Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages

 ,
,  and
and 
Abstract
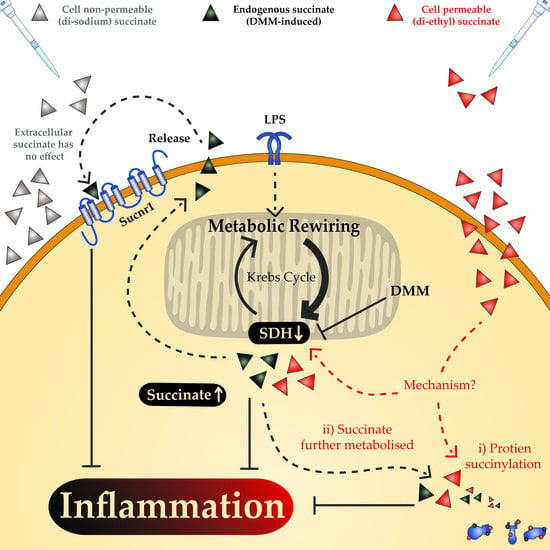
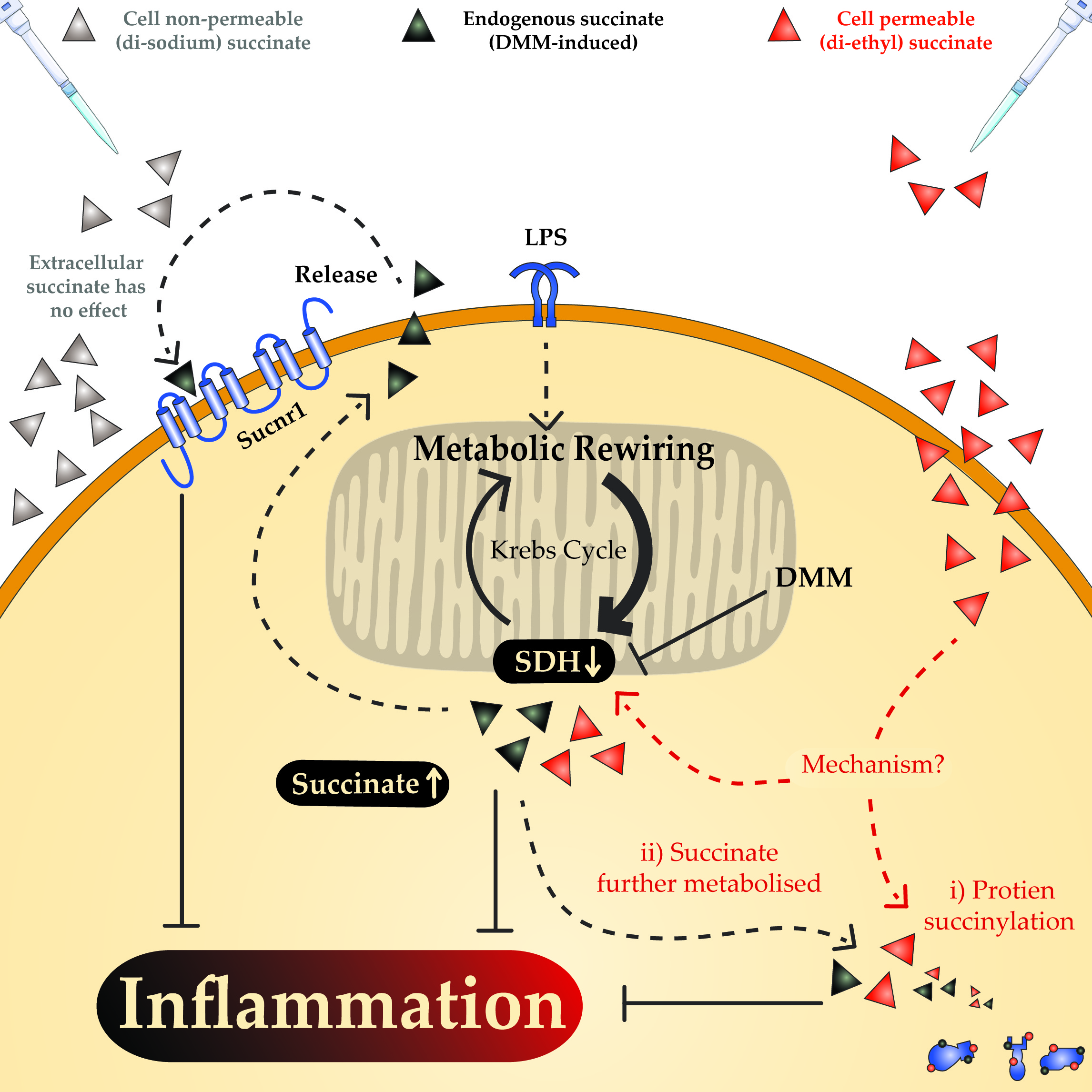
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
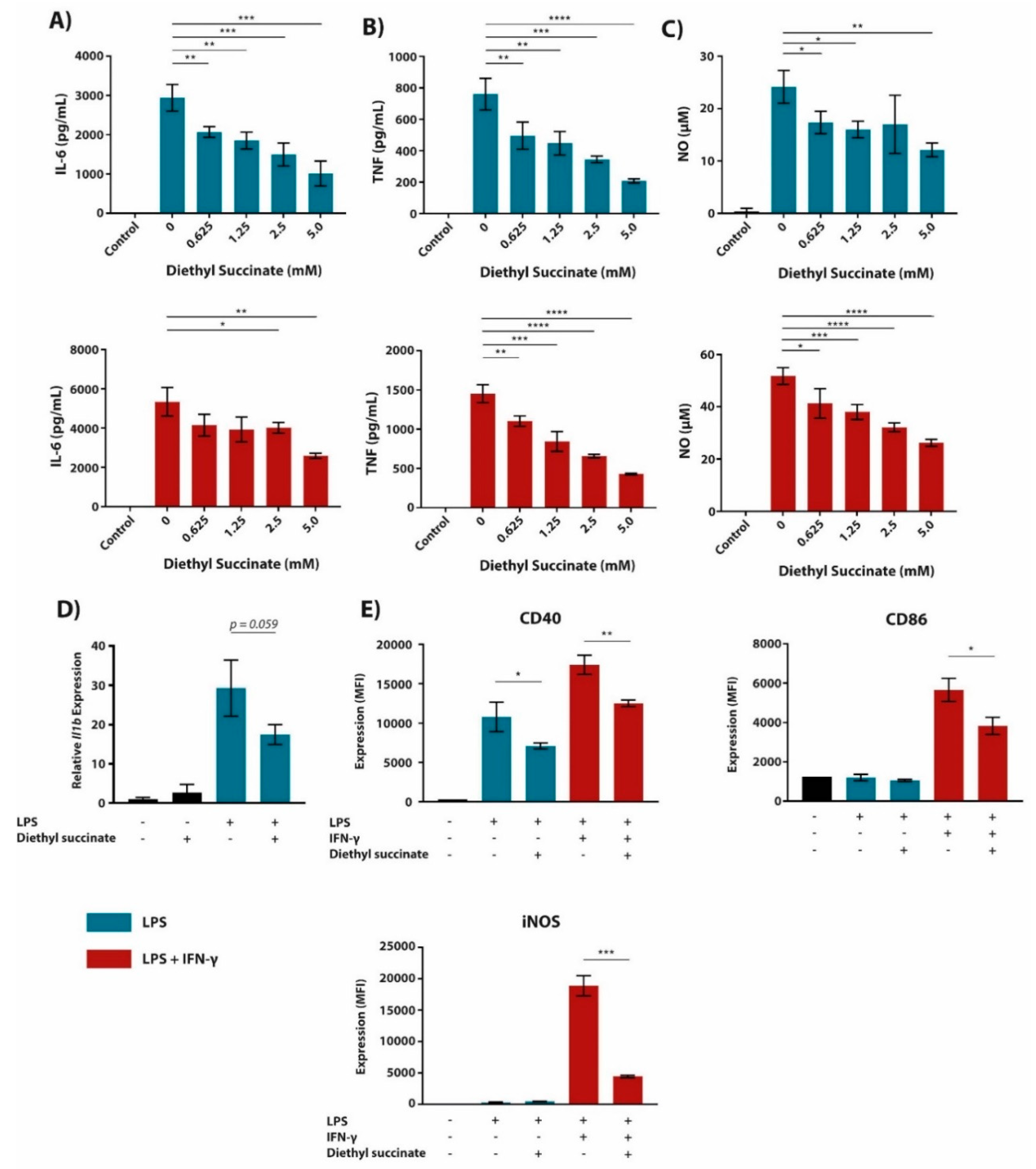
2.1. Cell-Permeable Diethyl Succinate Reduces Secretion and Expression of Inflammatory Mediators in Macrophages
2.2. The Succinate Receptor SUCNR1 Suppresses Classically Activated Macrophages
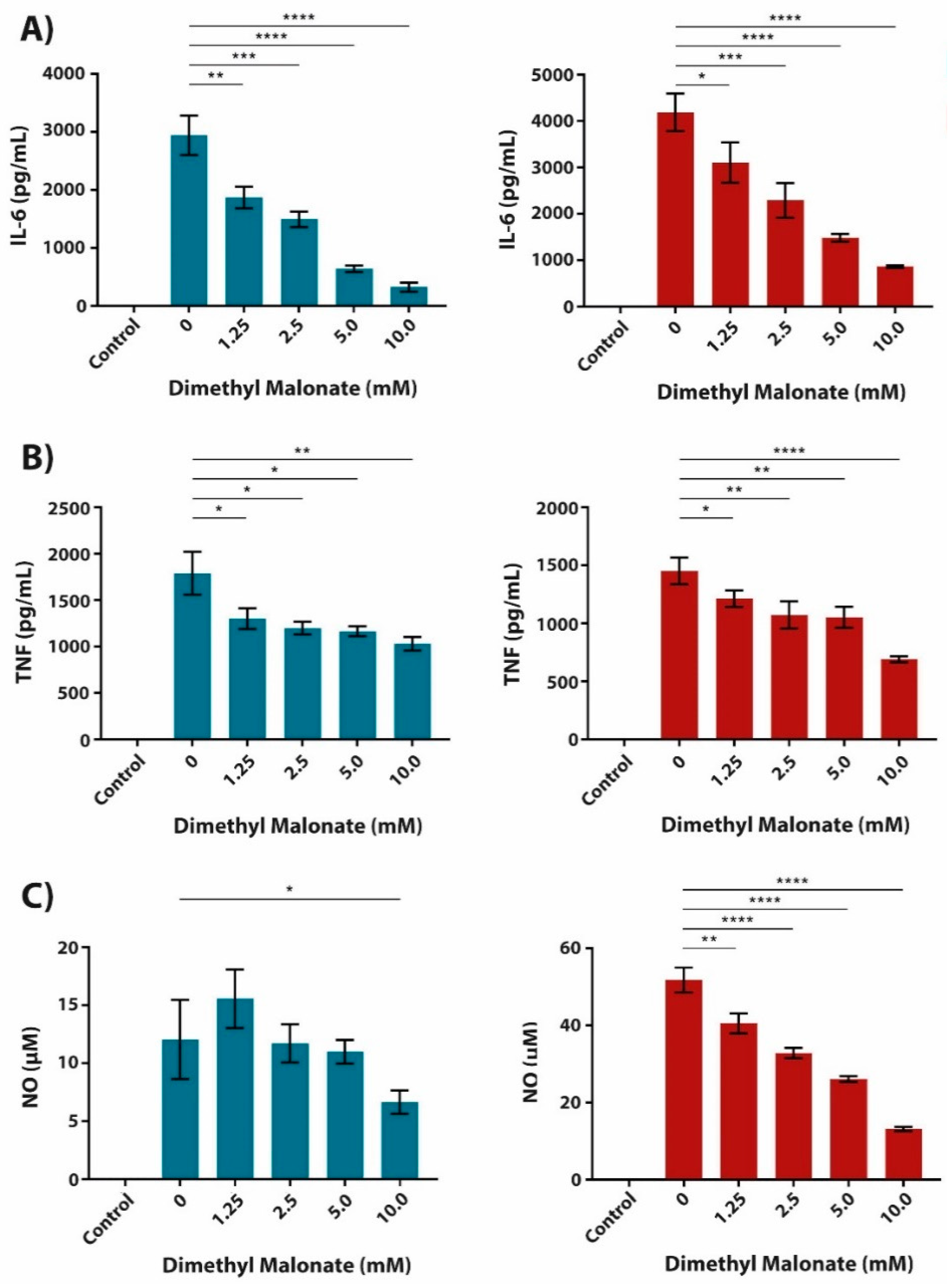
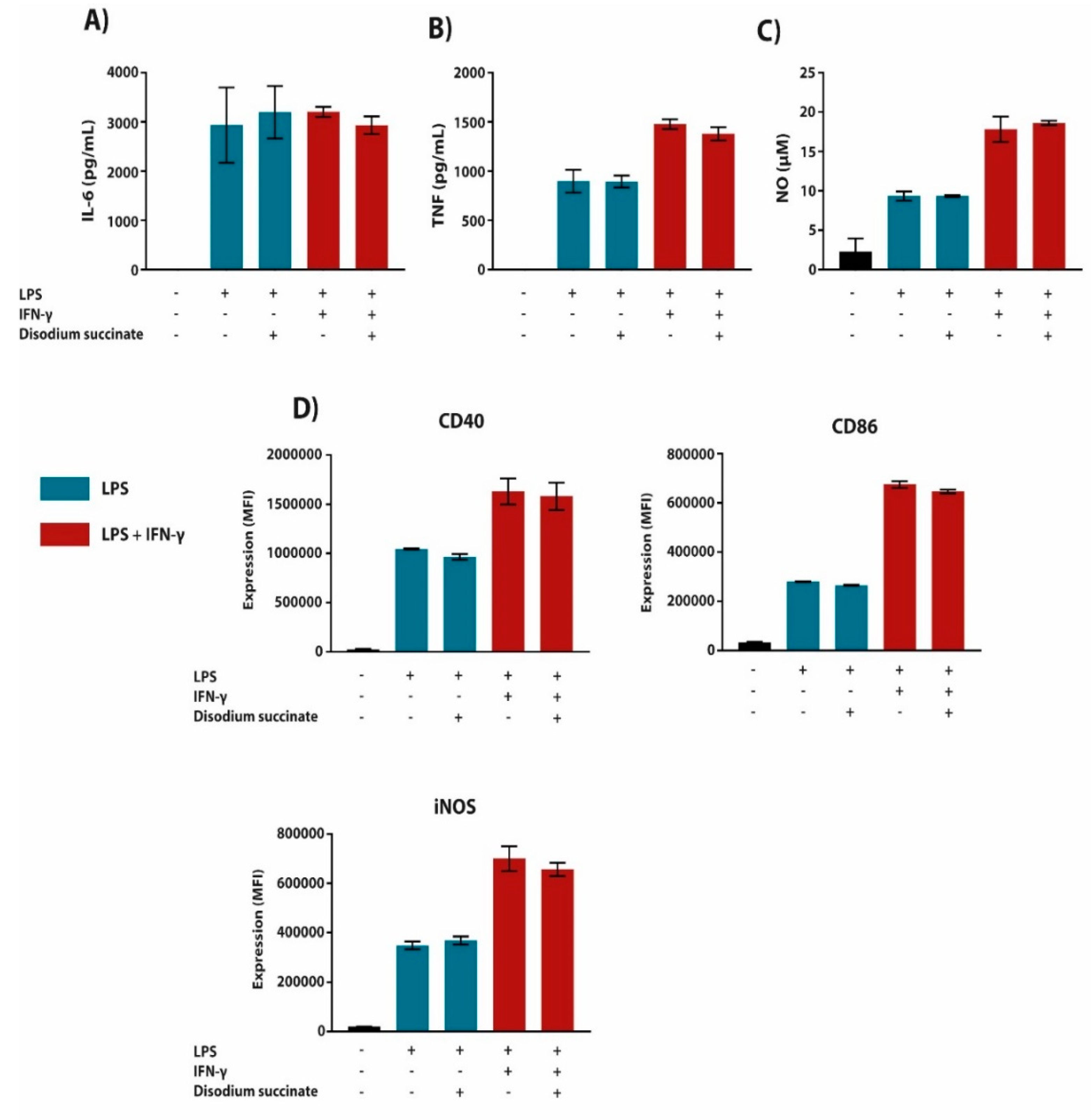
2.3. The Anti-Inflammatory Effect of Succinate is Mediated through SUCNR1-Independent Mechanisms
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Macrophage Cultures
4.3. Macrophage Function
4.4. Gene Expression Analysis
4.5. Flow Cytometry
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Neill, L.A. A critical role for citrate metabolism in LPS signalling. Biochem. J. 2011, 438, e5–e6. [Google Scholar] [CrossRef] [Green Version]
- Benit, P.; Letouze, E.; Rak, M.; Aubry, L.; Burnichon, N.; Favier, J.; Gimenez-Roqueplo, A.P.; Rustin, P. Unsuspected task for an old team: Succinate, fumarate and other krebs cycle acids in metabolic remodeling. Biochim. Biophys. Acta 2014, 1837, 1330–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvan-Pena, S.; O’Neill, L.A. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artyomov, M.N.; Sergushichev, A.; Schilling, J.D. Integrating immunometabolism and macrophage diversity. Semin. Immunol. 2016, 28, 417–424. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- O’Neill, L.A. A broken krebs cycle in macrophages. Immunity 2015, 42, 393–394. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Ryan, D.G.; O’Neill, L.A.J. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. 2017, 591, 2992–3006. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2018, 1, 16–33. [Google Scholar] [CrossRef]
- Murphy, M.P.; O’Neill, L.A.J. Krebs cycle reimagined: The emerging roles of succinate and itaconate as signal transducers. Cell 2018, 174, 780–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, E.L.; O’Neill, L.A. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur. J. Immunol. 2016, 46, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.; Cucchi, D.; Smith, J.; Pucino, V.; Macdougall, C.E.; Mauro, C. Intermediates of metabolism: From bystanders to signalling molecules. Trends Biochem. Sci. 2016, 41, 460–471. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwarzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef]
- Gilissen, J.; Jouret, F.; Pirotte, B.; Hanson, J. Insight into SUCNR1 (GPR91) structure and function. Pharmacol. Ther. 2016, 159, 56–65. [Google Scholar] [CrossRef]
- de Castro Fonseca, M.; Aguiar, C.J.; da Rocha Franco, J.A.; Gingold, R.N.; Leite, M.F. GPR91: Expanding the frontiers of Krebs cycle intermediates. Cell Commun. Signal 2016, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Huang, T.W.; Hsieh, Y.T.; Wang, Y.F.; Yen, C.C.; Lee, G.L.; Yeh, C.C.; Peng, Y.J.; Kuo, Y.Y.; Wen, H.T.; et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol. Cell 2020, 77, 213–227.e5. [Google Scholar] [CrossRef]
- Keiran, N.; Ceperuelo-Mallafre, V.; Calvo, E.; Hernandez-Alvarez, M.I.; Ejarque, M.; Nunez-Roa, C.; Horrillo, D.; Maymo-Masip, E.; Rodriguez, M.M.; Fradera, R.; et al. SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat. Immunol. 2019, 20, 581–592. [Google Scholar] [CrossRef]
- Baardman, J.; Verberk, S.G.S.; Prange, K.J.H.M.; van Weeghel, M.; van der Velden, S.; Ryan, D.G.; Wust, R.C.I.; Neele, A.E.; Speijer, D.; Denis, S.W.; et al. A defective pentose phosphate pathway reduces inflammatory macrophage responses during hypercholesterolemia. Cell Rep. 2018, 25, 2044–2052.e2045. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016, 167, 457–470.e413. [Google Scholar] [CrossRef] [Green Version]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Vicario, N.; Costa, A.S.H.; Kwok, C.K.; Leonardi, T.; Booty, L.M.; Bicci, I.; Balzarotti, B.; Volpe, G.; et al. Macrophage-derived extracellular succinate licenses neural stem cells to suppress chronic neuroinflammation. Cell Stem. Cell 2018, 22, 355–368.e313. [Google Scholar] [CrossRef] [Green Version]
- Infantino, V.; Iacobazzi, V.; Menga, A.; Avantaggiati, M.L.; Palmieri, F. A key role of the mitochondrial citrate carrier (SLC25A1) in TNFalpha- and IFNgamma-triggered inflammation. Biochim. Biophys. Acta 2014, 1839, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- He, W.H.; Miao, F.J.P.; Lin, D.C.H.; Schwandner, R.T.; Wang, Z.L.; Gao, J.H.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef]
- Robben, J.H.; Fenton, R.A.; Vargas, S.L.; Schweer, H.; Peti-Peterdi, J.; Deen, P.M.; Milligan, G. Localization of the succinate receptor in the distal nephron and its signaling in polarized MDCK cells. Kidney Int. 2009, 76, 1258–1267. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, K.; Xu, W.; Zhao, S.; Ye, D.; Wang, Y.; Xu, Y.; Zhou, L.; Chu, Y.; Zhang, C.; et al. SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1beta production and to prevent DSS-induced colitis in mice. Cell Rep. 2017, 19, 2331–2344. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Diepen, J.A.; Robben, J.H.; Hooiveld, G.J.; Carmone, C.; Alsady, M.; Boutens, L.; Bekkenkamp-Grovenstein, M.; Hijmans, A.; Engelke, U.F.H.; Wevers, R.A.; et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia 2017, 60, 1304–1313. [Google Scholar] [CrossRef] [Green Version]
- Van den Bossche, J.; Lamers, W.H.; Koehler, E.S.; Geuns, J.M.; Alhonen, L.; Uimari, A.; Pirnes-Karhu, S.; Van Overmeire, E.; Morias, Y.; Brys, L.; et al. Pivotal advance: Arginase-1-independent polyamine production stimulates the expression of IL-4-induced alternatively activated macrophage markers while inhibiting LPS-induced expression of inflammatory genes. J. Leukoc. Biol. 2012, 91, 685–699. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harber, K.J.; de Goede, K.E.; Verberk, S.G.S.; Meinster, E.; de Vries, H.E.; van Weeghel, M.; de Winther, M.P.J.; Van den Bossche, J. Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages. Metabolites 2020, 10, 372. https://doi.org/10.3390/metabo10090372
Harber KJ, de Goede KE, Verberk SGS, Meinster E, de Vries HE, van Weeghel M, de Winther MPJ, Van den Bossche J. Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages. Metabolites. 2020; 10(9):372. https://doi.org/10.3390/metabo10090372
Chicago/Turabian StyleHarber, Karl J., Kyra E. de Goede, Sanne G. S. Verberk, Elisa Meinster, Helga E. de Vries, Michel van Weeghel, Menno P. J. de Winther, and Jan Van den Bossche. 2020. "Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages" Metabolites 10, no. 9: 372. https://doi.org/10.3390/metabo10090372
APA StyleHarber, K. J., de Goede, K. E., Verberk, S. G. S., Meinster, E., de Vries, H. E., van Weeghel, M., de Winther, M. P. J., & Van den Bossche, J. (2020). Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages. Metabolites, 10(9), 372. https://doi.org/10.3390/metabo10090372