Metabolic Reprogramming of Cancer Cells during Tumor Progression and Metastasis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cancer Cell Adaptation to Tumor Microenvironments
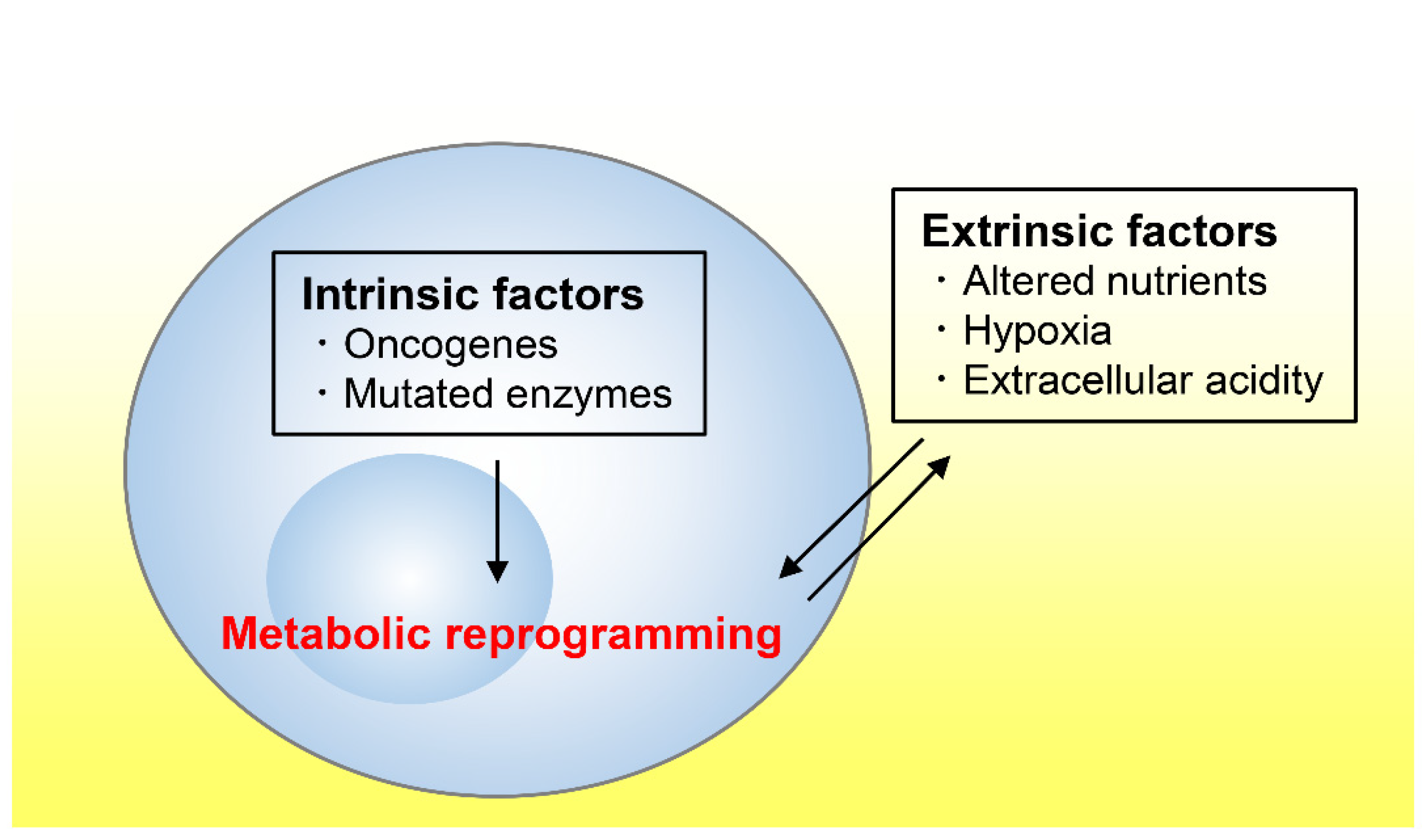
2.1. Cell-Intrinsic Factors Promoting Tumor Growth
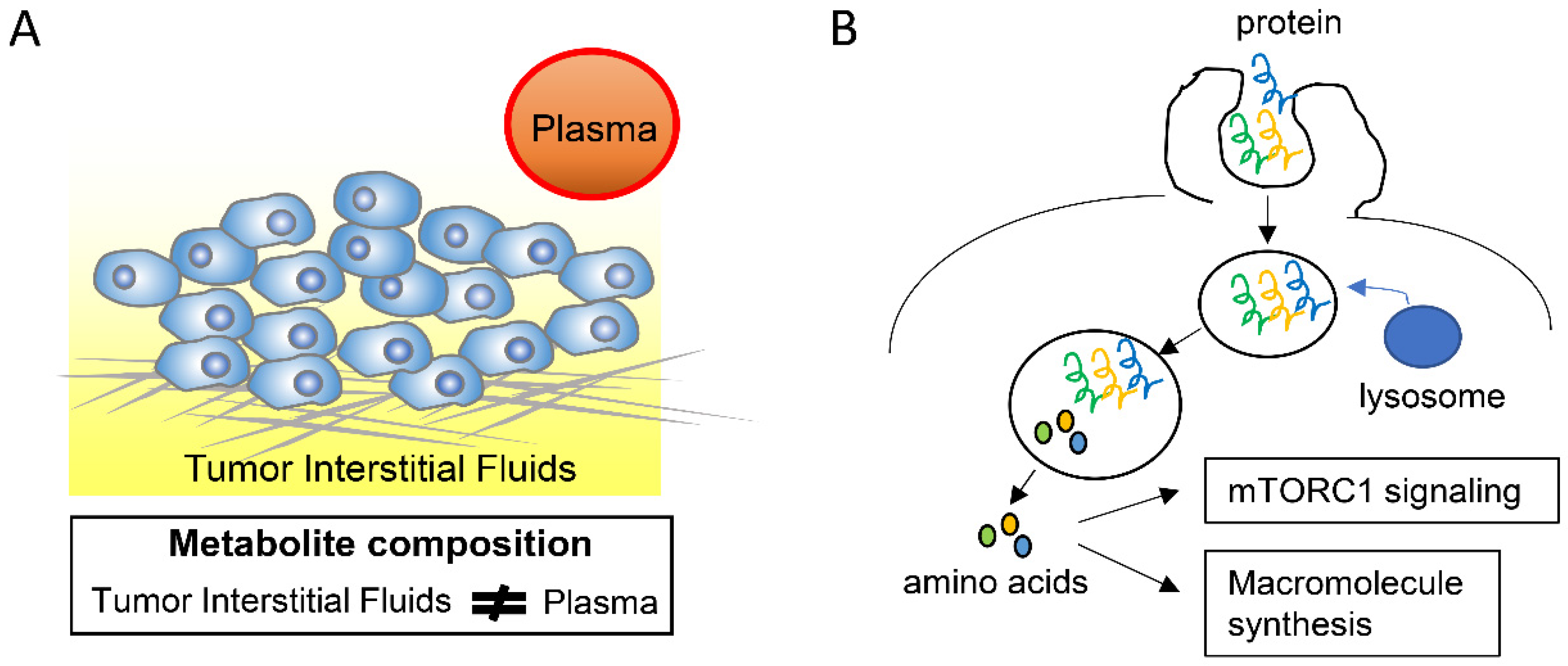
2.2. Adaptation to Nutrient-Altered Tumor Microenvironments
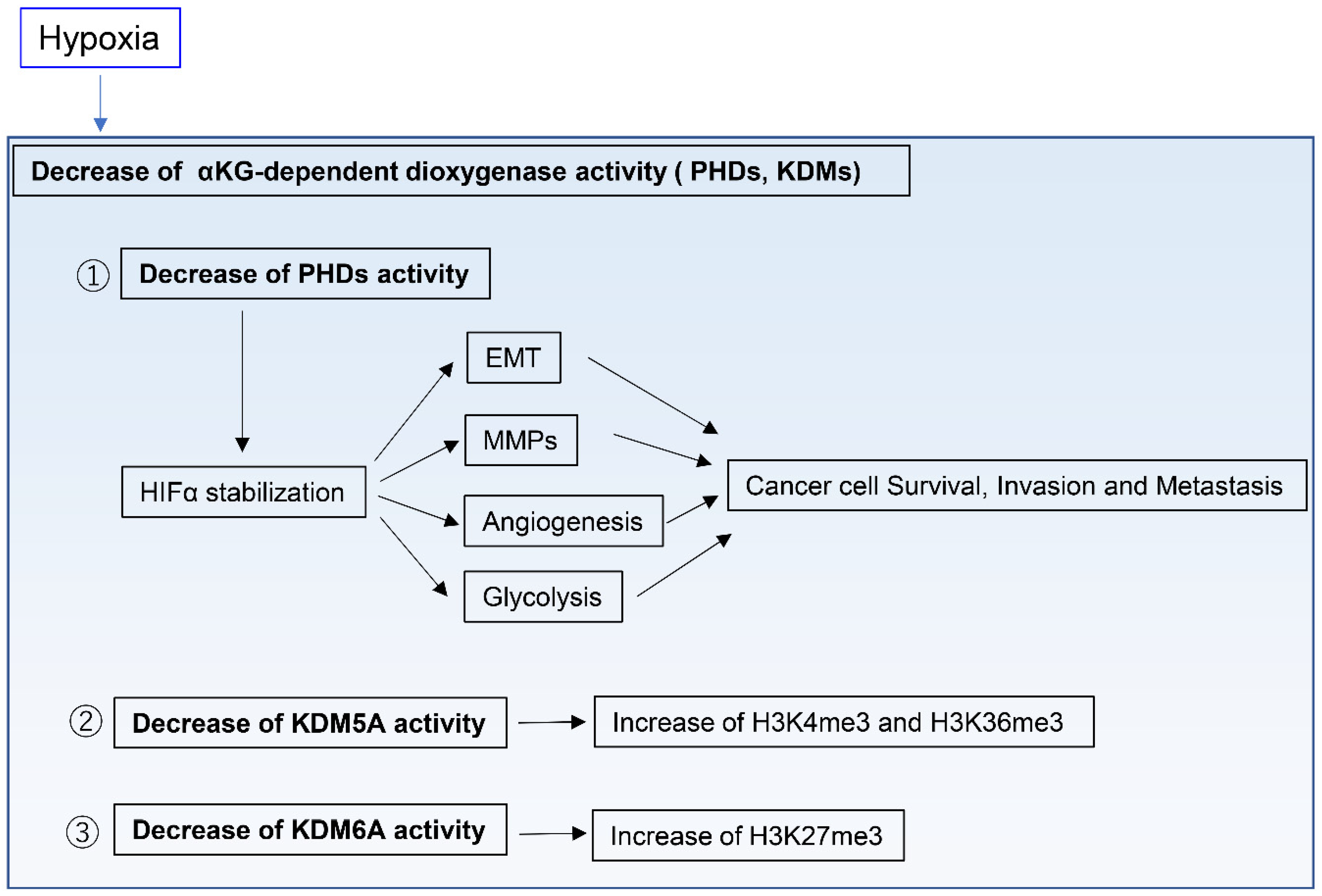
2.3. Adaptation to Hypoxic Tumor Microenvironments
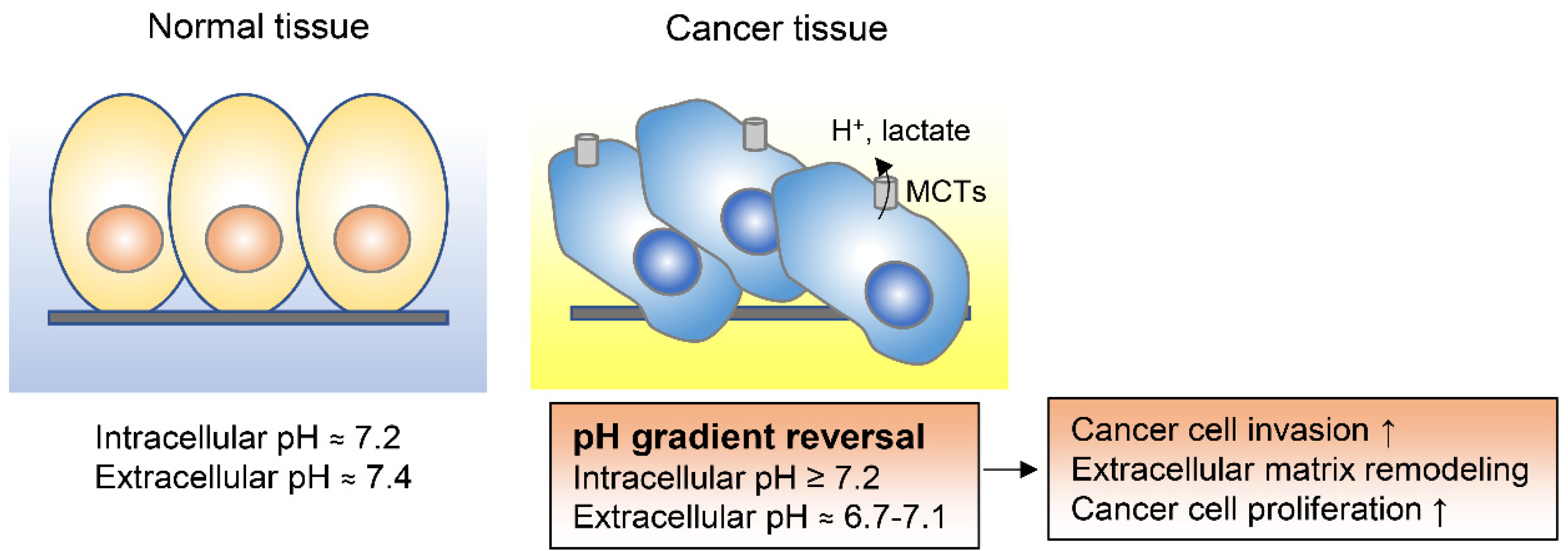
2.4. Adaptation to the Extracellular Acidification of Tumor Microenvironments
2.5. Metabolic Heterogeneity in the Tumor Tissues
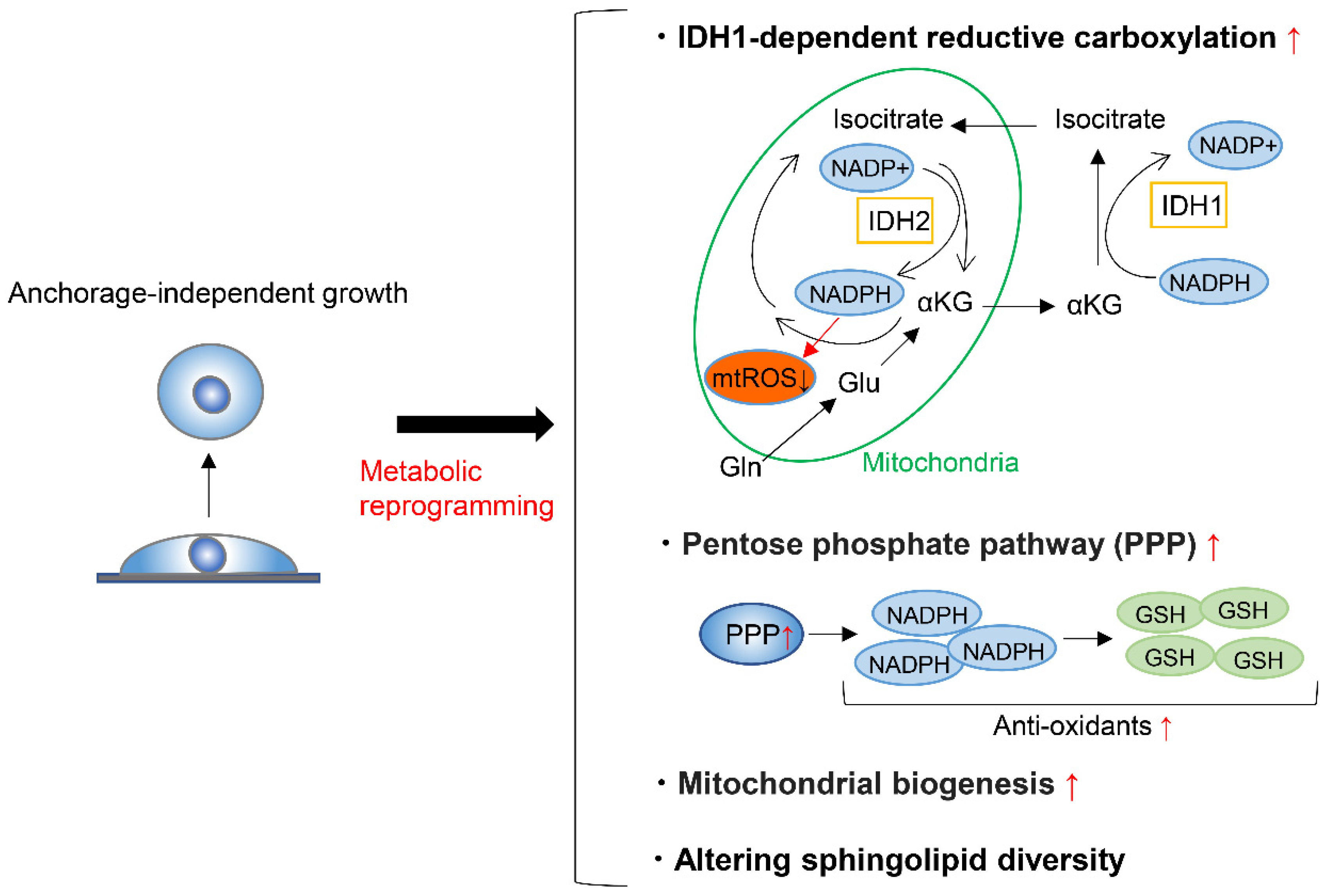
3. Metabolic Reprograming during Anchorage-Independent Growth
4. Metabolic Reprogramming to Form Metastatic Tumors
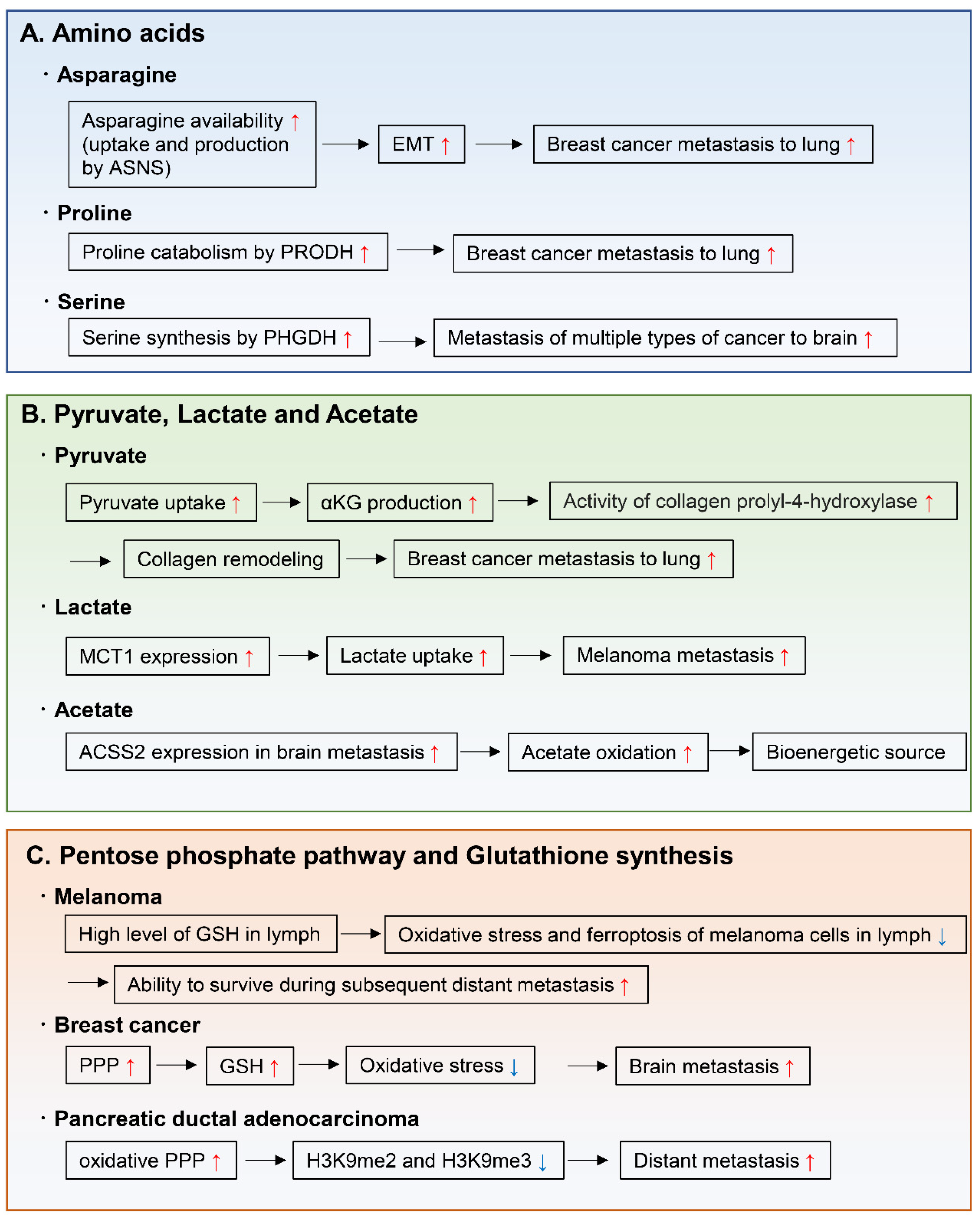
4.1. Amino Acids
4.2. Pyruvate, Lactate, and Acetate
4.3. Pentose Phosphate Pathway and Glutathione Synthesis
4.4. Lipid
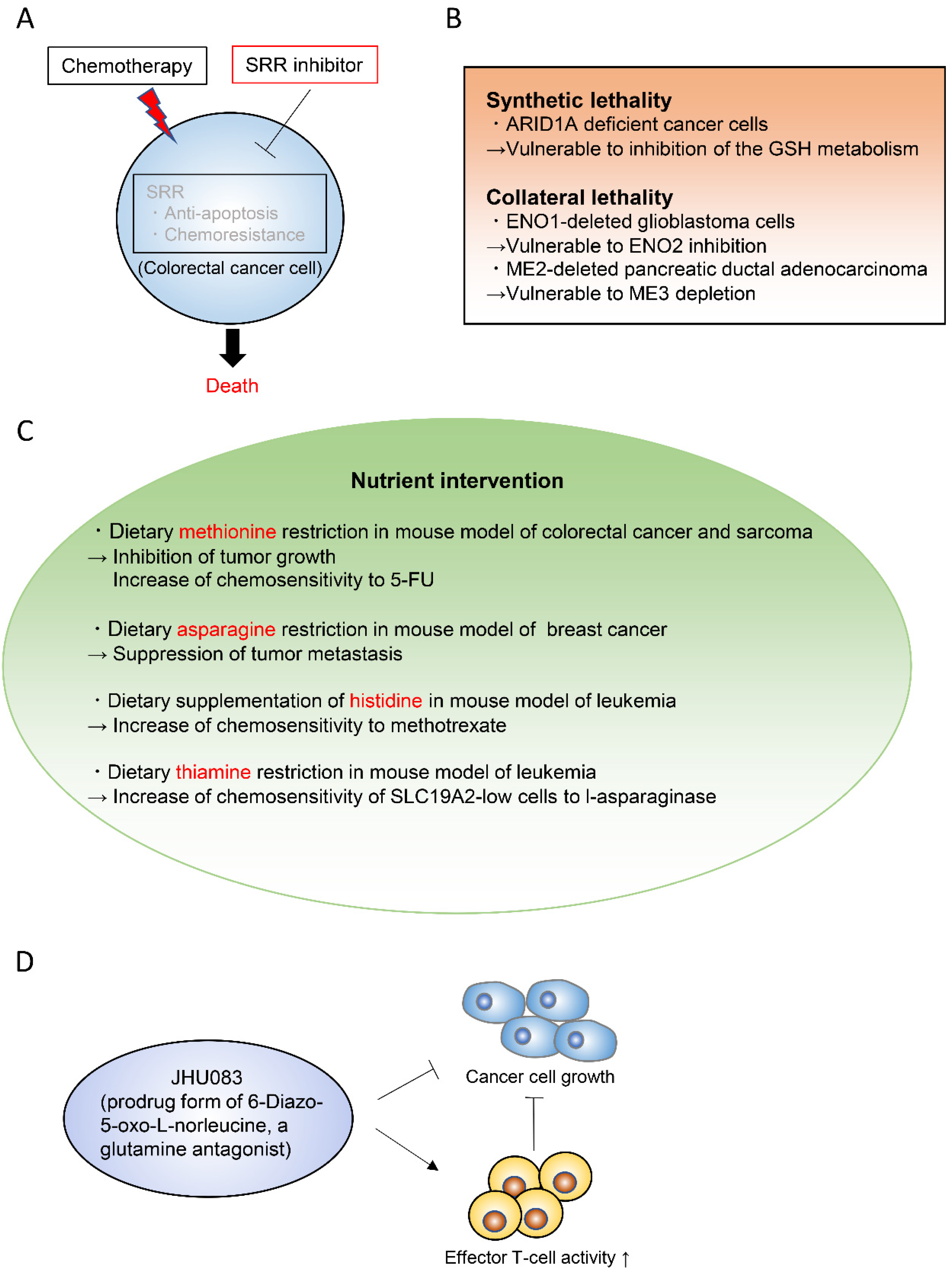
5. Emerging Therapeutic Strategies to Target Cancer Metabolism
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Schild, T.; Low, V.; Blenis, J.; Gomes, A.P. Unique Metabolic Adaptations Dictate Distal Organ-Specific Metastatic Colonization. Cancer Cell 2018, 33, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Berardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e160020. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Oncogene-Directed Alterations in Cancer Cell Metabolism. Trends Cancer 2016, 2, 365–377. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [Green Version]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matés, J.M.; Di Paola, F.J.; Campos-Sandoval, J.A.; Mazurek, S.; Márquez, J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin. Cell Dev. Biol. 2020, 98, 34–43. [Google Scholar]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Reyes, I.; Cardona, L.R.; Kong, H.; Vasan, K.; McElroy, G.S.; Werner, M.; Kihshen, H.; Reczek, C.R.; Weinberg, S.E.; Gao, P.; et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 2020, 585, 288–292. [Google Scholar] [CrossRef]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13C] glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.W.M.; Lane, A.N.; Higashi, R.M.; Farag, M.A.; Gao, H.; Bousamra, M.; Miller, D.M. Altered regulation of metabolic pathways in human lung cancer discerned by 13C stable isotope-resolved metabolomics (SIRM). Mol. Cancer 2009, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.N.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, K.G.; Mambetsariev, I.; Kulkarni, P.; Salgia, R. The Mitochondrion as an Emerging Therapeutic Target in Cancer. Trends Mol. Med. 2020, 26, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; Van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Sciacovelli, M.; Frezza, C. Fumarate hydratase in cancer: A multifaceted tumour suppressor. Semin. Cell Dev. Biol. 2020, 98, 15–25. [Google Scholar] [CrossRef]
- Bardella, C.; Pollard, P.J.; Tomlinson, I. SDH mutations in cancer. Biochim. Biophys. Acta-Bioenerg. 2011, 1807, 1432–1443. [Google Scholar] [CrossRef] [Green Version]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I.; Jahromi, M.S.; Xekouki, P.; et al. Succinate Dehydrogenase Mutation Underlies Global Epigenomic Divergence in Gastrointestinal Stromal Tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Pollard, P.J. Succinate: A new epigenetic hacker. Cancer Cell 2013, 23, 709–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; Da Costa, A.S.H.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.B.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Ohshima, K.; Nojima, S.; Tahara, S.; Kurashige, M.; Hori, Y.; Okuzaki, D.; Wada, N.; Ikeda, J.I.; Morii, E. Adenylosuccinate lyase enhances aggressiveness of endometrial cancer by increasing killer cell lectin-like receptor C3 expression by fumarate. Lab. Investig. 2018, 98, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnsworth, R.H.; Lackmann, M.; Achen, M.G.; Stacker, S.A. Vascular remodeling in cancer. Oncogene 2014, 33, 3496–3505. [Google Scholar] [CrossRef] [Green Version]
- Burgess, E.A.; Sylvén, B. Glucose, Lactate, and Lactic Dehydrogenase Activity in Normal Interstitial Fluid and That of Solid Mouse Tumors. Cancer Res. 1962, 22, 581–588. [Google Scholar]
- Gullino, P.M.; Grantham, F.H.; Courtney, A.H. Glucose Consumption by Transplanted Tumors in Vivo. Cancer Res. 1967, 27, 1031–1040. [Google Scholar]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Heiden, M.G.V.; Muir, A. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 2019, 8. [Google Scholar] [CrossRef]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Muir, A.; Danai, L.V.; Gui, D.Y.; Waingarten, C.Y.; Lewis, C.A.; Vander Heiden, M.G. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 2017, 6. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Vander Heiden, M.G. Determinants of nutrient limitation in cancer. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Lagziel, S.; Gottlieb, E.; Shlomi, T. Mind your media. Nat. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Abu-Remaileh, M.; Kanarek, N.; Freinkman, E.; Gao, X.; Louissaint, A.; Lewis, C.A.; Sabatini, D.M. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 2017, 169, 258–272.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savino, A.M.; Fernandes, S.I.; Olivares, O.; Zemlyansky, A.; Cousins, A.; Markert, E.K.; Barel, S.; Geron, I.; Frishman, L.; Birger, Y.; et al. Metabolic adaptation of acute lymphoblastic leukemia to the central nervous system microenvironment depends on stearoyl-CoA desaturase. Nat. Cancer 2020, 1, 998–1009. [Google Scholar] [CrossRef]
- Zeng, Q.; Michael, I.P.; Zhang, P.; Saghafinia, S.; Knott, G.; Jiao, W.; McCabe, B.D.; Galván, J.A.; Robinson, H.P.C.; Zlobec, I.; et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019, 573, 526–531. [Google Scholar] [CrossRef]
- Osawa, T.; Shimamura, T.; Saito, K.; Hasegawa, Y.; Ishii, N.; Nishida, M.; Ando, R.; Kondo, A.; Anwar, M.; Tsuchida, R.; et al. Phosphoethanolamine Accumulation Protects Cancer Cells under Glutamine Starvation through Downregulation of PCYT2. Cell Rep. 2019, 29, 89–103.e7. [Google Scholar] [CrossRef] [Green Version]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Palm, W.; Park, Y.; Wright, K.; Pavlova, N.N.; Tuveson, D.A.; Thompson, C.B. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 2015, 162, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M.; Jonas, O.; Keibler, M.A.; Hou, H.W.; Luengo, A.; Mayers, J.R.; Wyckoff, J.; Del Rosario, A.M.; Whitman, M.; Chin, C.R.; et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 2017, 23, 235–241. [Google Scholar] [CrossRef]
- Swanson, J.A.; Watts, C. Macropinocytosis. Trends Cell Biol. 1995, 5, 424–428. [Google Scholar] [CrossRef]
- Falk, S.J.; Ward, R.; Bleehen, N.M. The influence of carbogen breathing on tumour tissue oxygenation in man evaluated by computerised p02 histography. Br. J. Cancer 1992, 66, 919–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, Q.T.; Chen, E.; Salim, A.; Cao, H.; Kong, C.S.; Whyte, R.; Donington, J.; Cannon, W.; Wakelee, H.; Tibshirani, R.; et al. An evaluation of tumor oxygenation and gene expression in patients with early stage non-small cell lung cancers. Clin. Cancer Res. 2006, 12, 1507–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxidants Redox Signal. 2007, 9, 1221–1235. [Google Scholar] [CrossRef] [PubMed]
- Ast, T.; Mootha, V.K. Oxygen and mammalian cell culture: Are we repeating the experiment of Dr. Ox? Nat. Metab. 2019, 1, 858–860. [Google Scholar] [CrossRef] [PubMed]
- Mckeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmlinger, G.; Yuan, F.; Dellian, M.; Jain, R.K. Interstitial pH and pO2 gradients in solid tumors in vivo: High-resolution measurements reveal a lack of correlation. Nat. Med. 1997, 3, 177–182. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Rankin, E.B.; Nam, J.-M.; Giaccia, A.J. Hypoxia: Signaling the Metastatic Cascade. Trends Cancer 2016, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, X.; Li, J.; Zhang, C.; Hu, T.; Li, S.; He, S.; Yan, H.; Tan, Y.; Lei, M.; Wen, M.; et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017, 4, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Semenza, G.L. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 382–391. [Google Scholar] [CrossRef]
- Krishnamachary, B.; Berg-Dixon, S.; Kelly, B.; Agani, F.; Feldser, D.; Ferreira, G.; Iyer, N.; Larusch, J.; Pak, B.; Taghavi, P.; et al. Regulation of Colon Carcinoma Cell Invasion by Hypoxia-Inducible Factor 1. Cancer Res. 2003, 63, 1138–1143. [Google Scholar]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L.L. HIF-1 Regulates Cytochrome Oxidase Subunits to Optimize Efficiency of Respiration in Hypoxic Cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Cooper, C.E. The steady-state kinetics of cytochrome c oxidation by cytochrome oxidase. BBA-Bioenerg. 1990, 1017, 187–203. [Google Scholar] [CrossRef]
- Wilson, D.F.; Rumsey, W.L.; Green, T.J.; Vanderkooi, J.M. The oxygen dependence of mitochondrial oxidative phosphorylation measured by a new optical method for measuring oxygen concentration. J. Biol. Chem. 1988, 263, 2712–2718. [Google Scholar] [PubMed]
- Hollinshead, K.E.R.; Parker, S.J.; Eapen, V.V.; Encarnacion-Rosado, J.; Sohn, A.; Oncu, T.; Cammer, M.; Mancias, J.D.; Kimmelman, A.C. Respiratory Supercomplexes Promote Mitochondrial Efficiency and Growth in Severely Hypoxic Pancreatic Cancer. Cell Rep. 2020, 33, 108231. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.A.; Laukka, T.; Myllykoski, M.; Ringel, A.E.; Booker, M.A.; Tolstorukov, M.Y.; Meng, Y.J.; Meier, S.R.; Jennings, R.B.; Creech, A.L.; et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 2019, 363, 1217–1222. [Google Scholar] [CrossRef] [Green Version]
- Batie, M.; Frost, J.; Frost, M.; Wilson, J.W.; Schofield, P.; Rocha, S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 2019, 363, 1222–1226. [Google Scholar] [CrossRef]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130099. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.R.; Jain, R.K. Noninvasive Measurement of Interstitial pH Profiles in Normal and Neoplastic Tissue Using Fluorescence Ratio Imaging Microscopy. Cancer Res. 1994, 54, 5670–5674. [Google Scholar]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13. [Google Scholar] [CrossRef] [Green Version]
- Robertson-Tessi, M.; Gillies, R.J.; Gatenby, R.A.; Anderson, A.R.A. Impact of metabolic heterogeneity on tumor growth, invasion, and treatment outcomes. Cancer Res. 2015, 75, 1567–1579. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Smallbone, K.; Maini, P.K.; Rose, F.; Averill, J.; Nagle, R.B.; Worrall, L.; Gillies, R.J. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br. J. Cancer 2007, 97, 646–653. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, M.C.; Cunningham, J.J.; Bui, M.M.; Gillies, R.J.; Brown, J.S.; Gatenby, R.A. Darwinian dynamics of intratumoral heterogeneity: Not solely random mutations but also variable environmental selection forces. Cancer Res. 2016, 76, 3136–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rofstad, E.K.; Mathiesen, B.; Kindem, K.; Galappathi, K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006, 66, 6699–6707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtkowiak, J.W.; Verduzco, D.; Schramm, K.J.; Gillies, R.J. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol. Pharm. 2011, 8, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Yamamoto, S.; Nakaki, R.; Shimamura, T.; Hamakubo, T.; Sakai, J.; Kodama, T.; Yoshida, T.; Aburatani, H.; Osawa, T. Extracellular Acidic pH Activates the Sterol Regulatory Element-Binding Protein 2 to Promote Tumor Progression. Cell Rep. 2017, 18, 2228–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohani, N.; Hao, L.; Alexis, M.S.; Joughin, B.A.; Krismer, K.; Moufarrej, M.N.; Soltis, A.R.; Lauffenburger, D.A.; Yaffe, M.B.; Burge, C.B.; et al. Acidification of tumor at stromal boundaries drives transcriptome alterations associated with aggressive phenotypes. Cancer Res. 2019, 79, 1952–1966. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Persi, E.; Duran-Frigola, M.; Damaghi, M.; Roush, W.R.; Aloy, P.; Cleveland, J.L.; Gillies, R.J.; Ruppin, E. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat. Commun. 2018, 9, 2997. [Google Scholar] [CrossRef]
- Gerweck, L.E.; Seetharaman, K. Cellular pH Gradient in Tumor versus Normal Tissue: Potential Exploitation for the Treatment of Cancer. Cancer Res. 1996, 56, 1194–1198. [Google Scholar]
- Stüwe, L.; Müller, M.; Fabian, A.; Waning, J.; Mally, S.; Noël, J.; Schwab, A.; Stock, C. pH dependence of melanoma cell migration: Protons extruded by NHE1 dominate protons of the bulk solution. J. Physiol. 2007, 585, 351–360. [Google Scholar] [CrossRef]
- Busco, G.; Cardone, R.A.; Greco, M.R.; Bellizzi, A.; Colella, M.; Antelmi, E.; Mancini, M.T.; Dell’Aquila, M.E.; Casavola, V.; Paradiso, A.; et al. NHE1 promotes invadopodial ECM proteolysis through acidification of the peri-invadopodial space. FASEB J. 2010, 24, 3903–3915. [Google Scholar] [CrossRef]
- Koch, L.M.; Birkeland, E.S.; Battaglioni, S.; Helle, X.; Meerang, M.; Hiltbrunner, S.; Ibáñez, A.J.; Peter, M.; Curioni-Fontecedro, A.; Opitz, I.; et al. Cytosolic pH regulates proliferation and tumour growth by promoting expression of cyclin D1. Nat. Metab. 2020, 2, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Kessenbrock, K.; Davis, R.T.; Pervolarakis, N.; Werb, Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 2018, 20, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Dvorak, H.F. Heterogeneity of the tumor vasculature: The need for new tumor blood vessel type-specific targets. Clin. Exp. Metastasis 2012, 29, 657–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, A.; Vander Heiden, M.G. The nutrient environment affects therapy. Science 2018, 360, 962–963. [Google Scholar] [CrossRef] [PubMed]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Vander Heiden, M.G. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Ciocan-Cartita, C.A.; Jurj, A.; Buse, M.; Gulei, D.; Braicu, C.; Raduly, L.; Cojocneanu, R.; Pruteanu, L.L.; Iuga, C.A.; Coza, O.; et al. The relevance of mass spectrometry analysis for personalized medicine through its successful application in cancer “Omics”. Int. J. Mol. Sci. 2019, 20, 2576. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Li, T.; Song, X.; Huang, L.; Zang, Q.; Xu, J.; Bi, N.; Jiao, G.; Hao, Y.; Chen, Y.; et al. Spatially resolved metabolomics to discover tumor-associated metabolic alterations. Proc. Natl. Acad. Sci. USA 2019, 116, 52–57. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, D.; Pratx, G. Imaging metabolic heterogeneity in cancer. Mol. Cancer 2016, 15, 4. [Google Scholar] [CrossRef] [Green Version]
- Momcilovic, M.; Jones, A.; Bailey, S.T.; Waldmann, C.M.; Li, R.; Lee, J.T.; Abdelhady, G.; Gomez, A.; Holloway, T.; Schmid, E.; et al. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature 2019, 575, 380–384. [Google Scholar] [CrossRef]
- Gallagher, F.A.; Woitek, R.; McLean, M.A.; Gill, A.B.; Garcia, R.M.; Provenzano, E.; Riemer, F.; Kaggie, J.; Chhabra, A.; Ursprung, S.; et al. Imaging breast cancer using hyperpolarized carbon-13 MRI. Proc. Natl. Acad. Sci. USA 2020, 117, 2092–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Sharife, H.; Kreisel, T.; Mogilevsky, M.; Bar-Lev, L.; Grunewald, M.; Aizenshtein, E.; Karni, R.; Paldor, I.; Shlomi, T.; et al. Intra-Tumoral Metabolic Zonation and Resultant Phenotypic Diversification Are Dictated by Blood Vessel Proximity. Cell Metab. 2019, 30, 201–211.e6. [Google Scholar] [CrossRef] [PubMed]
- Commander, R.; Wei, C.; Sharma, A.; Mouw, J.K.; Burton, L.J.; Summerbell, E.; Mahboubi, D.; Peterson, R.J.; Konen, J.; Zhou, W.; et al. Subpopulation targeting of pyruvate dehydrogenase and GLUT1 decouples metabolic heterogeneity during collective cancer cell invasion. Nat. Commun. 2020, 11, 1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima, K.; Nojima, S.; Tahara, S.; Kurashige, M.; Hori, Y.; Hagiwara, K.; Okuzaki, D.; Oki, S.; Wada, N.; Ikeda, J.I.; et al. Argininosuccinate Synthase 1-Deficiency Enhances the Cell Sensitivity to Arginine through Decreased DEPTOR Expression in Endometrial Cancer. Sci. Rep. 2017, 7, 45504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stott, S.L.; Richard, L.; Nagrath, S.; Min, Y.; Miyamoto, D.T.; Ulkus, L.; Inserra, E.J.; Ulman, M.; Springer, S.; Nakamura, Z.; et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci. Transl. Med. 2010, 2, 25ra23. [Google Scholar] [CrossRef] [Green Version]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef] [Green Version]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bäuerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef]
- Bueno, M.J.; Jimenez-Renard, V.; Samino, S.; Capellades, J.; Junza, A.; López-Rodríguez, M.L.; Garcia-Carceles, J.; Lopez-Fabuel, I.; Bolaños, J.P.; Chandel, N.S.; et al. Essentiality of fatty acid synthase in the 2D to anchorage-independent growth transition in transforming cells. Nat. Commun. 2019, 10, 5011. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labuschagne, C.F.; Cheung, E.C.; Blagih, J.; Domart, M.C.; Vousden, K.H. Cell Clustering Promotes a Metabolic Switch that Supports Metastatic Colonization. Cell Metab. 2019, 30, 720–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; De Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef] [Green Version]
- Muthusamy, T.; Cordes, T.; Handzlik, M.K.; You, L.; Lim, E.W.; Gengatharan, J.; Pinto, A.F.M.; Badur, M.G.; Kolar, M.J.; Wallace, M.; et al. Serine restriction alters sphingolipid diversity to constrain tumour growth. Nature 2020, 586, 790–795. [Google Scholar] [CrossRef]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.; Grünewald, T.G.P.; Fendt, S.M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Mattaini, K.R.; Dennstedt, E.A.; Nguyen, A.A.; Sivanand, S.; Reilly, M.F.; Meeth, K.; Muir, A.; Darnell, A.M.; Bosenberg, M.W.; et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. 2019, 29, 1410–1421.e4. [Google Scholar] [CrossRef]
- Rainesalo, S.; Keränen, T.; Palmio, J.; Peltola, J.; Oja, S.S.; Saransaari, P. Plasma and Cerebrospinal Fluid Amino Acids in Epileptic Patients. Neurochem Res. 2004, 29, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Dolgodilina, E.; Imobersteg, S.; Laczko, E.; Welt, T.; Verrey, F.; Makrides, V. Brain interstitial fluid glutamine homeostasis is controlled by amino acid transporter. J. Cereb. Blood Flow Metab. 2016, 36, 1929–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, B.; Kim, E.; Osorio-Vasquez, V.; Doll, S.; Bustraan, S.; Liang, R.J.; Luengo, A.; Davidson, S.M.; Ali, A.; Ferraro, G.B.; et al. Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH Inhibition. Cancer Discov. 2020, 10, 1352–1373. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Chaturvedi, P.; Bajpai, S.; Wong, C.C.; Wei, H.; Pitcairn, S.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Collagen Prolyl Hydroxylases Are Essential for Breast Cancer Metastasis. Cancer Res. 2013, 73, 3285–3297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elia, I.; Rossi, M.; Stegen, S.; Broekaert, D.; Doglioni, G.; van Gorsel, M.; Boon, R.; Escalona-Noguero, C.; Torrekens, S.; Verfaillie, C.; et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 2019, 568, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Christen, S.; Lorendeau, D.; Schmieder, R.; Christen, S.; Lorendeau, D.; Schmieder, R.; Broekaert, D.; Metzger, K.; Veys, K. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. 2016, 17, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate metabolism in human lung tumors. Cell 2017, 171, 358–371. [Google Scholar] [CrossRef] [Green Version]
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcdonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Iii, J.R.Y.; Felding-habermann, B. Adaptation of Energy Metabolism in Breast Cancer Brain Metastases. Cancer Res. 2007, 67, 1472–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020, 585, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 76. [Google Scholar] [CrossRef] [Green Version]
- Peck, B.; Schulze, A. Lipid Metabolism at the Nexus of Diet and Tumor Microenvironment. Trends Cancer 2019, 5, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Wen, J.; Min, X.; Shen, M.; Hua, Q.; Han, Y.; Zhao, L.; Liu, L.; Huang, G.; Liu, J.; Zhao, X. ACLY facilitates colon cancer cell metastasis by CTNNB1. J. Exp. Clin. Cancer Res. 2019, 38, 401. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.W.; Lin, Y.H.; Pai, M.H.; Lo, A.C.; Lee, Y.C.; Fang, I.C.; Lin, J.; Hsieh, R.K.; Chang, Y.F.; Chen, C.L. Association between phosphorylated AMP-activated protein kinase and acetyl-CoA carboxylase expression and outcome in patients with squamous cell carcinoma of the head and neck. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.D.; Wu, H.; Fu, G.B.; Zhang, H.L.; Zhou, X.; Tang, L.; Dong, L.W.; Qin, C.J.; Huang, S.; Zhao, L.H.; et al. Acetyl-coenzyme A carboxylase alpha promotion of glucose-mediated fatty acid synthesis enhances survival of hepatocellular carcinoma in mice and patients. Hepatology 2016, 63, 1272–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Wang, H.; Li, J.; Fang, X.; Pan, H.; Yuan, X.; Zhang, P. Up-regulated FASN expression promotes transcoelomic metastasis of ovarian cancer cell through epithelial-mesenchymal transition. Int. J. Mol. Sci. 2014, 15, 11539–11554. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.J.; Hou, J.; Xu, B.; Cortez, M.; Potma, E.O.; Tromberg, B.J.; Razorenova, O.V. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc. Natl. Acad. Sci. USA 2017, 114, E6556–E6565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef] [Green Version]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef]
- Pan, J.; Fan, Z.; Wang, Z.; Dai, Q.; Xiang, Z.; Yuan, F.; Yan, M.; Zhu, Z.; Liu, B.; Li, C. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef] [Green Version]
- Farber, S.; Diamond, L.K.; Mercer, R.D.; Sylvester, R.F.; Wolff, J.A. Temporary Remissions in Acute Leukemia in Children Produced by Folic Acid Antagonist, 4-Aminopteroyl-Glutamic Acid (Aminopterin). N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef]
- Newman, A.C.; Maddocks, O.D.K. Serine and Functional Metabolites in Cancer. Trends Cell Biol. 2017, 27, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.; Zhao, Y. Dysregulated metabolic enzymes and metabolic reprogramming in cancer cells. Biomed. Rep. 2018, 8, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Foltyn, V.N.; Bendikov, I.; De Miranda, J.; Panizzutti, R.; Dumin, E.; Shleper, M.; Li, P.; Toney, M.D.; Kartvelishvily, E.; Wolosker, H. Serine racemase modulates intracellular D-serine levels through an α,β-elimination activity. J. Biol. Chem. 2005, 280, 1754–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Miranda, J.; Panizzutti, R.; Foltyn, V.N.; Wolosker, H. Cofactors of serine racemase that physiologically stimulate the synthesis of the N-methyl-D-aspartate (NMDA) receptor coagonist D-serine. Proc. Natl. Acad. Sci. USA 2002, 99, 14542–14547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolosker, H.; Blackshaw, S.; Snyder, S.H. Serine racemase: A glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc. Natl. Acad. Sci. USA 1999, 96, 13409–13414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima, K.; Nojima, S.; Tahara, S.; Kurashige, M.; Kawasaki, K.; Hori, Y.; Taniguchi, M.; Umakoshi, Y.; Okuzaki, D.; Wada, N.; et al. Serine racemase enhances growth of colorectal cancer by producing pyruvate from serine. Nat. Metab. 2020, 2, 81–96. [Google Scholar] [CrossRef]
- Muller, F.L.; Aquilanti, E.A.; Depinho, R.A. Collateral Lethality: A New Therapeutic Strategy in Oncology. Trends Cancer 2015, 1, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Zecchini, V.; Frezza, C. Metabolic synthetic lethality in cancer therapy. Biochim. Biophys. Acta-Bioenerg. 2017, 1858, 723–731. [Google Scholar] [CrossRef]
- Caumanns, J.J.; Wisman, G.B.A.; Berns, K.; van der Zee, A.G.J.; de Jong, S. ARID1A mutant ovarian clear cell carcinoma: A clear target for synthetic lethal strategies. Biochim. Biophys. Acta-Rev. Cancer 2018, 1870, 176–184. [Google Scholar] [CrossRef]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190.e8. [Google Scholar] [CrossRef] [Green Version]
- Muller, F.L.; Colla, S.; Aquilanti, E.; Manzo, V.E.; Genovese, G.; Lee, J.; Eisenson, D.; Narurkar, R.; Deng, P.; Nezi, L.; et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 2012, 488, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, P.; Baddour, J.; Muller, F.; Wu, C.C.; Wang, H.; Liao, W.T.; Lan, Z.; Chen, A.; Gutschner, T.; Kang, Y.; et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 2017, 542, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Tajan, M.; Vousden, K.H. Dietary Approaches to Cancer Therapy. Cancer Cell 2020, 37, 767–785. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Allen, A.E.; Locasale, J.W. The Molecular Link from Diet to Cancer Cell Metabolism. Mol. Cell 2020, 78, 1034–1044. [Google Scholar] [CrossRef]
- Lien, E.C.; Vander Heiden, M.G. A framework for examining how diet impacts tumour metabolism. Nat. Rev. Cancer 2019, 19, 651–661. [Google Scholar] [CrossRef]
- Gao, X.; Sanderson, S.M.; Dai, Z.; Reid, M.A.; Cooper, D.E.; Lu, M.; Richie, J.P.; Ciccarella, A.; Calcagnotto, A.; Mikhael, P.G.; et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 2019, 572, 397–401. [Google Scholar] [CrossRef]
- Kanarek, N.; Petrova, B.; Sabatini, D.M. Dietary modifications for enhanced cancer therapy. Nature 2020, 579, 507–517. [Google Scholar] [CrossRef]
- Kanarek, N.; Keys, H.R.; Cantor, J.R.; Lewis, C.A.; Chan, S.H.; Kunchok, T.; Abu-Remaileh, M.; Freinkman, E.; Schweitzer, L.D.; Sabatini, D.M. Histidine catabolism is a major determinant of methotrexate sensitivity. Nature 2018, 559, 632–636. [Google Scholar] [CrossRef]
- Guarecuco, R.; Williams, R.T.; Baudrier, L.; La, K.; Passarelli, M.C.; Ekizoglu, N.; Mestanoglu, M.; Alwaseem, H.; Rostandy, B.; Fidelin, J.; et al. Dietary thiamine influences l-asparaginase sensitivity in a subset of leukemia cells. Sci. Adv. 2020, 6, eabc7120. [Google Scholar] [CrossRef]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouidhi, S.; Ben Ayed, F.; Benammar Elgaaied, A. Targeting tumor metabolism: A new challenge to improve immunotherapy. Front. Immunol. 2018, 9, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevchenko, I.; Bazhin, A.V. Metabolic checkpoints: Novel avenues for immunotherapy of cancer. Front. Immunol. 2018, 9, 1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J. Tumor Microenvironment, Metabolism, and Immunotherapy. N. Engl. J. Med. 2020, 382, 869–871. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohshima, K.; Morii, E. Metabolic Reprogramming of Cancer Cells during Tumor Progression and Metastasis. Metabolites 2021, 11, 28. https://doi.org/10.3390/metabo11010028
Ohshima K, Morii E. Metabolic Reprogramming of Cancer Cells during Tumor Progression and Metastasis. Metabolites. 2021; 11(1):28. https://doi.org/10.3390/metabo11010028
Chicago/Turabian StyleOhshima, Kenji, and Eiichi Morii. 2021. "Metabolic Reprogramming of Cancer Cells during Tumor Progression and Metastasis" Metabolites 11, no. 1: 28. https://doi.org/10.3390/metabo11010028
APA StyleOhshima, K., & Morii, E. (2021). Metabolic Reprogramming of Cancer Cells during Tumor Progression and Metastasis. Metabolites, 11(1), 28. https://doi.org/10.3390/metabo11010028