The ABRF Metabolomics Research Group 2016 Exploratory Study: Investigation of Data Analysis Methods for Untargeted Metabolomics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Untargeted Metabolomics: An Emerging Technology for Biomarker Discovery
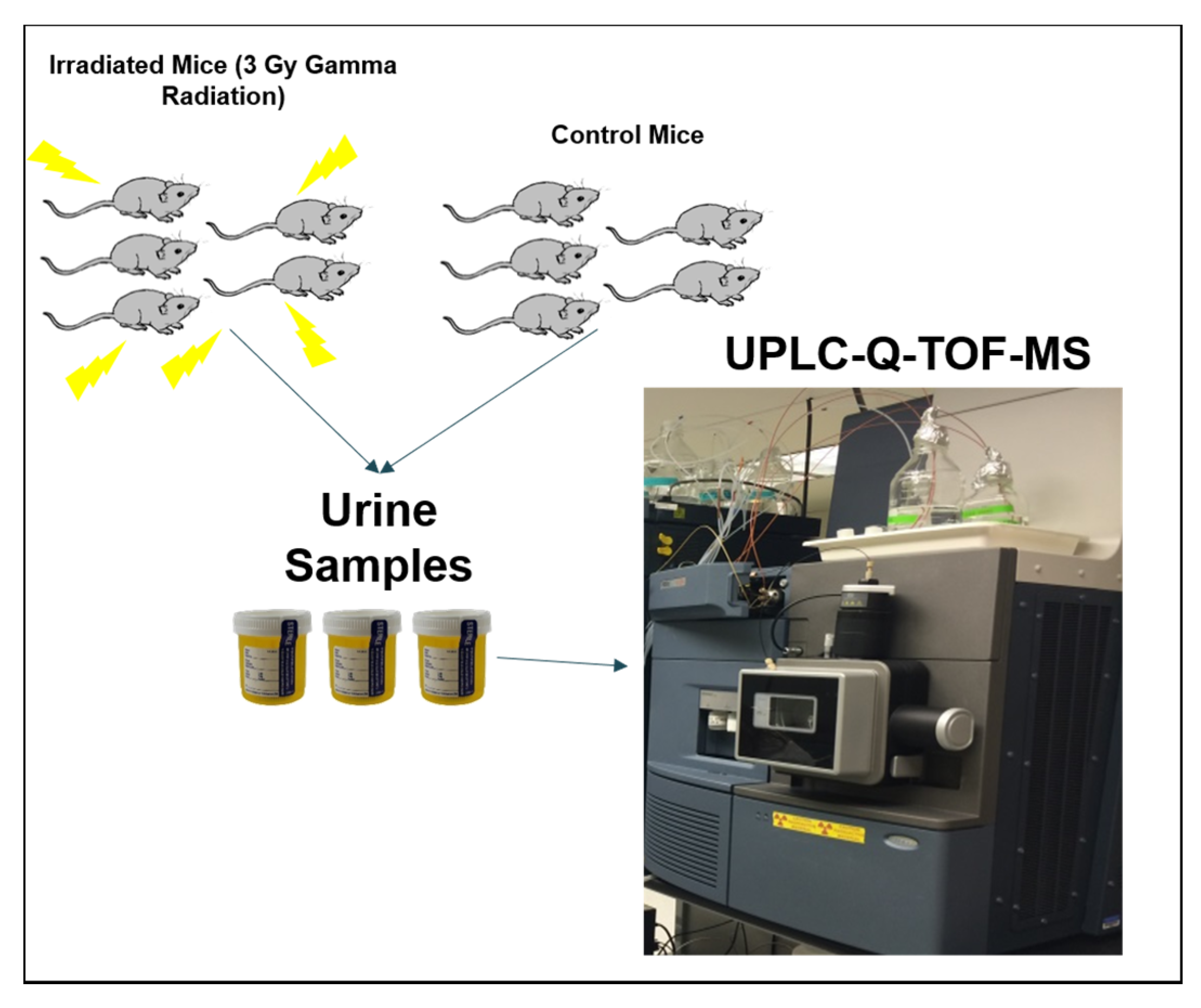
2. Association of Biomolecular Research Facilities (ABRF) Metabolomics Research Group (MRG) 2016 Study Design
- Deconvolute the raw data using a preprocessing software of their choice and provide a data matrix consisting of m/z, retention time, and ion intensity (e.g., peak area);
- Postprocess the data using statistical tools of their choice and determine the top 50 most statistically significant perturbed (e.g., p-value) mouse urinary spectral features postexposure to 5 Gy external beam irradiation;
- Assign putative identification to these urinary spectral features using various online databases.
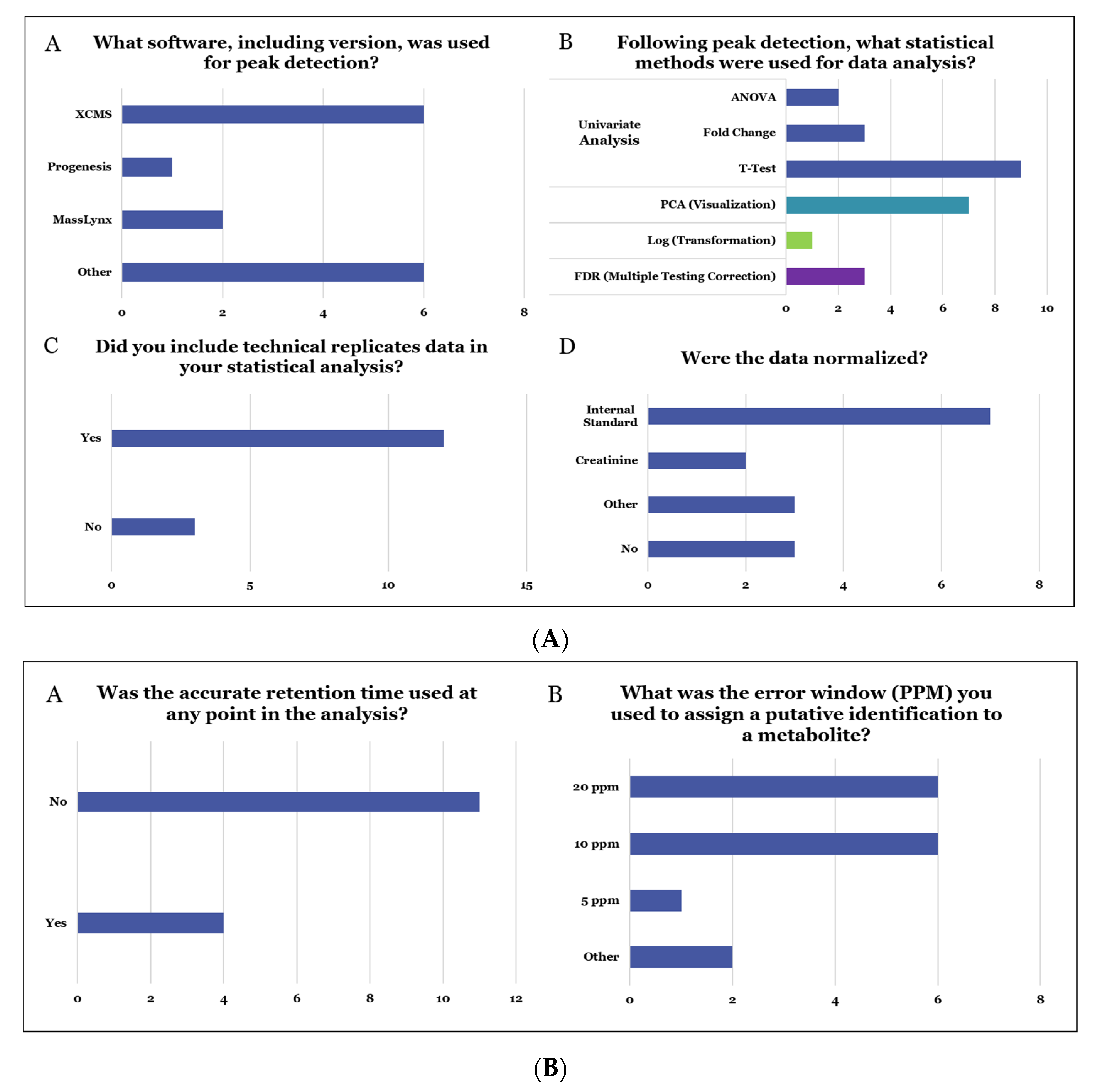
- Methods used for the determination of relative quantitative metabolite differences across the two sample types (groups).
- The effects of different computational techniques on the determination of significantly altered metabolites in the two groups.
- The level of confidence and consistency in the results obtained from unique computational and chemometric approaches.
- The ability of software to determine differences across samples or help analyze data from metabolomics experiments.
- Databases used for assigning metabolite ID.
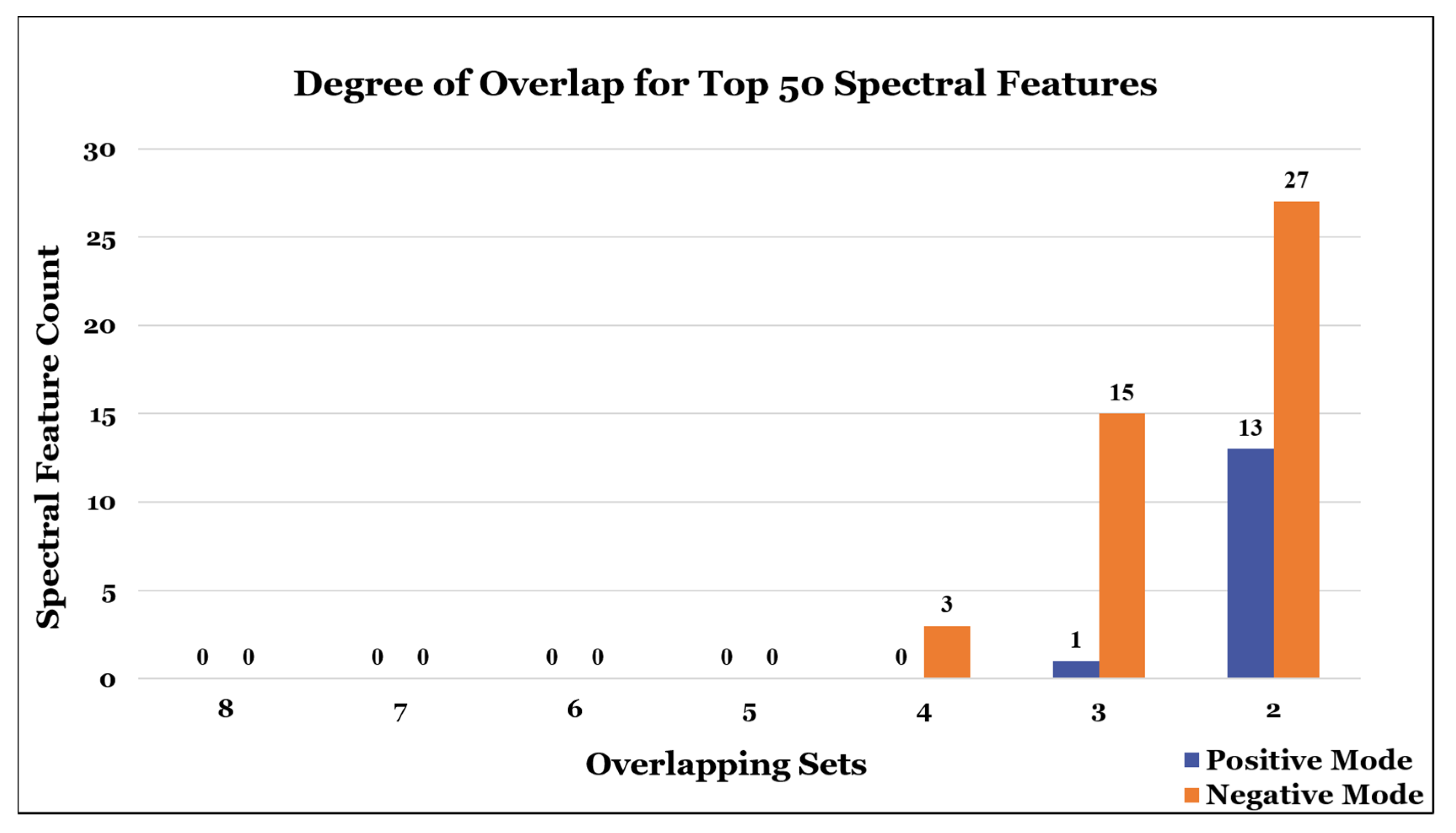
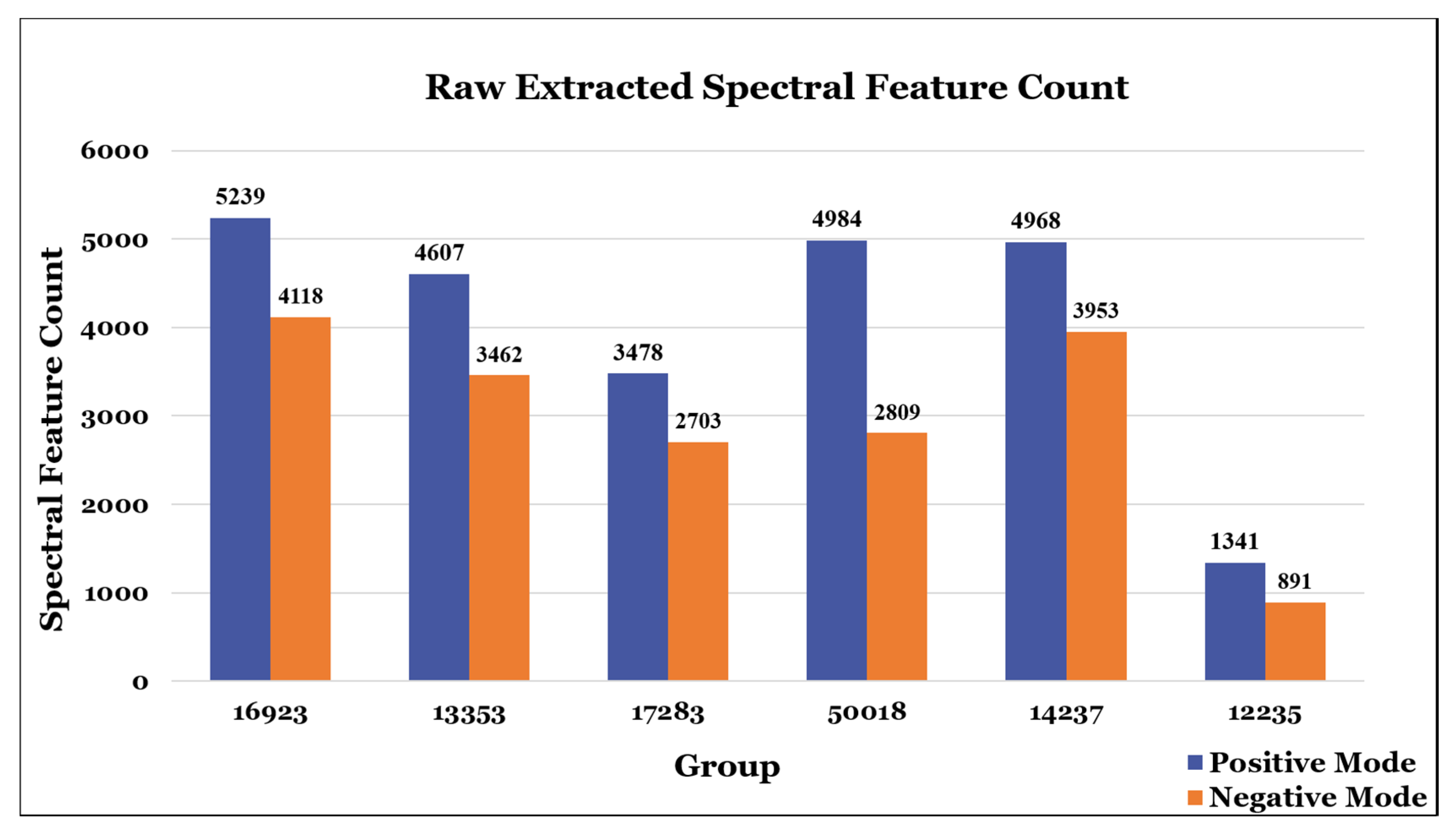
3. Meta-Analysis of Study Participant Results
4. MRG 2016 Study: Lessons Learned and Future Directions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mahieu, N.G.; Spalding, J.L.; Patti, G.J. Warpgroup: Increased precision of metabolomic data processing by consensus integration bound analysis. Bioinformatics 2016, 32, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klupczynska, A.; Derezinski, P.; Kokot, Z.J. Metabolomics in Medical Sciences—Trends, Challenges and Perspectives. Acta Pol. Pharm. 2015, 72, 629–641. [Google Scholar] [PubMed]
- Kessler, N.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. ALLocator: An interactive web platform for the analysis of metabolomic LC-ESI-MS datasets, enabling semi-automated, user-revised compound annotation and mass isotopomer ratio analysis. PLoS ONE 2014, 9, e113909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Lewis, I.A.; Hegeman, A.D.; Anderson, M.E.; Li, J.; Schulte, C.F.; Westler, W.M.; Eghbalnia, H.R.; Sussman, M.R.; Markley, J.L. Metabolite identification via the Madison Metabolomics Consortium Database. Nat. Biotechnol. 2008, 26, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Gil-de-la-Fuente, A.; Godzien, J.; Saugar, S.; Garcia-Carmona, R.; Badran, H.; Wishart, D.S.; Barbas, C.; Otero, A. CEU Mass Mediator 3.0: A Metabolite Annotation Tool. J. Proteome Res. 2019, 18, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A web-based platform to process untargeted metabolomic data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffman, C.; Petrick, L.; Perttula, K.; Yano, Y.; Carlsson, H.; Whitehead, T.; Metayer, C.; Hayes, J.; Rappaport, S.; Dudoit, S. Filtering procedures for untargeted LC-MS metabolomics data. BMC Bioinform. 2019, 20, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.; Marsal, S.; Julià, A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front. Bioeng. Biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.C.; Maillot, M.; Mazerolles, G.; Verdu, A.; Lyan, B.; Migne, C.; Defoort, C.; Canlet, C.; Junot, C.; Guillou, C.; et al. Can we trust untargeted metabolomics? Results of the metabo-ring initiative, a large-scale, multi-instrument inter-laboratory study. Metabolomics 2015, 11, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, X.; Lu, X.; Lin, X.; Xu, G. A data preprocessing strategy for metabolomics to reduce the mask effect in data analysis. Front. Mol. Biosci. 2015, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaman, I. Preprocessing and Pretreatment of Metabolomics Data for Statistical Analysis. In Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2017; pp. 145–161. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turck, C.W.; Mak, T.D.; Goudarzi, M.; Salek, R.M.; Cheema, A.K. The ABRF Metabolomics Research Group 2016 Exploratory Study: Investigation of Data Analysis Methods for Untargeted Metabolomics. Metabolites 2020, 10, 128. https://doi.org/10.3390/metabo10040128
Turck CW, Mak TD, Goudarzi M, Salek RM, Cheema AK. The ABRF Metabolomics Research Group 2016 Exploratory Study: Investigation of Data Analysis Methods for Untargeted Metabolomics. Metabolites. 2020; 10(4):128. https://doi.org/10.3390/metabo10040128
Chicago/Turabian StyleTurck, Christoph W., Tytus D Mak, Maryam Goudarzi, Reza M Salek, and Amrita K Cheema. 2020. "The ABRF Metabolomics Research Group 2016 Exploratory Study: Investigation of Data Analysis Methods for Untargeted Metabolomics" Metabolites 10, no. 4: 128. https://doi.org/10.3390/metabo10040128
APA StyleTurck, C. W., Mak, T. D., Goudarzi, M., Salek, R. M., & Cheema, A. K. (2020). The ABRF Metabolomics Research Group 2016 Exploratory Study: Investigation of Data Analysis Methods for Untargeted Metabolomics. Metabolites, 10(4), 128. https://doi.org/10.3390/metabo10040128