Integrative Analysis of Metabolomic and Transcriptomic Profiles Uncovers Biological Pathways of Feed Efficiency in Pigs


Abstract
:1. Introduction
2. Results
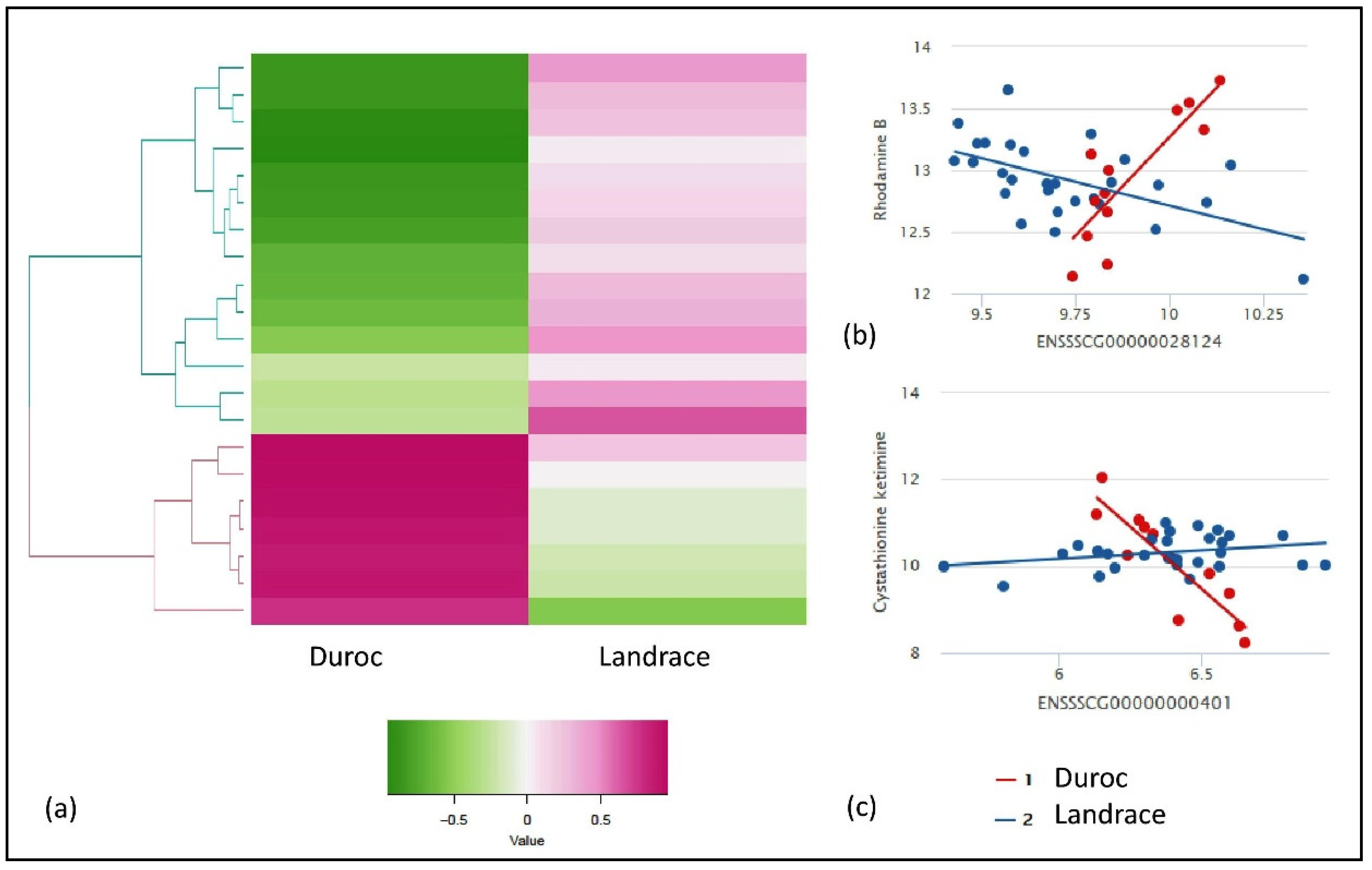
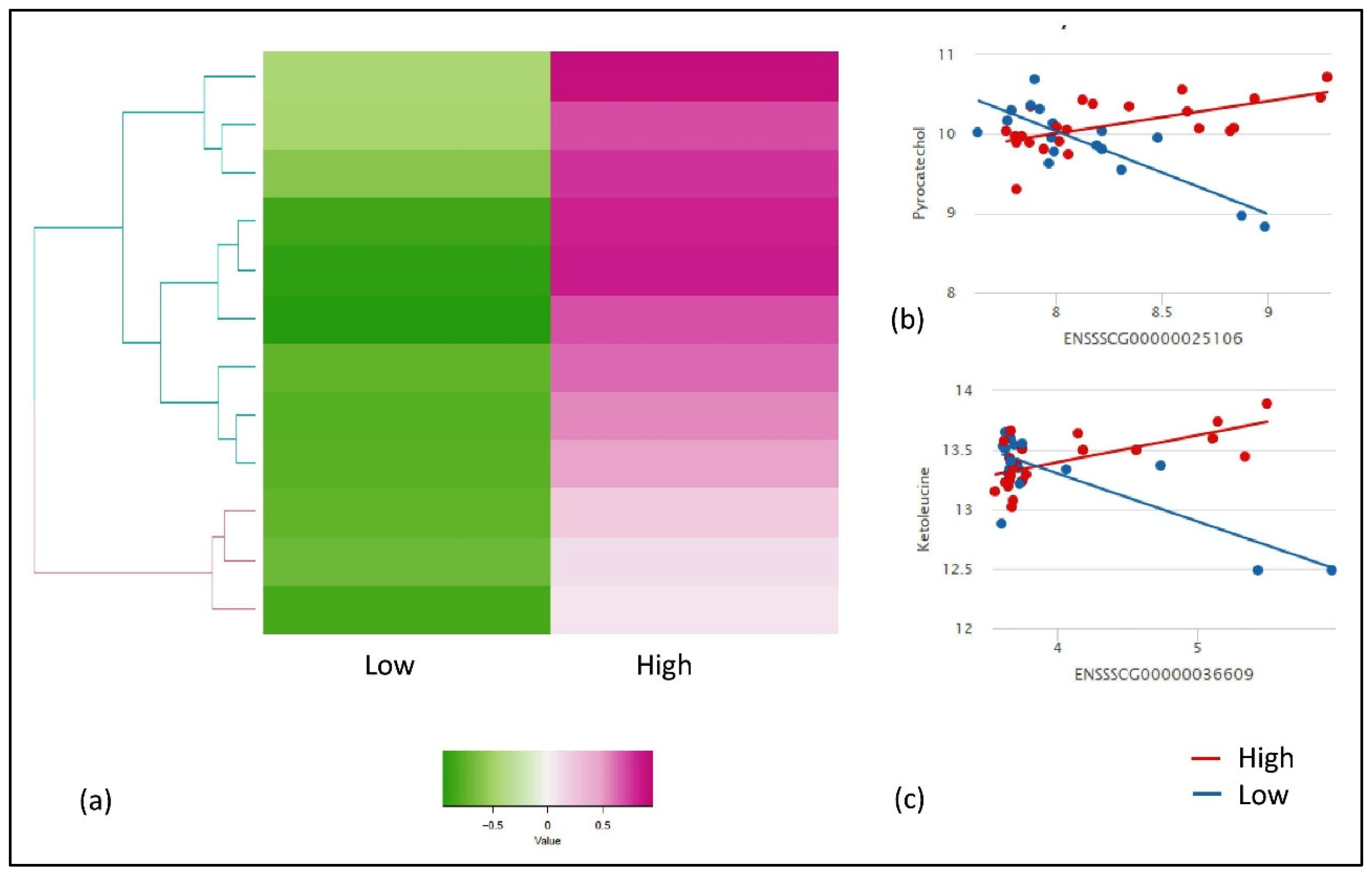
2.1. Descriptive Statistics and Linear Model Analysis for Genes and Metabolites
2.2. Gene Metabolite Interaction of Breed-Specific and FE-Specific Groups
2.3. Pathway and Gene Ontology Over-Representation Analysis
3. Discussion
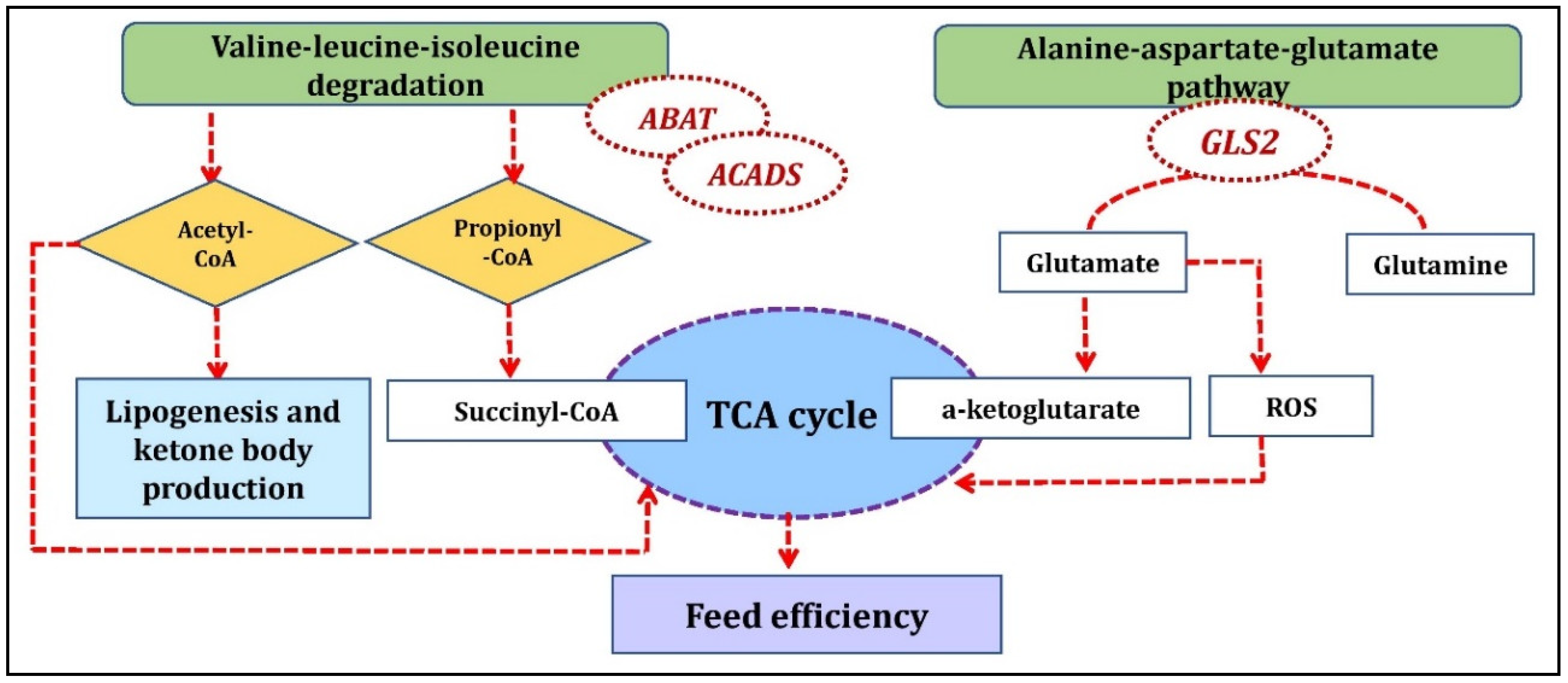
3.1. Breed-Specific Pathway Analysis
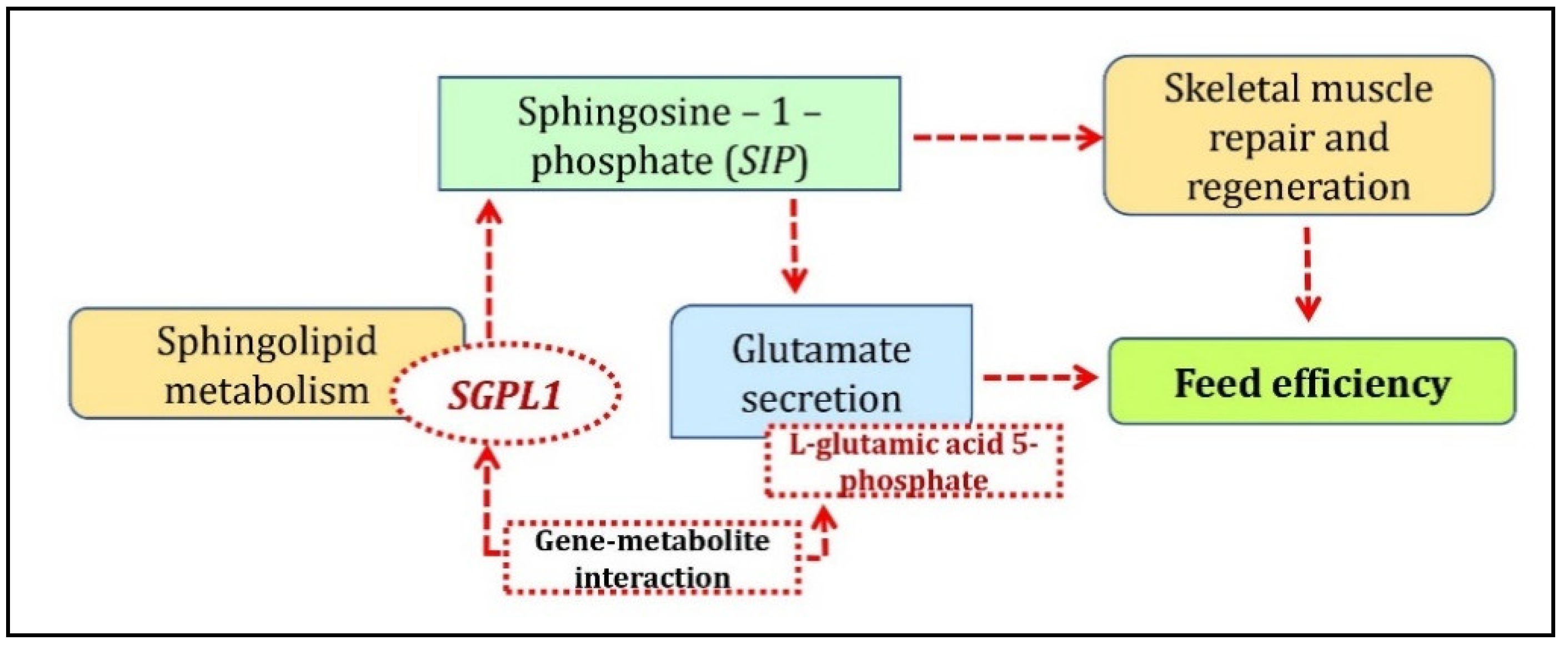
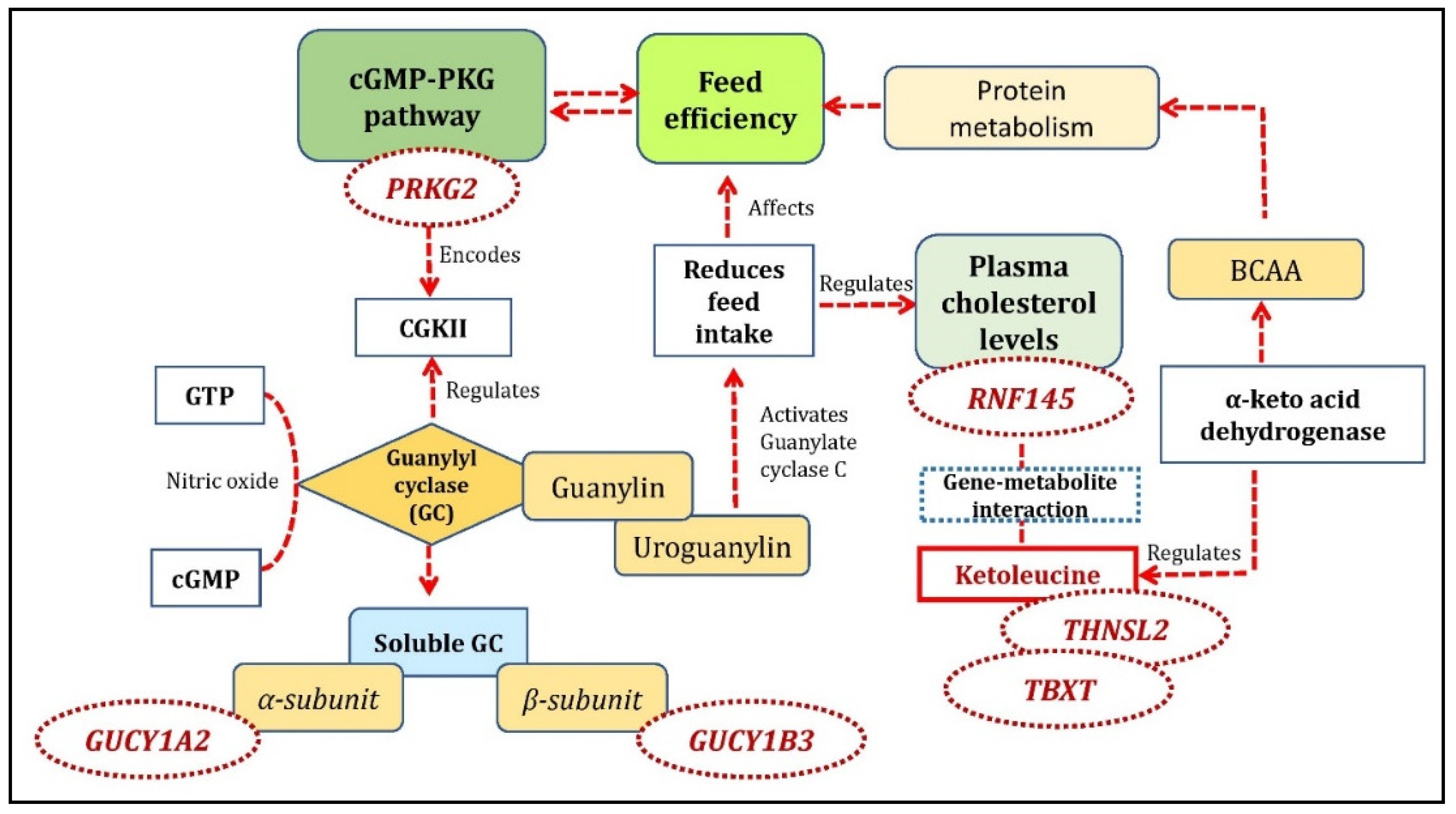
3.2. cGMP-PKG Pathway Involved with FE-Specific Analysis
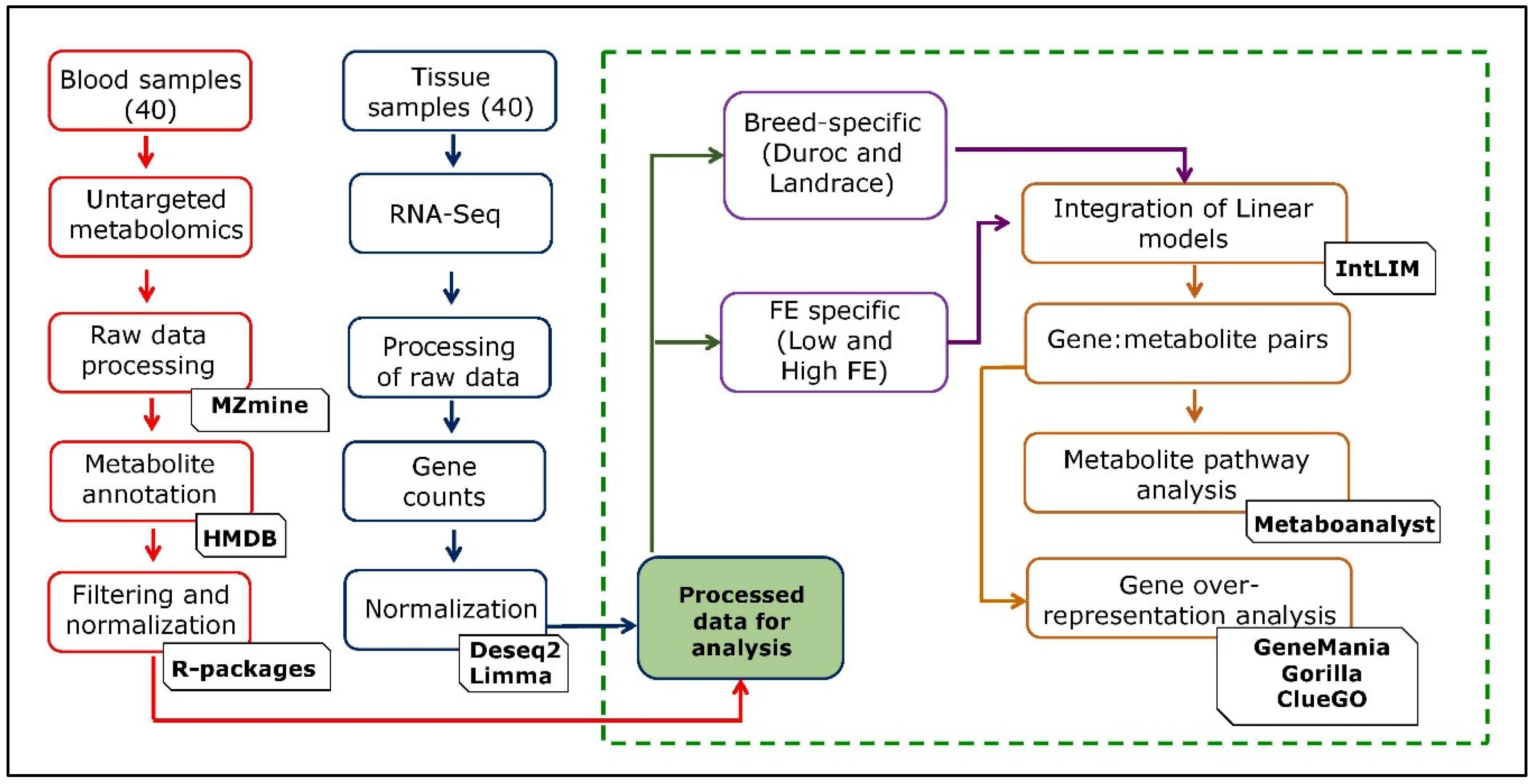
4. Materials and Methods
4.1. Data Resource and Phenotype Generation
4.2. Gene Expression Profile, Metabolite Profile, and Data Analyses
4.3. Integration of Transcriptomic and Metabolomic Data Based on the Linear Model
4.4. Pathway Over-Representation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Consent for Publication
Availability of Data and Material
References
- Patience, J.F.; Rossoni-Serão, M.C.; Gutiérrez, N.A. A review of feed efficiency in swine: biology and application. J. Anim. Sci. Biotechnol. 2015, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Li, T.; Wang, W.; Gao, H.; Bai, Y.; Zhang, S.; Zang, J.; Li, D.; Wang, J. Metabolic characteristics and nutrient utilization in high-feed-efficiency pigs selected using different feed conversion ratio models. Sci. China Life Sci. 2019, 62, 959–970. [Google Scholar] [CrossRef]
- Carmelo, V.A.O.; Banerjee, P.; da Silva Diniz, W.J.; Kadarmideen, H.N. Metabolomic networks and pathways associated with feed efficiency and related-traits in Duroc and Landrace pigs. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godinho, R.M.; Bergsma, R.; Silva, F.F.; Sevillano, C.A.; Knol, E.F.; Lopes, M.S.; Lopes, P.S.; Bastiaansen, J.W.M.; Guimarães, S.E.F. Genetic correlations between feed efficiency traits, and growth performance and carcass traits in purebred and crossbred pigs. J. Anim. Sci. 2018, 96, 817–829. [Google Scholar] [CrossRef]
- Ren, P.; Yang, X.J.; Cui, S.Q.; Kim, J.S.; Menon, D.; Baidoo, S.K. Effects of different feeding levels during three short periods of gestation on gilt and litter performance, nutrient digestibility, and energy homeostasis in gilts. J. Anim. Sci. 2017, 95, 1232–1242. [Google Scholar] [CrossRef] [PubMed]
- Do, D.N.; Strathe, A.B.; Ostersen, T.; Pant, S.D.; Kadarmideen, H.N. Genome-wide association and pathway analysis of feed efficiency in pigs reveal candidate genes and pathways for residual feed intake. Front. Genet. 2014, 5, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novais, F.J.; Dromms, R.A.; Alexandre, P.A.; Pires, P.R.L.; Styczynski, M.P.-W.; Ferraz, J.B.S.; Fukumasu, H.; Iglesias, A.H. Identification of a metabolomic signature associated with feed efficiency in beef cattle. BMC Genom. 2019, 20, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohart, F.; Paris, A.; Laurent, B.; Canlet, C.; Molina, J.; Mercat, M.J.; Tribout, T.; Muller, N.; Iannuccelli, N.; Villa-Vialaneix, N.; et al. Phenotypic prediction based on metabolomic data for growing pigs from three main european breeds. J. Anim. Sci. 2012, 90, 4729–4740. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Marrocco, C.; Zolla, V.; D’Andrea, M.; Zolla, L. Meat quality of the longissimus lumborum muscle of Casertana and Large White pigs: Metabolomics and proteomics intertwined. J. Proteom. 2011, 75, 610–627. [Google Scholar] [CrossRef]
- Bertram, H.C.; Oksbjerg, N.; Young, J.F. NMR-based metabonomics reveals relationship between pre-slaughter exercise stress, the plasma metabolite profile at time of slaughter, and water-holding capacity in pigs. Meat Sci. 2010, 84, 108–113. [Google Scholar] [CrossRef]
- Jing, L.; Hou, Y.; Wu, H.; Miao, Y.; Li, X.; Cao, J.; Michael Brameld, J.; Parr, T.; Zhao, S. Transcriptome analysis of mRNA and miRNA in skeletal muscle indicates an important network for differential Residual Feed Intake in pigs. Sci. Rep. 2015, 5, 11953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, A.; Louveau, I.; Gondret, F.; Tréfeu, C.; Gilbert, H.; Lefaucheur, L. Divergent selection for residual feed intake affects the transcriptomic and proteomic profiles of pig skeletal muscle. J. Anim. Sci. 2015, 93, 2745–2758. [Google Scholar] [CrossRef] [Green Version]
- Horodyska, J.; Hamill, R.M.; Reyer, H.; Trakooljul, N.; Lawlor, P.G.; Mccormack, U.M.; Wimmers, K. RNA-Seq of Liver From Pigs Divergent in Feed Efficiency Highlights Shifts in Macronutrient Metabolism, Hepatic Growth and Immune Response. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Carmelo, V.A.O.; Kadarmideen, H.N. Genome Regulation and Gene Interaction Networks Inferred From Muscle Transcriptome Underlying Feed Efficiency in Pigs. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Kadarmideen, H.N. Genomics to systems biology in animal and veterinary sciences: Progress, lessons and opportunities. Livest. Sci. 2014, 166, 232–248. [Google Scholar] [CrossRef] [Green Version]
- Suravajhala, P.; Kogelman, L.J.A.; Kadarmideen, H.N. Multi-omic data integration and analysis using systems genomics approaches: methods and applications in animal production, health and welfare. Genet. Sel. Evol. 2016, 48, 38. [Google Scholar] [CrossRef] [Green Version]
- Carrillo, J.A.; He, Y.; Li, Y.; Liu, J.; Erdman, R.A.; Sonstegard, T.S.; Song, J. Integrated metabolomic and transcriptome analyses reveal finishing forage affects metabolic pathways related to beef quality and animal welfare. Sci. Rep. 2016, 6, 25948. [Google Scholar] [CrossRef]
- Cavill, R.; Jennen, D.; Kleinjans, J.; Briedé, J.J. Transcriptomic and metabolomic data integration. Brief. Bioinform. 2016, 17, 891–901. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, J.M. Re-defining efficiency of feed use by livestock. Animal 2011, 5, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Do, D.N.; Strathe, A.B.; Jensen, J.; Mark, T.; Kadarmideen, H.N. Genetic parameters for different measures of feed efficiency and related traits in boars of three pig breeds. J. Anim. Sci. 2013, 91, 4069–4079. [Google Scholar] [CrossRef]
- Morales, P.E.; Bucarey, J.L.; Espinosa, A. Muscle Lipid Metabolism: Role of Lipid Droplets and Perilipins. J. Diabetes Res. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. Muscle as a Secretory Organ. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Siddiqui, J.K.; Baskin, E.; Liu, M.; Cantemir-Stone, C.Z.; Zhang, B.; Bonneville, R.; McElroy, J.P.; Coombes, K.R.; Mathé, E.A. IntLIM: integration using linear models of metabolomics and gene expression data. BMC Bioinform. 2018, 19, 81. [Google Scholar] [CrossRef] [PubMed]
- Taylor, V.A.; Stone, H.K.; Schuh, M.P.; Zhao, X.; Setchell, K.D.; Erkan, E. Disarranged Sphingolipid Metabolism From Sphingosine-1-Phosphate Lyase Deficiency Leads to Congenital Nephrotic Syndrome. Kidney Int. Rep. 2019, 4, 1763–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donati, C.; Cencetti, F.; Bruni, P. Sphingosine 1-phosphate axis: a new leader actor in skeletal muscle biology. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Lefaucheur, L.; Lebret, B.; Ecolan, P.; Louveau, I.; Damon, M.; Prunier, A.; Billon, Y.; Sellier, P.; Gilbert, H. Muscle characteristics and meat quality traits are affected by divergent selection on residual feed intake in pigs1. J. Anim. Sci. 2011, 89, 996–1010. [Google Scholar] [CrossRef]
- Kajimoto, T.; Okada, T.; Yu, H.; Goparaju, S.K.; Jahangeer, S.; Nakamura, S. Involvement of Sphingosine-1-Phosphate in Glutamate Secretion in Hippocampal Neurons. Mol. Cell. Biol. 2007, 27, 3429–3440. [Google Scholar] [CrossRef] [Green Version]
- Santos, L.S.; Miassi, G.M.; Tse, M.L.P.; Gomes, L.M.; Berto, P.N.; Denadai, J.C.; Caldara, F.R.; Dalto, D.B.; Berto, D.A. Growth performance and intestinal replacement time of 13C in newly weaned piglets supplemented with nucleotides or glutamic acid. Livest. Sci. 2019, 227, 160–165. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, X.; Kang, L.; Jiang, C.; Jiang, Y. Molecular characterization of porcine NECD, SNRPN and UBE3A genes and imprinting status in the skeletal muscle of neonate pigs. Mol. Biol. Rep. 2012, 39, 9415–9422. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [Green Version]
- Bottje, W.G.; Lassiter, K.; Piekarski-Welsher, A.; Dridi, S.; Reverter, A.; Hudson, N.J.; Kong, B.-W. Proteogenomics Reveals Enriched Ribosome Assembly and Protein Translation in Pectoralis major of High Feed Efficiency Pedigree Broiler Males. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Zhang, S.; Zeng, X.; Ren, M.; Mao, X.; Qiao, S. Novel metabolic and physiological functions of branched chain amino acids: a review. J. Anim. Sci. Biotechnol. 2017, 8, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, A.E.; Miller, R.H.; Block, K.P. Branched-chain amino acid metabolism. Annu. Rev. Nutr. 1984, 4, 409–454. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Xu, Y.; Hou, Y.; Qi, X.; Zhou, L.; Liu, H.; Luan, Y.; Jing, L.; Miao, Y.; Zhao, S.; et al. Proteomic analysis indicates that mitochondrial energy metabolism in skeletal muscle tissue is negatively correlated with feed efficiency in pigs. Sci. Rep. 2017, 7, 45291. [Google Scholar] [CrossRef]
- Freund, H.R.; Hanani, M. The metabolic role of branched-chain amino acids. Nutrition 2002, 18, 287–288. [Google Scholar] [CrossRef]
- Duarte, D.A.S.; Newbold, C.J.; Detmann, E.; Silva, F.F.; Freitas, P.H.F.; Veroneze, R.; Duarte, M.S. Genome-wide association studies pathway-based meta-analysis for residual feed intake in beef cattle. Anim. Genet. 2019, 50, 150–153. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampiga, M.; Flees, J.; Meluzzi, A.; Dridi, S.; Sirri, F. Application of omics technologies for a deeper insight into quali-quantitative production traits in broiler chickens: A review. J. Anim. Sci. Biotechnol. 2018, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, J.K.; Fritchen, A.N.; Huff-Lonergan, E.; Dekkers, J.C.M.; Gabler, N.K.; Lonergan, S.M. Divergent genetic selection for residual feed intake impacts mitochondria reactive oxygen species production in pigs1. J. Anim. Sci. 2013, 91, 2133–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, P.; Carmelo, V.A.O.; Kadarmideen, H.N. Genome-Wide Epistatic Interaction Networks Affecting Feed Efficiency in Duroc and Landrace Pigs. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Higgins, M.G.; Fitzsimons, C.; McClure, M.C.; McKenna, C.; Conroy, S.; Kenny, D.A.; McGee, M.; Waters, S.M.; Morris, D.W. GWAS and eQTL analysis identifies a SNP associated with both residual feed intake and GFRA2 expression in beef cattle. Sci. Rep. 2018, 8, 14301. [Google Scholar] [CrossRef] [Green Version]
- Bijvelds, M.J.C.; Tresadern, G.; Hellemans, A.; Smans, K.; Nieuwenhuijze, N.D.A.; Meijsen, K.F.; Bongartz, J.-P.; Ver Donck, L.; de Jonge, H.R.; Schuurkes, J.A.J.; et al. Selective inhibition of intestinal guanosine 3′,5′-cyclic monophosphate signaling by small-molecule protein kinase inhibitors. J. Biol. Chem. 2018, 293, 8173–8181. [Google Scholar] [CrossRef] [Green Version]
- Valente, T.S.; Baldi, F.; Sant’Anna, A.C.; Albuquerque, L.G.; Paranhos da Costa, M.J.R. Genome-Wide Association Study between Single Nucleotide Polymorphisms and Flight Speed in Nellore Cattle. PLoS ONE 2016, 11, e0156956. [Google Scholar] [CrossRef] [PubMed]
- Valentino, M.A.; Lin, J.E.; Snook, A.E.; Li, P.; Kim, G.W.; Marszalowicz, G.; Magee, M.S.; Hyslop, T.; Schulz, S.; Waldman, S.A. A uroguanylin-GUCY2C endocrine axis regulates feeding in mice. J. Clin. Investig. 2011, 121, 3578–3588. [Google Scholar] [CrossRef] [Green Version]
- Rauw, W.M.; Portolés, O.; Corella, D.; Soler, J.; Reixach, J.; Tibau, J.; Prat, J.M.; Diaz, I.; Gómez-Raya, L. Behaviour influences cholesterol plasma levels in a pig model. Animal 2007, 1, 865–871. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Rajbhandari, P.; Priest, C.; Sandhu, J.; Wu, X.; Temel, R.; Castrillo, A.; de Aguiar Vallim, T.Q.; Sallam, T.; Tontonoz, P. Inhibition of cholesterol biosynthesis through RNF145-dependent ubiquitination of SCAP. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Holeček, M. Effect of starvation on branched-chain α-keto acid dehydrogenase activity in rat heart and skeletal muscle. Physiol. Res. 2001, 50, 19–24. [Google Scholar] [PubMed]
- Duan, Y.; Duan, Y.; Li, F.; Li, Y.; Guo, Q.; Ji, Y.; Tan, B.; Li, T.; Yin, Y. Effects of supplementation with branched-chain amino acids to low-protein diets on expression of genes related to lipid metabolism in skeletal muscle of growing pigs. Amino Acids 2016, 48, 2131–2144. [Google Scholar] [CrossRef] [PubMed]
- Mukiibi, R.; Vinsky, M.; Keogh, K.; Fitzsimmons, C.; Stothard, P.; Waters, S.M.; Li, C. Liver transcriptome profiling of beef steers with divergent growth rate, feed intake, or metabolic body weight phenotypes1. J. Anim. Sci. 2019, 97, 4386–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, Y.J.; Pérusse, L.; Sarzynski, M.A.; Fornage, M.; Sidney, S.; Sternfeld, B.; Rice, T.; Terry, J.G.; Jacobs, D.R.; Katzmarzyk, P.; et al. Genome-wide association studies suggest sex-specific loci associated with abdominal and visceral fat. Int. J. Obes. 2016, 40, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Valadkhan, S.; Gunawardane, L.S. Role of small nuclear RNAs in eukaryotic gene expression. Essays Biochem. 2013, 54, 79–90. [Google Scholar] [CrossRef]
- Sun, J.S.; Manley, J.L. A novel U2-U6 snRNA structure is necessary for mammalian mRNA splicing. Genes Dev. 1995, 9, 843–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, S.; Schilder, R.J.; Kimball, S.R. Role of Precursor mRNA Splicing in Nutrient-Induced Alterations in Gene Expression and Metabolism. J. Nutr. 2015, 145, 841–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansouri, E.; Khorsandi, L.; Moaiedi, M.Z. Grape seed proanthocyanidin extract improved some of biochemical parameters and antioxidant disturbances of red blood cells in diabetic rats. Iran. J. Pharm. Res. 2015, 14, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.R.; Sexten, W.J.; Kerley, M.S.; Hansen, S.L. Relationship between antioxidant capacity, oxidative stress, and feed efficiency in beef steers. J. Anim. Sci. 2016, 94, 2942–2953. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Mostafavi, S.; Ray, D.; Warde-Farley, D.; Grouios, C.; Morris, Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008, 9, S4. [Google Scholar] [CrossRef] [Green Version]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite_Name | Ensembl ID | Gene Name | Duroc_cor | Landrace_cor | Abs diff.corr | Pval | FDRadjPval |
|---|---|---|---|---|---|---|---|
| Rhodamine B | ENSSSCG00000028124 | SNRPN | 0.776224 | −0.54242 | −1.31864 | 3.11 × 10−7 | 0.1 |
| L-glutamic acid 5-phosphate | ENSSSCG00000010274 | SGPL1 | 0.888112 | −0.21456 | −1.10267 | 1.57 × 10−7 | 0.09 |
| Cystathionine ketimine | ENSSSCG00000010274 | SGPL1 | 0.874126 | −0.18719 | −1.06132 | 2.67 × 10−7 | 0.1 |
| Cystathionine ketimine | ENSSSCG00000040110 | Novel_gene | 0.923077 | −0.11385 | −1.03692 | 1.08 × 10−8 | 0.02 |
| L-glutamic acid 5-phosphate | ENSSSCG00000038948 | ETS2 | 0.895105 | −0.11002 | −1.00512 | 2.66 × 10−8 | 0.02 |
| L-glutamic acid 5-phosphate | ENSSSCG00000040110 | Novel_gene | 0.937063 | 0.010947 | −0.92612 | 1.48 × 10−9 | 0.01 |
| L-glutamic acid 5-phosphate | ENSSSCG00000014632 | FAM160A2 | 0.951049 | 0.229885 | −0.72116 | 1.09 × 10−7 | 0.08 |
| Aloesol | ENSSSCG00000004128 | ZC2HC1B | −0.22767 | 0.048714 | 0.276385 | 4.33 × 10−7 | 0.1 |
| Theogallin | ENSSSCG00000026442 | FAM163B | −0.29772 | 0.45156 | 0.749284 | 1.69 × 10−7 | 0.09 |
| Fenamiphos | ENSSSCG00000018649 | Novel_gene | −0.68652 | 0.093596 | 0.780112 | 3.44 × 10−7 | 0.1 |
| Taraxacolide 1-o-b-d-glucopyranoside | ENSSSCG00000026442 | FAM163B | −0.27321 | 0.648057 | 0.921262 | 1.55 × 10−7 | 0.09 |
| Proanthocyanidin a2 | ENSSSCG00000033688 | ZDHHC22 | −0.63047 | 0.32567 | 0.956144 | 4.20 × 10−7 | 0.1 |
| Fenamiphos | ENSSSCG00000040467 | Novel_gene | −0.67251 | 0.288998 | 0.961504 | 2.25 × 10−8 | 0.02 |
| L-glutamic acid 5-phosphate | ENSSSCG00000000401 | GLS2 | −0.85315 | 0.109469 | 0.962616 | 1.80 × 10−7 | 0.09 |
| Paracetamol sulfate | ENSSSCG00000000401 | GLS2 | −0.94406 | 0.038314 | 0.98237 | 2.87 × 10−7 | 0.1 |
| L-glutamic acid 5-phosphate | ENSSSCG00000034989 | LRRTM2 | −0.83916 | 0.15052 | 0.989681 | 2.17 × 10−8 | 0.02 |
| Cystathionine ketimine | ENSSSCG00000034989 | LRRTM2 | −0.79021 | 0.204707 | 0.994917 | 2.53 × 10−8 | 0.02 |
| Ketoleucine | ENSSSCG00000026442 | FAM163B | −0.5289 | 0.466886 | 0.995783 | 1.19 × 10−8 | 0.02 |
| Ganoderenic acid e | ENSSSCG00000034200 | SEC22C | −0.85315 | 0.288998 | 1.142145 | 3.47 × 10−7 | 0.1 |
| Cystathionine ketimine | ENSSSCG00000000401 | GLS2 | −0.93007 | 0.249042 | 1.179112 | 2.92 × 10−9 | 0.01 |
| Ganoderenic acid e | ENSSSCG00000037595 | Novel_gene | −0.85315 | 0.449371 | 1.302517 | 2.26 × 10−7 | 0.1 |
| Metabolite_Name | Ensembl ID | Gene Name | High_cor | Low_cor | Abs diff.corr | Pval | FDRadjPval |
|---|---|---|---|---|---|---|---|
| Pyrocatechol | ENSSSCG00000025106 | THNSL2 | 0.66996 | −0.73284 | −1.4028 | 4.72 × 10−8 | 0.06 |
| 2-pyrocatechuic acid | ENSSSCG00000025106 | THNSL2 | 0.645257 | −0.68137 | −1.32663 | 7.13 × 10−8 | 0.08 |
| Ketoleucine | ENSSSCG00000017043 | RNF145 | 0.548419 | −0.75735 | −1.30577 | 2.17 × 10−7 | 0.1 |
| Ketoleucine | ENSSSCG00000025106 | THNSL2 | 0.498024 | −0.58088 | −1.07891 | 1.19 × 10−7 | 0.10 |
| Theogallin | ENSSSCG00000036609 | TBXT | 0.727273 | −0.35049 | −1.07776 | 2.87 × 10−8 | 0.05 |
| Neodiospyrin | ENSSSCG00000029077 | TUBAL3 | 0.608696 | −0.46814 | −1.07683 | 2.63 × 10−8 | 0.05 |
| Theogallin | ENSSSCG00000025106 | THNSL2 | 0.4417 | −0.6152 | −1.0569 | 2.52 × 10−7 | 0.1 |
| Proanthocyanidin a2 | ENSSSCG00000008938 | ENAM | 0.37954 | −0.60539 | −0.98493 | 1.99 × 10−7 | 0.1 |
| Ketoleucine | ENSSSCG00000036609 | TBXT | 0.557312 | −0.3701 | −0.92741 | 8.78 × 10−8 | 0.08 |
| Adrenochrome | ENSSSCG00000038441 | Novel_gene | 0.171485 | −0.58088 | −0.75237 | 1.71 × 10−8 | 0.05 |
| Proanthocyanidin a2 | ENSSSCG00000019329 | U2 | 0.052384 | −0.66176 | −0.71415 | 1.11 × 10−8 | 0.05 |
| Levulinic acid | ENSSSCG00000009250 | PRKG2 | 0.075117 | −0.54902 | −0.62414 | 1.93 × 10−7 | 0.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, P.; Carmelo, V.A.O.; Kadarmideen, H.N. Integrative Analysis of Metabolomic and Transcriptomic Profiles Uncovers Biological Pathways of Feed Efficiency in Pigs. Metabolites 2020, 10, 275. https://doi.org/10.3390/metabo10070275
Banerjee P, Carmelo VAO, Kadarmideen HN. Integrative Analysis of Metabolomic and Transcriptomic Profiles Uncovers Biological Pathways of Feed Efficiency in Pigs. Metabolites. 2020; 10(7):275. https://doi.org/10.3390/metabo10070275
Chicago/Turabian StyleBanerjee, Priyanka, Victor Adriano Okstoft Carmelo, and Haja N. Kadarmideen. 2020. "Integrative Analysis of Metabolomic and Transcriptomic Profiles Uncovers Biological Pathways of Feed Efficiency in Pigs" Metabolites 10, no. 7: 275. https://doi.org/10.3390/metabo10070275
APA StyleBanerjee, P., Carmelo, V. A. O., & Kadarmideen, H. N. (2020). Integrative Analysis of Metabolomic and Transcriptomic Profiles Uncovers Biological Pathways of Feed Efficiency in Pigs. Metabolites, 10(7), 275. https://doi.org/10.3390/metabo10070275