Current Concepts in Pharmacometabolomics, Biomarker Discovery, and Precision Medicine



Abstract
:1. Introduction
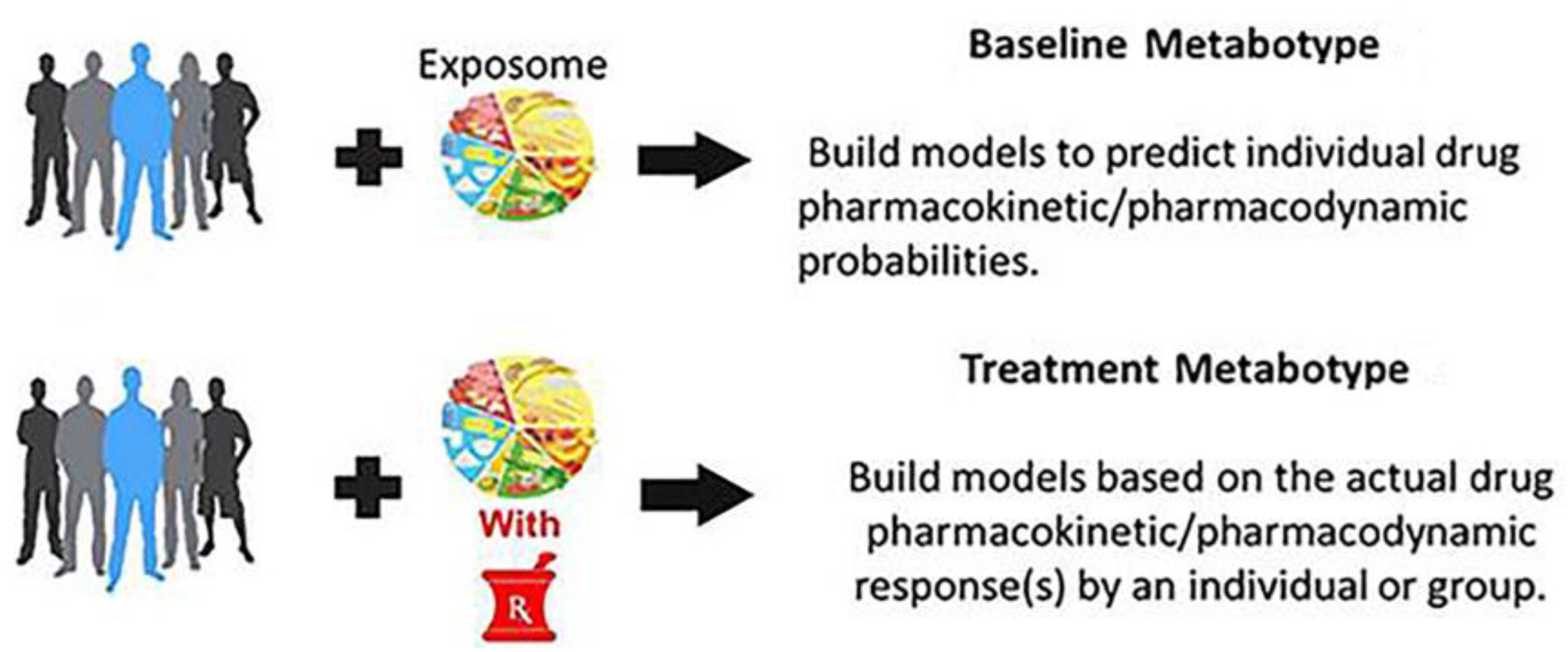
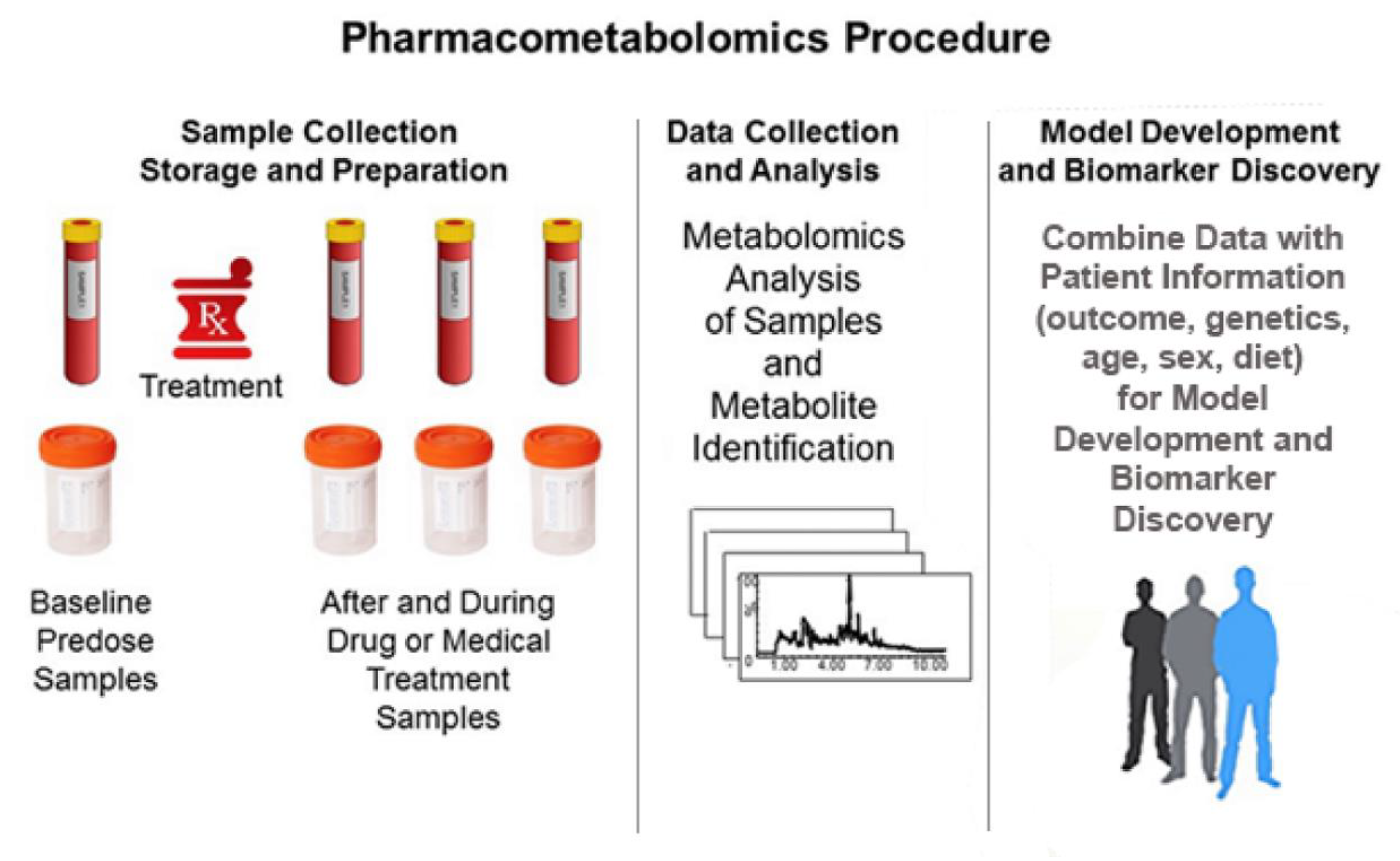
2. Pharmacometabolomics and “Metabotypes”
2.1. PMx Data Alone
2.2. PMx Data and PGx Data
2.3. PMx Data and Gut Flora Metagenomics Data
2.4. PMx Data and Multi-Scale Omics Data
3. Gut Microflora Metagenome and Drug Metabolism
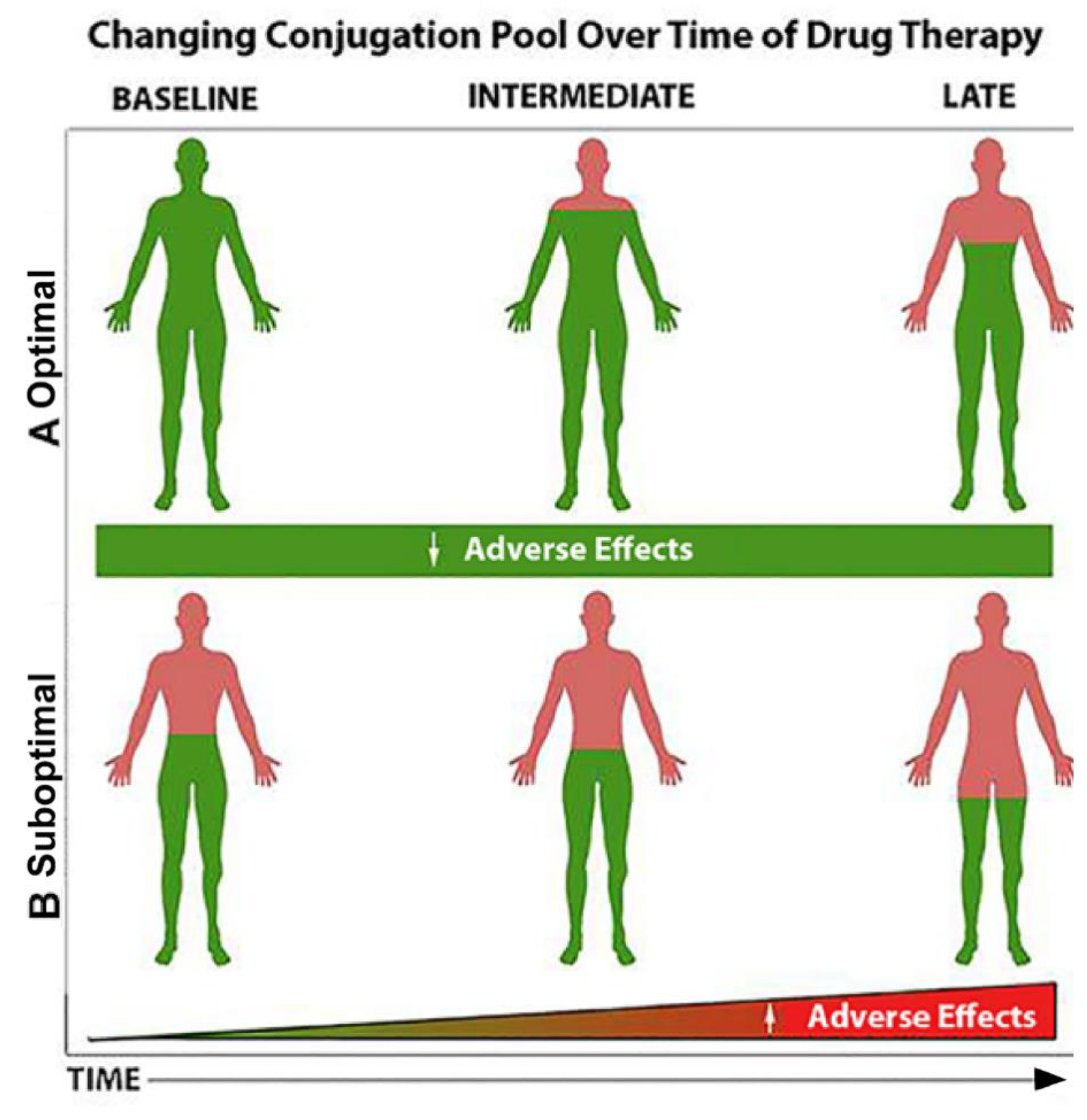
4. Where We are Today and the Future of PMx
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wilkinson, G.R. Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 2005, 352, 2211–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, J.; Pomeranz, B.H.; Corey, P.N. Incidence of adverse drug reactions in hospitalized patientsa meta-analysis of prospective studies. JAMA 1998, 279, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, G.; Mohorn, P.; Yacoub, K.; May, D.W. Adverse drug reaction deaths reported in United States vital statistics, 1999–2006. Ann. Pharmacother. 2012, 46, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Mancinelli, L.; Cronin, M.; Sadée, W. Pharmacogenomics: The promise of personalized medicine. AAPS Pharmsci. 2000, 2, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Pirmohamed, M. Pharmacogenetics and pharmacogenomics. Br. J. Clin. Pharmacol. 2001, 52, 345–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, A.K. Genome-wide association studies in pharmacogenomics. Nat. Rev. Genet. 2010, 11, 241–246. [Google Scholar] [CrossRef]
- Roden, D.M.; Wilke, R.A.; Kroemer, H.K.; Stein, C.M. Pharmacogenomics. Circulation 2011, 123, 1661–1670. [Google Scholar] [CrossRef] [Green Version]
- Table of Pharmacogenomic Biomarkers in Drug Labelin. Available online: https://www.fda.gov/drugs/science-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling (accessed on 1 March 2020).
- Ventola, C.L. Role of pharmacogenomic biomarkers in predicting and improving drug response: Part 1: The clinical significance of pharmacogenetic variants. P T A Peer-Rev. J. Formul. Manag. 2013, 38, 545–560. [Google Scholar]
- Clayton, T.A.; Lindon, J.C.; Cloarec, O.; Antti, H.; Charuel, C.; Hanton, G.; Provost, J.P.; Le Net, J.L.; Baker, D.; Walley, R.J.; et al. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature 2006, 440, 1073–1077. [Google Scholar] [CrossRef]
- Lindon, J.C.; Nicholson, J.K.; Holmes, E.; Antti, H.; Bollard, M.E.; Keun, H.; Beckonert, O.; Ebbels, T.M.; Reily, M.D.; Robertson, D.; et al. Contemporary issues in toxicology the role of metabonomics in toxicology and its evaluation by the COMET project. Toxicol. Appl. Pharmacol. 2003, 187, 137–146. [Google Scholar] [CrossRef]
- Ebbels, T.M.; Keun, H.C.; Beckonert, O.P.; Bollard, M.E.; Lindon, J.C.; Holmes, E.; Nicholson, J.K. Prediction and classification of drug toxicity using probabilistic modeling of temporal metabolic data: The consortium on metabonomic toxicology screening approach. J. Proteome Res. 2007, 6, 4407–4422. [Google Scholar] [CrossRef] [PubMed]
- Kaddurah-Daouk, R.; Kristal, B.S.; Weinshilboum, R.M. Metabolomics: A global biochemical approach to drug response and disease. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 653–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaddurah-Daouk, R.; McEvoy, J.; Baillie, R.A.; Lee, D.; Yao, J.K.; Doraiswamy, P.M.; Krishnan, K.R. Metabolomic mapping of atypical antipsychotic effects in schizophrenia. Mol. Psychiatry 2007, 12, 934–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pharmacometabolomics Research Network. Available online: http://pharmacometabolomics.duhs.duke.edu/ (accessed on 1 March 2020).
- Pharmacogenomics Research Network. Available online: https://www.pgrn.org (accessed on 1 March 2020).
- Kaddurah-Daouk, R.; Weinshilboum, R.M.; Network, P.R. Pharmacometabolomics: Implications for clinical pharmacology and systems pharmacology. Clin. Pharmacol. Ther. 2014, 95, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Kaddurah-Daouk, R.; Weinshilboum, R. Pharmacometabolomics research network. metabolomic signatures for drug response phenotypes: Pharmacometabolomics enables precision medicine. Clin. Pharmacol. Ther. 2015, 98, 71–75. [Google Scholar]
- Kaddurah-Daouk, R.; Baillie, R.A.; Zhu, H.; Zeng, Z.B.; Wiest, M.M.; Nguyen, U.T.; Wojnoonski, K.; Watkins, S.M.; Trupp, M.; Krauss, R.M. Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS ONE 2011, 6, e25482. [Google Scholar] [CrossRef] [Green Version]
- Kaddurah-Daouk, R.; Hankemeier, T.; Scholl, E.H.; Baillie, R.; Harms, A.; Stage, C.; Dalhoff, K.P.; Jűrgens, G.; Taboureau, O.; Nzabonimpa, G.S.; et al. Pharmacometabolomics Informs About Pharmacokinetic Profile of Methylphenidate. CPT Pharmacomet. Syst. Pharm. 2018, 7, 525–533. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Frye, R.F.; Zhu, H.; Gong, Y.; Boyle, S.; Churchill, E.; Cooper-Dehoff, R.M.; Beitelshees, A.L.; Chapman, A.B.; Fiehn, O.; et al. Pharmacometabolomics reveals racial differences in response to atenolol treatment. PLoS ONE 2013, 8, e57639. [Google Scholar] [CrossRef] [Green Version]
- Elbadawi-Sidhu, M.; Baillie, R.A.; Zhu, H.; Chen, Y.D.; Goodarzi, M.O.; Rotter, J.I.; Krauss, R.M.; Fiehn, O.; Kaddurah-Daouk, R. Pharmacometabolomic signature links simvastatin therapy and insulin resistance. Metab. Off. J. Metab. Soc. 2017, 13, 11. [Google Scholar] [CrossRef] [Green Version]
- Wilson, I.D. Drugs, bugs, and personalized medicine: Pharmacometabonomics enters the ring. Proc. Natl. Acad. Sci. USA 2009, 106, 14187–14188. [Google Scholar] [CrossRef] [Green Version]
- Everett, J.R. Pharmacometabonomics in humans: A new tool for personalized medicine. Pharmacogenomics 2015, 16, 737–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beger, R.D.; Dunn, W.; Schmidt, M.A.; Gross, S.S.; Kirwan, J.A.; Cascante, M.; Brennan, L.; Wishart, D.S.; Oresic, M.; Hankemeier, T.; et al. Metabolomics enables precision medicine: “A White Paper, Community Perspective”. Metab. Off. J. Metab. Soc. 2016, 12, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattray, N.J.W.; Daouk, R.K. Pharmacometabolomics and precision medicine special issue editorial. Metabolomics 2017, 13, 59. [Google Scholar] [CrossRef] [Green Version]
- Jain, D.; Ahmad, T.; Cairo, M.; Aronow, W. Cardiotoxicity of cancer chemotherapy: Identification, prevention and treatment. Ann. Transl. Med. 2017, 5, 348. [Google Scholar] [CrossRef] [Green Version]
- Mosedale, M.; Watkins, P.B. Drug-induced liver injury: Advances in mechanistic understanding that will inform risk management. Clin. Pharmacol. Ther. 2017, 101, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasool, M.; Malik, A.; Naseer, M.I.; Manan, A.; Ansari, S.A.; Begum, I.; Qazi, M.H.; Pushparaj, P.N.; Abuzenadah, A.M.; Al-Qahtani, M.H.; et al. The role of epigenetics in personalized medicine: Challenges and opportunities. BMC Med Genom. 2015, 8, S5. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.K. Global systems biology, personalized medicine and molecular epidemiology. Mol. Syst. Biol. 2006, 2, 52. [Google Scholar] [CrossRef]
- Drug Development Tool Qualification Process. Available online: https://www.fda.gov/drugs/drug-development-tool-ddt-qualification-programs/drug-development-tool-qualification-process-transparency-provisions (accessed on 1 March 2020).
- Leptak, C.; Menetski, J.P.; Wagner, J.A.; Aubrecht, J.; Brady, L.; Brumfield, M.; Chin, W.W.; Hoffmann, S.; Kelloff, G.; Lavezzari, G.; et al. What evidence do we need for biomarker qualification? Sci. Transl. Med. 2017, 9, eaal4599. [Google Scholar] [CrossRef]
- Resources for Biomarker Requestors. Available online: https://www.fda.gov/drugs/cder-biomarker-qualification-program/resources-biomarker-requestors (accessed on 1 March 2020).
- Context. Available online: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/BiomarkerQualificationProgram/ucm535395.htm (accessed on 1 March 2020).
- BEST (Biomarkers, EndpointS, and other Tools) Resource. Available online: https://www.ncbi.nlm.nih.gov/books/NBK338448/?report=reader (accessed on 1 March 2020).
- Wishart, D.S.; Xia, J. MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar]
- Pazos, F.; Chagoyen, M. Tools for the functional interpretation of metabolomic experiments. Brief. Bioinform. 2012, 14, 737–744. [Google Scholar]
- Everett, J.R.; Loo, R.L.; Pullen, F.S. Pharmacometabonomics and personalized medicine. Ann. Clin. Biochem. 2013, 50, 523–545. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.P.; Yerges-Armstrong, L.M.; Ellero-Simatos, S.; Georgiades, A.; Kaddurah-Daouk, R.; Hankemeier, T. Integration of pharmacometabolomic and pharmacogenomic approaches reveals novel insights into antiplatelet therapy. Clin. Pharmacol. Ther. 2013, 94, 570–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotroff, D.M.; Corum, D.G.; Motsinger-Reif, A.; Fiehn, O.; Bottrel, N.; Drevets, W.C.; Singh, J.; Salvadore, G.; Kaddurah-Daouk, R. Metabolomic signatures of drug response phenotypes for ketamine and esketamine in subjects with refractory major depressive disorder: New mechanistic insights for rapid acting antidepressants. Transl. Psychiatry 2016, 6, e894. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Ahmed, A.T.; Arnold, M.; Liu, D.; Luo, C.; Zhu, H.; Mahmoudiandehkordi, S.; Neavin, D.; Louie, G.; Dunlop, B.W.; et al. Metabolomic signature of exposure and response to citalopram/escitalopram in depressed outpatients. Transl. Psychiatry 2019, 9, 173. [Google Scholar] [CrossRef] [Green Version]
- Beger, R.D.; Flynn, T.J. Pharmacometabolomics in drug safety and drug-exposome interactions. Metabolomics 2016, 12, 123. [Google Scholar] [CrossRef]
- Palleria, C.; Di Paolo, A.; Giofrè, C.; Caglioti, C.; Leuzzi, G.; Siniscalchi, A.; De Sarro, G.; Gallelli, L. Pharmacokinetic drug-drug interaction and their implication in clinical management. J. Res. Med Sci. Off. J. Isfahan Univ. Med Sci. 2013, 18, 601–610. [Google Scholar]
- Bushra, R.; Aslam, N.; Khan, A.Y. Food-drug interactions. Oman Med. J. 2011, 26, 77–83. [Google Scholar] [CrossRef]
- Swanson, H.I. Drug Metabolism by the host and gut microbiota: A partnership or rivalry? Drug Metab. Dispos. Biol. Fate Chem. 2015, 43, 1499–1504. [Google Scholar] [CrossRef] [Green Version]
- Holbrook, A.M.; Pereira, J.A.; Labiris, R.; McDonald, H.; Douketis, J.D.; Crowther, M.; Wells, P.S. Systematic overview of warfarin and its drug and food interactions. JAMA Intern. Med. 2005, 165, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Kamlage, B.; Wagner-Golbs, A.; Maisha, M.; Sun, J.; Schnackenberg, L.K.; Pence, L.; Schmitt, T.C.; Daniels, J.R.; Rogstad, S.; et al. An integrated analysis of metabolites, peptides, and inflammation biomarkers for assessment of preanalytical variability of human plasma. J. Proteome Res. 2019, 18, 2411–2421. [Google Scholar] [CrossRef]
- Szymańska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics 2012, 8, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults. Circulation 2014, 129, S1–S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.; Ewy, W.; Corrigan, B.W.; Ouellet, D.; Hermann, D.; Kowalski, K.G.; Lockwood, P.; Koup, J.R.; Donevan, S.; El-Kattan, A.; et al. How modeling and simulation have enhanced decision making in new drug development. J. Pharmacokinet. Pharmacodyn. 2005, 32, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.; Zhou, Z.; Zhou, J.; Chen, S.Q. Pharmacogenomics of drug metabolizing enzymes and transporters: Relevance to precision medicine. Genom. Proteom. Bioinform. 2016, 14, 298–313. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.M.; Park, B.K.; Pirmohamed, M. Parsing interindividual drug variability: An emerging role for systems pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 221–241. [Google Scholar] [CrossRef]
- Kantae, V.; Krekels, E.H.; Van Esdonk, M.J.; Lindenburg, P.; Harms, A.C.; Knibbe, C.A.; Van der Graaf, P.H.; Hankemeier, T. Integration of pharmacometabolomics with pharmacokinetics and pharmacodynamics: Towards personalized drug therapy. Metab. Off. J. Metab. Soc. 2017, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Shuker, N.; Shuker, L.; van Rosmalen, J.; Roodnat, J.I.; Borra, L.C.; Weimar, W.; Hesselink, D.A.; van Gelder, T. A high intrapatient variability in tacrolimus exposure is associated with poor long-term outcome of kidney transplantation. Transpl. Int. 2016, 29, 1158–1167. [Google Scholar] [CrossRef]
- Goldsmith, P.M.; Bottomley, M.J.; Okechukwu, O.; Ross, V.C.; Ghita, R.; Wandless, D.; Falconer, S.J.; Papachristos, S.; Nash, P.; Androshchuk, V.; et al. Impact of intrapatient variability (IPV) in tacrolimus trough levels on long-term renal transplant function: Multicentre collaborative retrospective cohort study protocol. BMJ Open 2017, 7, e016144. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, D.; Gervasoni, C.; Meraviglia, P.; Landonio, S.; Fucile, S.; Cozzi, V.; Baldelli, S.; Pellegrini, M.; Galli, M.; Clementi, E. Inter- and intra-patient variability of raltegravir pharmacokinetics in HIV-1-infected subjects. J. Antimicrob. Chemother. 2011, 67, 460–464. [Google Scholar] [CrossRef] [Green Version]
- Siccardi, M.; D’Avolio, A.; Rodriguez-Novoa, S.; Cuenca, L.; Simiele, M.; Baietto, L.; Calcagno, A.; Moss, D.; Bonora, S.; Soriano, V.; et al. Intrapatient and interpatient pharmacokinetic variability of raltegravir in the clinical setting. Ther. Drug Monit. 2012, 34, 232–235. [Google Scholar] [CrossRef]
- MetaboLights. Available online: https://www.ebi.ac.uk/metabolights (accessed on 1 March 2020).
- Metabolomics Workbench. Available online: http://www.metabolomicsworkbench.org/ (accessed on 1 March 2020).
- COnsortium of METabolomics Studies. Available online: https://epi.grants.cancer.gov/comets/ (accessed on 1 March 2020).
- Clinical Trials. Available online: https://clinicaltrials.gov/ (accessed on 3 September 2019).
- van Roekel, E.H.; Loftfield, E.; Kelly, R.S.; Zeleznik, O.A.; Zanetti, K.A. Metabolomics in epidemiologic research: Challenges and opportunities for early-career epidemiologists. Metabolomics 2019, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, L.G.; Inouye, M. Metabolomics in epidemiology: From metabolite concentrations to integrative reaction networks. Int. J. Epidemiol. 2016, 45, 1319–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, T.A.; Baker, D.; Lindon, J.C.; Everett, J.R.; Nicholson, J.K. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc. Natl. Acad. Sci. USA 2009, 106, 14728–14733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhrez, K.; Benz-de Bretagne, I.; Nadal-Desbarats, L.; Blasco, H.; Gyan, E.; Choquet, S.; Montigny, F.; Emond, P.; Barin-Le Guellec, C. Endogenous metabolites that are substrates of organic anion transporter’s (OATs) predict methotrexate clearance. Pharmacol. Res. 2017, 118, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Krausz, K.W.; Idle, J.R.; Gonzalez, F.J. Identification of novel toxicity-associated metabolites by metabolomics and mass isotopomer analysis of acetaminophen metabolism in wild-type and Cyp2e1-null mice. J. Biol. Chem. 2008, 283, 4543–4559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Yan, K.; Pence, L.; Simpson, P.M.; Gill, P.; Letzig, L.G.; Beger, R.D.; Sullivan, J.E.; Kearns, G.L.; Reed, M.D.; et al. Targeted liquid chromatography–mass spectrometry analysis of serum acylcarnitines in acetaminophen toxicity in children. Biomark. Med. 2014, 8, 147–159. [Google Scholar] [CrossRef] [Green Version]
- McEvoy, J.; Baillie, R.A.; Zhu, H.; Buckley, P.; Keshavan, M.S.; Nasrallah, H.A.; Dougherty, G.G.; Yao, J.K.; Kaddurah-Daouk, R. Lipidomics reveals early metabolic changes in subjects with schizophrenia: Effects of atypical antipsychotics. PLoS ONE 2013, 8, e68717. [Google Scholar] [CrossRef] [Green Version]
- Kaddurah-Daouk, R.; Baillie, R.A.; Zhu, H.; Zeng, Z.B.; Wiest, M.M.; Nguyen, U.T.; Watkins, S.M.; Krauss, R.M. Lipidomic analysis of variation in response to simvastatin in the Cholesterol and Pharmacogenetics Study. Metab. Off. J. Metab. Soc. 2010, 6, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Trupp, M.; Zhu, H.; Wikoff, W.R.; Baillie, R.A.; Zeng, Z.B.; Karp, P.D.; Fiehn, O.; Krauss, R.M.; Kaddurah-Daouk, R. Metabolomics reveals amino acids contribute to variation in response to simvastatin treatment. PLoS ONE 2012, 7, e38386. [Google Scholar] [CrossRef] [Green Version]
- Cooper-DeHoff, R.M.; Hou, W.; Weng, L.; Baillie, R.A.; Beitelshees, A.L.; Gong, Y.; Shahin, M.H.; Turner, S.T.; Chapman, A.; Gums, J.G.; et al. Is diabetes mellitus-linked amino acid signature associated with β-blocker-induced impaired fasting glucose? Circ. Cardiovasc. Genet. 2014, 7, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Rotroff, D.M.; Shahin, M.H.; Gurley, S.B.; Zhu, H.; Motsinger-Reif, A.; Meisner, M.; Beitelshees, A.L.; Fiehn, O.; Johnson, J.A.; Elbadawi-Sidhu, M.; et al. Pharmacometabolomic assessments of atenolol and hydrochlorothiazide treatment reveal novel drug response phenotypes. CPT Pharmacomet. Syst. Pharmacol. 2015, 4, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Backshall, A.; Sharma, R.; Clarke, S.J.; Keun, H.C. Pharmacometabonomic profiling as a predictor of toxicity in patients with inoperable colorectal cancer treated with capecitabine. Clin. Cancer Res. 2011, 17, 3019–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miolo, G.; Muraro, E.; Caruso, D.; Crivellari, D.; Ash, A.; Scalone, S.; Lombardi, D.; Rizzolio, F.; Giordano, A.; Corona, G. Pharmacometabolomics study identifies circulating spermidine and tryptophan as potential biomarkers associated with the complete pathological response to trastuzumab-paclitaxel neoadjuvant therapy in HER-2 positive breast cancer. Oncotarget 2016, 7, 39809–39822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerges-Armstrong, L.M.; Ellero-Simatos, S.; Georgiades, A.; Zhu, H.; Lewis, J.P.; Horenstein, R.B.; Beitelshees, A.L.; Dane, A.; Reijmers, T.; Hankemeier, T.; et al. Purine pathway implicated in mechanism of resistance to aspirin therapy: Pharmacometabolomics-informed pharmacogenomics. Clin. Pharmacol. Ther. 2013, 94, 525–532. [Google Scholar] [CrossRef]
- Ellero-Simatos, S.; Lewis, J.P.; Georgiades, A.; Yerges-Armstrong, L.M.; Beitelshees, A.L.; Horenstein, R.B.; Dane, A.; Harms, A.C.; Ramaker, R.; Vreeken, R.J.; et al. Pharmacometabolomics reveals that serotonin is implicated in aspirin response variability. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, 125. [Google Scholar] [CrossRef]
- Lanznaster, D.; de Assis, D.R.; Corcia, P.; Pradat, P.F.; Blasco, H. Metabolomics biomarkers: A strategy toward therapeutics improvement in ALS. Front. Neurol. 2018, 9, 1126. [Google Scholar] [CrossRef] [Green Version]
- de Jager, J.; Kooy, A.; Lehert, P.; Wulffelé, M.G.; Van der Kolk, J.; Bets, D.; Verburg, J.; Donker, A.J.; Stehouwer, C.D. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: Randomised placebo controlled trial. BMJ (Clin. Res. Ed.), 2010; 340, c2181. [Google Scholar]
- Reinstatler, L.; Qi, Y.P.; Williamson, R.S.; Garn, J.V.; Oakley, G.P., Jr. Association of biochemical B₁₂ deficiency with metformin therapy and vitamin B₁₂ supplements: The national health and nutrition examination survey, 1999–2006. Diabetes Care 2012, 35, 327–333. [Google Scholar] [CrossRef] [Green Version]
- Ko, S.H.; Ko, S.H.; Ahn, Y.B.; Song, K.H.; Han, K.D.; Park, Y.M.; Ko, S.H.; Kim, H.S. Association of vitamin B12 deficiency and metformin use in patients with type 2 diabetes. J. Korean Med Sci. 2014, 29, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Aroda, V.R.; Edelstein, S.L.; Goldberg, R.B.; Knowler, W.C.; Marcovina, S.M.; Orchard, T.J.; Bray, G.A.; Schade, D.S.; Temprosa, M.G.; White, N.H.; et al. Long-term metformin use and vitamin B12 deficiency in the diabetes prevention program outcomes study. J. Clin. Endocrinol. Metab. 2016, 101, 1754–1761. [Google Scholar] [CrossRef]
- Ahmed, M.A.; Muntingh, G.; Rheeder, P. Vitamin B12 deficiency in metformin-treated type-2 diabetes patients, prevalence and association with peripheral neuropathy. BMC Pharmacol. Toxicol. 2016, 17, 44. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, M.; Rincon, O.; Saavedra, G.; Moreno, S.M. Vitamin B12 deficiency and diabetic neuropathy in patients taking metformin: A cross-sectional study. Endocr. Connect. 2019, 8, 1324–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alharbi, T.J.; Tourkmani, A.M.; Abdelhay, O.; Alkhashan, H.I.; Al-Asmari, A.K.; Rsheed, A.M.; Abuhaimed, S.N.; Mohammed, N.; AlRasheed, A.N.; AlHarbi, N.G. The association of metformin use with vitamin B12 deficiency and peripheral neuropathy in Saudi individuals with type 2 diabetes mellitus. PLoS ONE 2018, 13, e0204420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, L.E.; Darling, A.L.; Brown, J.E. Association between metformin dose and vitamin B12 deficiency in patients with type 2 diabetes. Medicine 2019, 98, e17918. [Google Scholar]
- Orlenko, A.; Moore, J.H.; Orzechowski, P.; Olson, R.S.; Cairns, J.; Caraballo, P.J.; Weinshilboum, R.M.; Wang, L.; Breitenstein, M.K. Considerations for automated machine learning in clinical metabolic profiling: Altered homocysteine plasma concentration associated with metformin exposure. Pac. Symp. Biocomput. Pac. Symp. Biocomput. 2018, 23, 460–471. [Google Scholar] [PubMed] [Green Version]
- Out, M.; Kooy, A.; Lehert, P.; Schalkwijk, C.A.; Stehouwer, C.D. Long-term treatment with metformin in type 2 diabetes and methylmalonic acid: Post hoc analysis of a randomized controlled 4.3year trial. J. Diabetes Its Complicat. 2018, 32, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Abo, R.; Hebbring, S.; Ji, Y.; Zhu, H.; Zeng, Z.B.; Batzler, A.; Jenkins, G.D.; Biernacka, J.; Snyder, K.; Drews, M.; et al. Merging pharmacometabolomics with pharmacogenomics using ‘1000 Genomes’ single-nucleotide polymorphism imputation: Selective serotonin reuptake inhibitor response pharmacogenomics. Pharm. Genom. 2012, 22, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Hebbring, S.; Zhu, H.; Jenkins, G.D.; Biernacka, J.; Snyder, K.; Drews, M.; Fiehn, O.; Zeng, Z.; Schaid, D.; et al. Glycine and a glycine dehydrogenase (GLDC) SNP as citalopram/escitalopram response biomarkers in depression: Pharmacometabolomics-informed pharmacogenomics. Clin. Pharmacol. Ther. 2011, 89, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Shahin, M.H.; Gong, Y.; Frye, R.F.; Rotroff, D.M.; Beitelshees, A.L.; Baillie, R.A.; Chapman, A.B.; Gums, J.G.; Turner, S.T.; Boerwinkle, E.; et al. Sphingolipid metabolic pathway impacts thiazide diuretics blood pressure response: Insights from genomics, metabolomics, and lipidomics. J. Am. Heart Assoc. 2017, 7, e006656. [Google Scholar] [CrossRef] [Green Version]
- Neavin, D.; Kaddurah-Daouk, R.; Weinshilboum, R. Pharmacometabolomics informs pharmacogenomics. Metab. Off. J. Metab. Soc. 2016, 12, 121. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, F.A.; Shahin, M.H.; Gong, Y.; McDonough, C.W.; Beitelshees, A.L.; Gums, J.G.; Chapman, A.B.; Boerwinkle, E.; Turner, S.T.; Frye, R.F.; et al. Novel plasma biomarker of atenolol-induced hyperglycemia identified through a metabolomics-genomics integrative approach. Metab. Off. J. Metab. Soc. 2016, 12, 129. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia (N. Y.) 2017, 19, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Morais, S.M.; Silva, K.A.; Araujo, H.; Vieira, I.G.; Alves, D.R.; Fontenelle, R.O.; Silva, A. Anacardic acid constituents from cashew nut shell liquid: NMR characterization and the effect of unsaturation on its biological activities. Pharmaceuticals 2017, 10, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollands, A.; Corriden, R.; Gysler, G.; Dahesh, S.; Olson, J.; Ali, S.R.; Kunkel, M.T.; Lin, A.E.; Forli, S.; Newton, A.C.; et al. Natural product anacardic acid from cashew nut shells stimulates neutrophil extracellular trap production and bactericidal activity. J. Biol. Chem. 2016, 291, 13964–13973. [Google Scholar] [CrossRef] [Green Version]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4554–4561. [Google Scholar] [CrossRef] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2013, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.A.; Schmidt, C.M.; Goodwin, T.J. Pharmacogenomics in Spaceflight. In Handbook of Space Pharmaceuticals; Pathak, Y., Araújo dos Santos, M., Zea, L., Eds.; Springer International Publishing: Cham/Basel, Switzerland, 2018; pp. 1–39. [Google Scholar]
- Garrett-Bakelman, F.E.; Darshi, M.; Green, S.J.; Gur, R.C.; Lin, L.; Macias, B.R.; McKenna, M.J.; Meydan, C.; Mishra, T.; Nasrini, J.; et al. The NASA twins study: A multidimensional analysis of a year-long human spaceflight. Science 2019, 364, eaau8650. [Google Scholar] [PubMed]
- Koppel, N.; Rekdal, V.M.; Balskus, E.P. Chemical transformation of xenobiotics by the human gut microbiota. Science 2017, 356, eaag2770. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. Plos Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Qin, N.; Yang, R. Human microbiota: A neglected “organ” in precision medicine. Infect. Dis. Transl. Med. 2015, 1, 63–72. [Google Scholar]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207. [Google Scholar]
- Heintz-Buschart, A.; Wilmes, P. Human gut microbiome: Function matters. Trends Microbiol. 2018, 26, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Sandhu, K.V.; Griffin, B.T.; Dinan, T.G.; Cryan, J.F.; Hyland, N.P. Gut reactions: Breaking down xenobiotic-microbiome interactions. Pharmacol. Rev. 2019, 71, 198. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Southall, N.; Wang, Y.; Yasgar, A.; Shinn, P.; Jadhav, A.; Nguyen, D.T.; Austin, C.P. The NCGC pharmaceutical collection: A comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci. Transl. Med. 2011, 3, 80ps16. [Google Scholar] [CrossRef] [Green Version]
- Haiser, H.J.; Turnbaugh, P.J. Is it time for a metagenomic basis of therapeutics? Science 2012, 336, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Yip, L.Y.; Chan, E.C.Y. Investigation of host-gut microbiota modulation of therapeutic outcome. Drug Metab. Dispos. 2015, 43, 1619–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, I.D.; Nicholson, J.K. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl. Res. 2017, 179, 204–222. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Sato, T.; Nomoto, K.; Tsuji, H. Identification of phenol- and p-cresol-producing intestinal bacteria by using media supplemented with tyrosine and its metabolites. Fems Microbiol. Ecol. 2018, 94, fiy125. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Davis, D.C.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J. Pharmacol. Exp. Ther. 1973, 187, 185. [Google Scholar]
- Ben-Shachar, R.; Chen, Y.; Luo, S.; Hartman, C.; Reed, M.; Nijhout, H.F. The biochemistry of acetaminophen hepatotoxicity and rescue: A mathematical model. Theor. Biol. Med Model. 2012, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Heruth, D.P.; Shortt, K.; Zhang, N.; Li, D.Y.; Zhang, L.Q.; Ye, S.Q. Genetic association of single nucleotide polymorphisms with acetaminophen-induced hepatotoxicity. J. Pharmacol. Exp. Ther. 2018, 367, 95–100. [Google Scholar] [CrossRef]
- Moyer, A.M.; Fridley, B.L.; Jenkins, G.D.; Batzler, A.J.; Pelleymounter, L.L.; Kalari, K.R.; Ji, Y.; Chai, Y.; Nordgren, K.K.; Weinshilboum, R.M. Acetaminophen-NAPQI hepatotoxicity: A cell line model system genome-wide association study. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 120, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Mohn, E.S.; Kern, H.J.; Saltzman, E.; Mitmesser, S.H.; McKay, D.L. Evidence of drug-nutrient interactions with chronic use of commonly prescribed medications: An update. Pharmaceutics 2018, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, S.M.; Gao, X.; Dai, Z.; Locasale, J.W. Methionine metabolism in health and cancer: A nexus of diet and precision medicine. Nat. Rev. Cancer 2019, 19, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drozda, K.; Pacanowski, M.A.; Grimstein, C.; Zineh, I. Pharmacogenetic labeling of FDA-approved drugs: A regulatory retrospective. Jacc. Basic Transl. Sci. 2018, 3, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Burt, T.; Dhillon, S. Pharmacogenomics in early-phase clinical development. Pharmacogenomics 2013, 14, 1085–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Yi, S.; Gu, N.; Shin, D.; Yu, K.S.; Yoon, S.H.; Cho, J.Y.; Jang, I.J. Utility of integrated analysis of pharmacogenomics and pharmacometabolomics in early phase clinical trial: A case study of a new molecular entity. Genom. Inform. 2018, 16, 52–58. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| BEST Biomarker Category | Relationship between Metabolites and Biomarker Category | Potential Context of Use (COU) in a Clinical Study |
|---|---|---|
| Prognosis Biomarker | Metabolites that indicate a likelihood of a future clinical event | Stratify Patients Enrichment: Inclusion/Exclusion Data |
| Diagnostic Biomarker | Metabolites that detect the presence of a disease or identify individuals with a subtype of the disease | Patient Selection |
| Monitoring Biomarker | Metabolites that are measured continually over time to assess status of a disease or medical condition or for evidence of exposure to (or effect of) a medical product or an environmental agent | Indicate Toxicity or assess safety Provide evidence of exposure |
| Predictive Biomarker | Metabolites that predict outcome | Identify individuals based on effect from a specific intervention or exposure |
| Safety Biomarker | Metabolites that are related to adverse and safety events | Indicate the presence or extent of toxicity related to an intervention or exposure |
| Pharmacodynamic Response Biomarker | Metabolites that are related to response in an individual or group of individuals who have been exposed to a medical product or an environmental agent | Efficacy biomarkers/surrogate endpoint Show biological response related to an intervention or exposure |
| Susceptibility/Risk Biomarker | Metabolites related to developing a disease or medical condition in an patient that does not currently have clinically apparent disease or medical condition | Indicate the potential for developing a disease or sensitivity to an exposure |
| Provisional Biomarker | Metabolites that are in discovery and show potential as biomarkers, although they have not been validated as true biomarkers | Discovery-associated analytes that assist in identification of signals with potential biological meaning. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beger, R.D.; Schmidt, M.A.; Kaddurah-Daouk, R. Current Concepts in Pharmacometabolomics, Biomarker Discovery, and Precision Medicine. Metabolites 2020, 10, 129. https://doi.org/10.3390/metabo10040129
Beger RD, Schmidt MA, Kaddurah-Daouk R. Current Concepts in Pharmacometabolomics, Biomarker Discovery, and Precision Medicine. Metabolites. 2020; 10(4):129. https://doi.org/10.3390/metabo10040129
Chicago/Turabian StyleBeger, Richard D., Michael A Schmidt, and Rima Kaddurah-Daouk. 2020. "Current Concepts in Pharmacometabolomics, Biomarker Discovery, and Precision Medicine" Metabolites 10, no. 4: 129. https://doi.org/10.3390/metabo10040129
APA StyleBeger, R. D., Schmidt, M. A., & Kaddurah-Daouk, R. (2020). Current Concepts in Pharmacometabolomics, Biomarker Discovery, and Precision Medicine. Metabolites, 10(4), 129. https://doi.org/10.3390/metabo10040129