Using Metabolic Networks. Metabolites, 10(3), 85. https://doi.org/10.3390/metabo10030085