
Perspective on the Structural Basis for Human Aldo-Keto Reductase 1B10 Inhibition



Abstract
:1. Introduction
2. AKR1B10 Inhibition Strategies
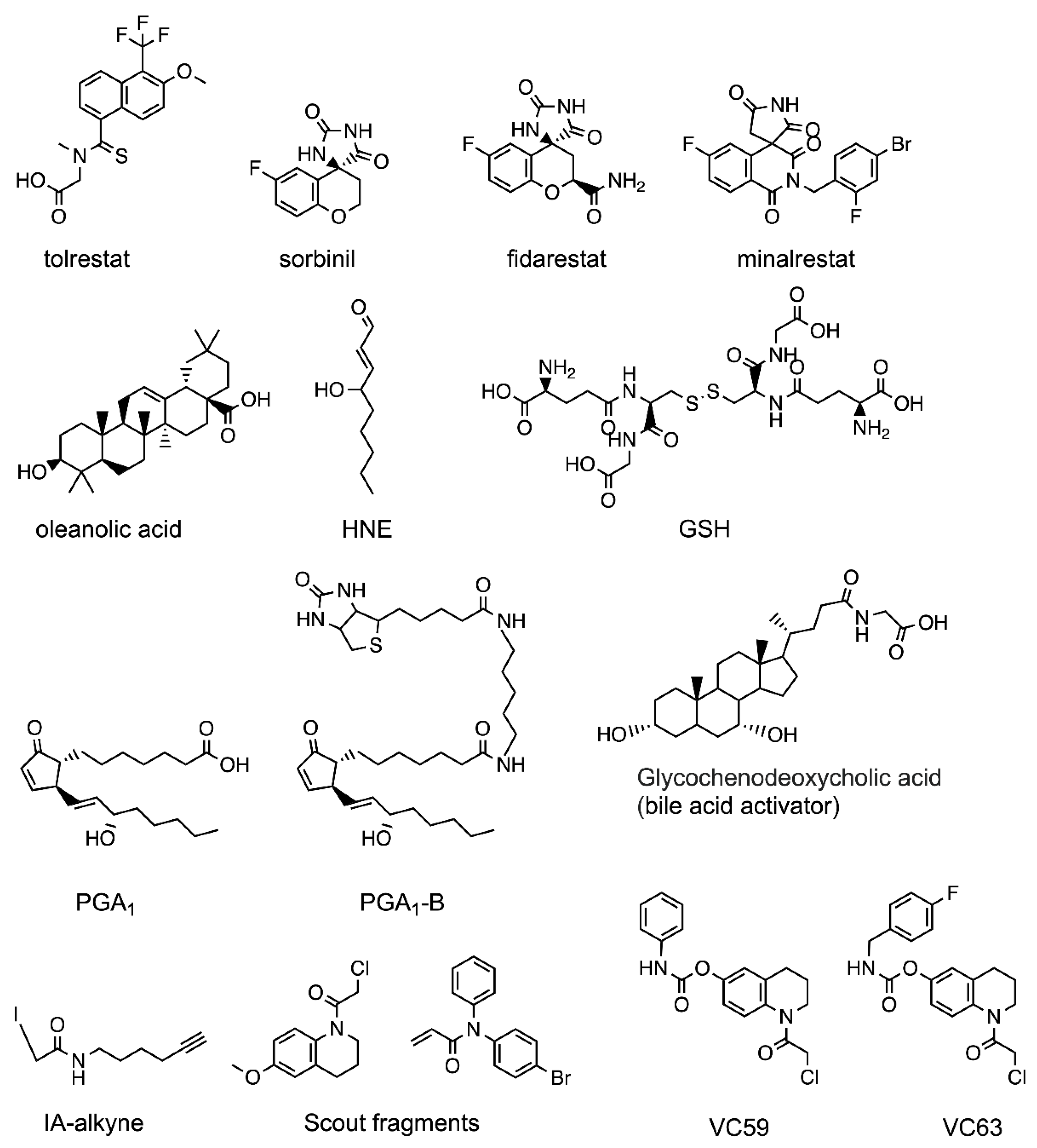
2.1. AKR1B10 Reversible Inhibitors
2.2. AKR1B10 Covalent Inhibitors
2.3. Potential for AKR1B10 Catalytic Activators
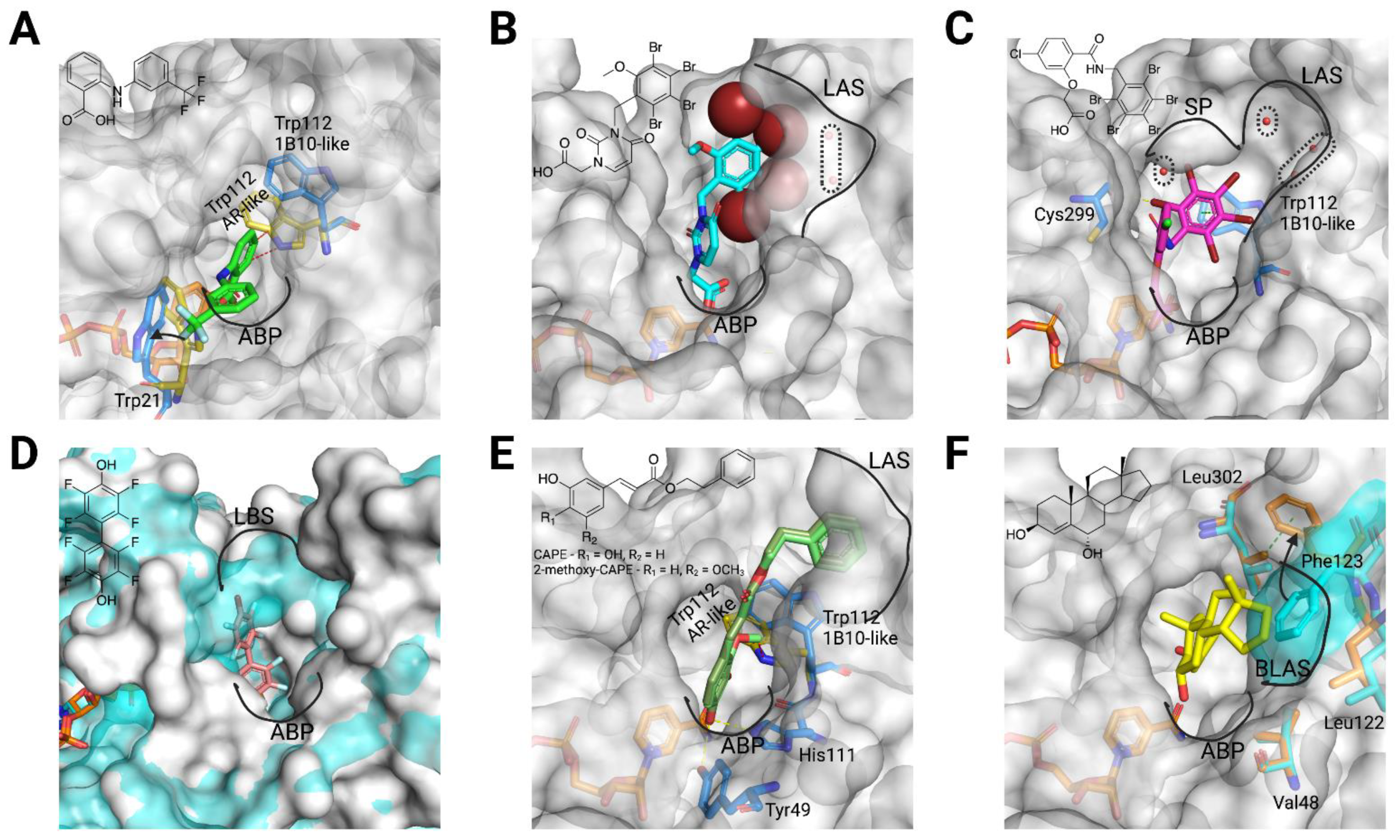
3. What We Have Learnt from 3D Structures
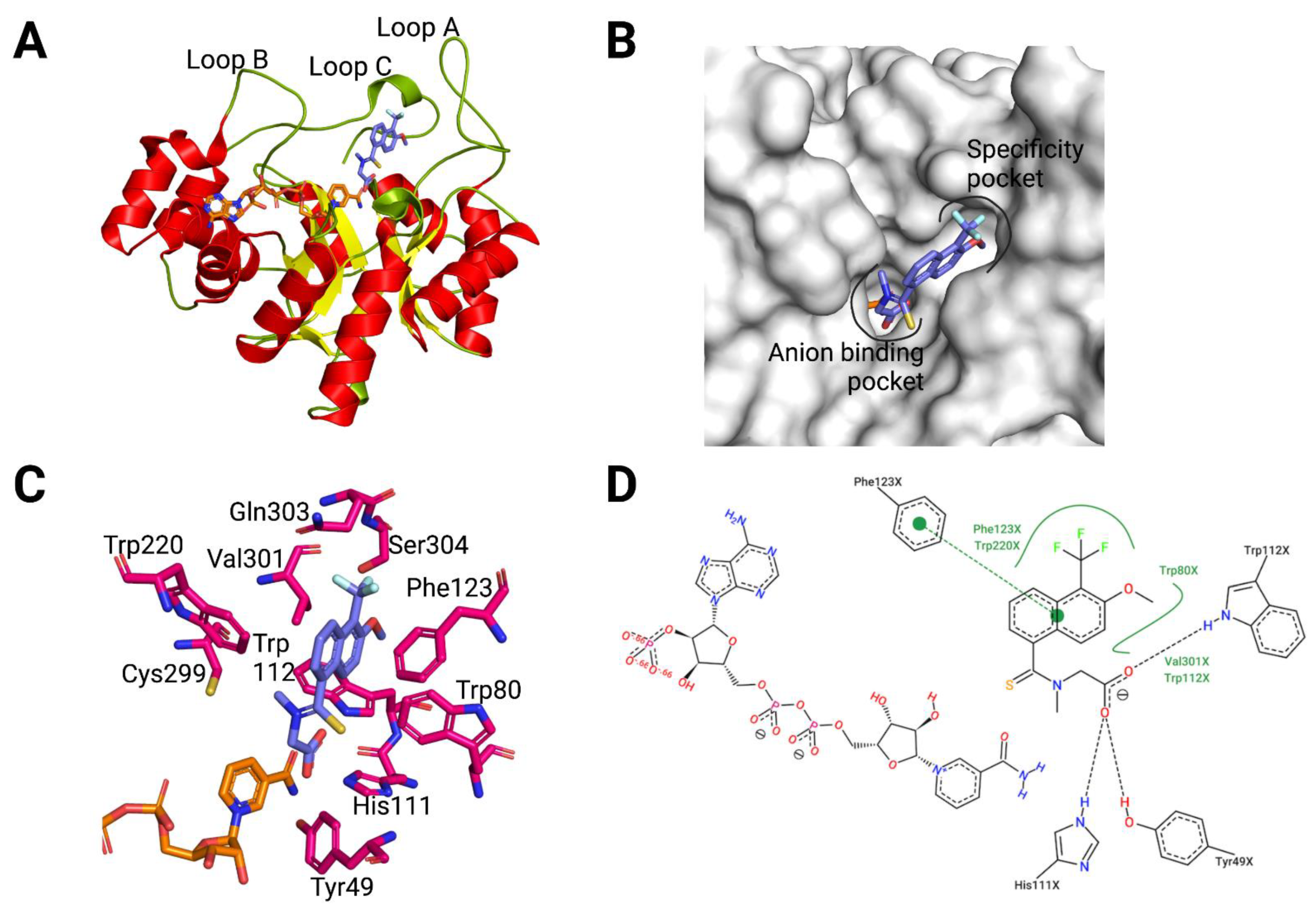
3.1. AKR1B10 Structure: Overview and Specific Features
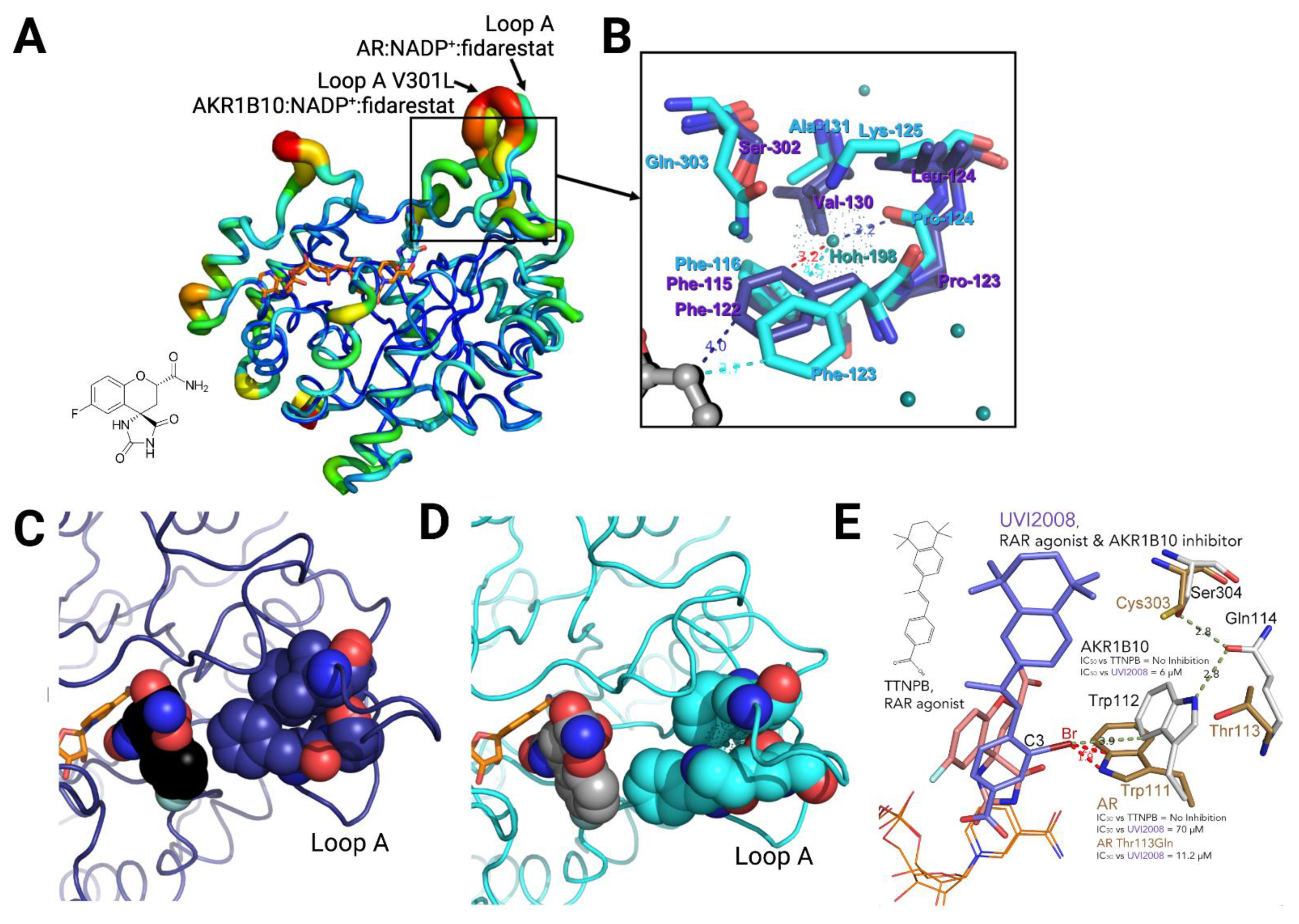
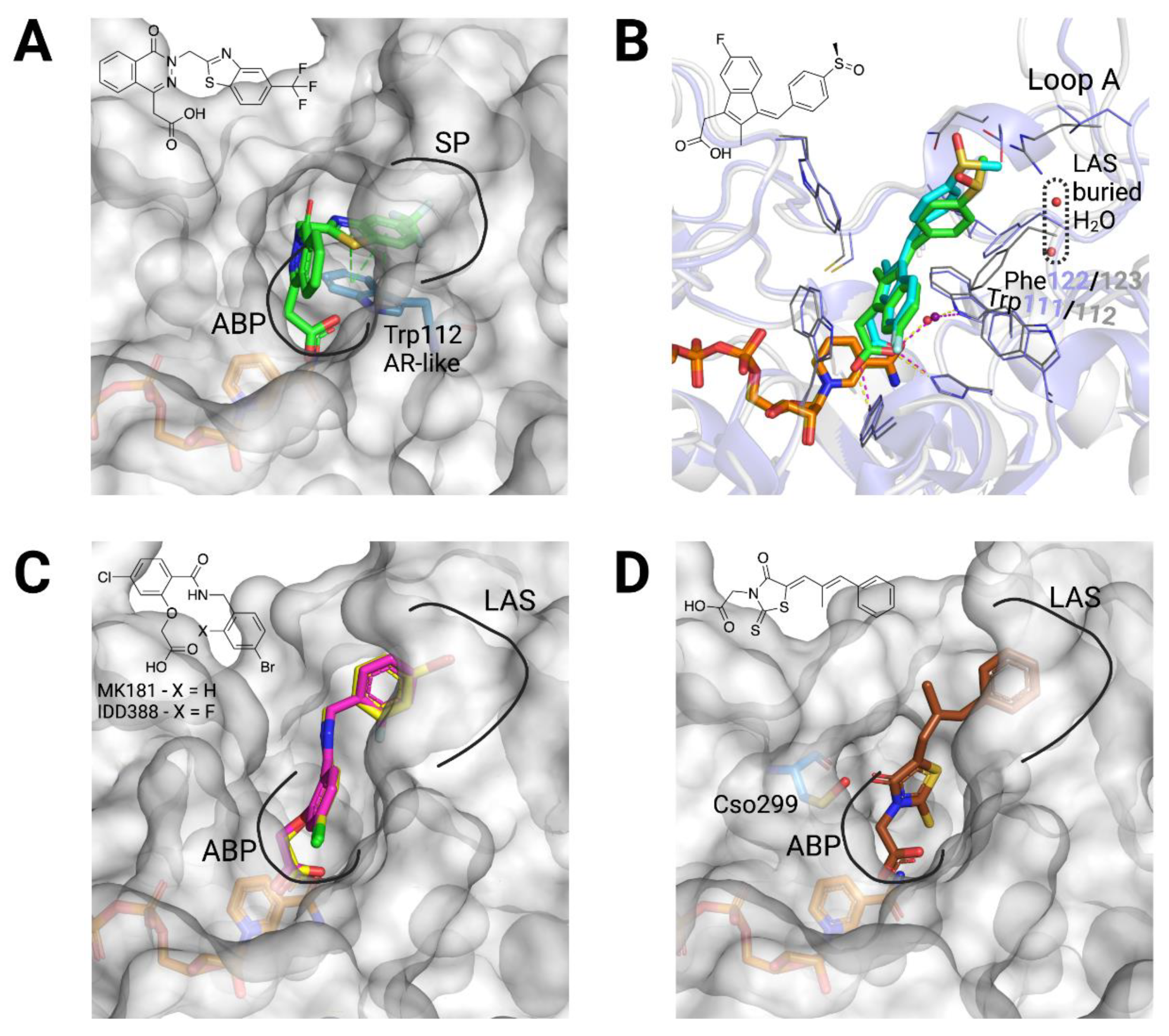
3.2. Structural Bases for AKR1B10 Selectivity
4. Conclusions
- (i)
- An anchoring moiety: Common to ARIs, an AKR1B10 inhibitor must have an anchoring moiety, with a carboxylic acid or an acidic hydroxyl as best choices. Cyclic imides, without the addition of an aryl moiety for binding to either the SP or the LAS (as it may be with minalrestat), are poor AKR1B10 inhibitors.
- (ii)
- Keeping Trp112 in its native conformation (AKR1B10-like): Substituents or ligand conformations that are not compatible with the Trp112 flipped (AR-like) conformation—e.g., flufenamic acid, UVI2008−, and/or aryl moieties that provide an “optimal filling” of the LAS are required for specificity. That is to displace the buried water molecule(s) in the LAS, an adequate shape complementarity, and to have interactions that are more favorable that those in the bulk water [50,51].
- (iii)
- Not opening the SP in AR: Another recurrent feature of selectivity for AKR1B10 over AR, is the inability of an inhibitor to induce the opening of the SP of AR, which normally occurs in ligands with a bulky aryl moiety as in JF0049 or MK204. This can be observed in Table A1, as, in the solved structures of AKR1B10 holoenzyme with inhibitors, no specific AKR1B10 inhibitor is able to open the SP in AR.
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Ligand Complexed to AKR1B10-NADP+ | Molecular Formula of Ligand | Selectivity (Fold) # | Pocket(s) Conformation | Trp112 in AKR1B10- or AR-Like Conformation | Loop A Subpocket (LAS) Occupation | Other Subpocket Occupation | Specificity Pocket (SP) in AR |
|---|---|---|---|---|---|---|---|---|
| 1ZUA | tolrestat |  | AR (39) | Open SP Open LAS | AR-like | Trapped water molecule | - | Yes |
| 4GA8 * | sorbinil |  | AR (18) | Partially closed SP & LAS | AR-like | Trapped water molecule | - | No |
| 4GAB * | fidarestat |  | AR (1270) | Closed SP & LAS | AR-like | Trapped water molecule | - | No |
| 4GQ0 | caffeic acid phenethyl ester |  | AKR1B10 (8) | Open SP Open LAS | AKR1B10-like | Yes | - | No |
| 4GQG | apo | - | - | - | AKR1B10-like | Trapped water molecule | - | - |
| 4I5X | flufenamic acid |  | AKR1B10 (54) | Partially closed SP & LAS | AKR1B10-like | No | - | No |
| 4ICC ** | JF0064 |  | AR (3.4) | Open loop B subpocket (LBS) | AR-like | No | LBS | No |
| 4JIH | epalrestat |  | AR (16) | Open SP Open LAS | AKR1B10-like | Yes? (partially modeled ligand in PDB) | - | No |
| 4JII | zopolrestat |  | AR (35) | Open SP Partially closed LAS | AR-like | Trapped water molecule | - | Yes |
| 4WEV ** | sulindac |  | AR (7.5) | Open SP Open LAS | AR-like (with interstitial water molecule) | Trapped water molecule | - | Yes |
| 4XZL ** | JF0049 |  | AKR1B10 (8) | Open SP Open LAS | AR-like | Yes | - | No |
| 4XZM | apo (methylated) | - | - | - | AKR1B10-like | Trapped water molecule | - | - |
| 4XZN ** | apo AKME2MU (methylated) | - | - | - | AKR1B10-like | Trapped water molecule | - | - |
| 5LIK ** | MK181 |  | AR (6) | Open SP Open LAS | AR-like (with interstitial water molecule) | Yes | - | Yes |
| 5LIU ** | IDD388 |  | AR (11) | Open SP Open LAS | AR-like (with interstitial water molecule) | Yes | - | Yes |
| 5LIW ** | MK319 |  | AR (23) | Open hyperextended SP Open LAS | AKR1B10-like | Trapped water molecule | - | Yes |
| 5LIX ** | MK184 |  | AKR1B10 (21) | Open extended SP Open LAS | AKR1B10-like | No | - | No |
| 5LIY ** | MK204 |  | AKR1B10 (271) | Open hyperextended SP Open LAS | AKR1B10-like | Trapped water molecule | - | No |
| 5M2F ** | UVI2008 |  | AKR1B10 (11) | Open SP Open LAS | AKR1B10-like | Yes | - | No |
| 5Y7N | Androst-4-ene-3-beta-6-alpha-diol |  | AKR1B10 (136) | Open base of loop A subpocket (BLAS) | AKR1B10-like | No | BLAS | No |
References
- Barski, O.A.; Tipparaju, S.M.; Bhatnagar, A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab. Rev. 2008, 40, 553–624. [Google Scholar] [CrossRef] [Green Version]
- Penning, T.M. Aldo-Keto Reductase Regulation by the Nrf2 System: Implications for Stress Response, Chemotherapy Drug Resistance, and Carcinogenesis. Chem. Res. Toxicol. 2017, 30, 162–176. [Google Scholar] [CrossRef]
- Penning, T.M.; Jonnalagadda, S.; Trippier, P.C.; Rizner, T.L. Aldo-Keto Reductases and Cancer Drug Resistance. Pharmacol. Rev. 2021, 73, 1150–1171. [Google Scholar] [CrossRef]
- Romero-Romero, S.; Costas, M.; Silva Manzano, D.A.; Kordes, S.; Rojas-Ortega, E.; Tapia, C.; Guerra, Y.; Shanmugaratnam, S.; Rodriguez-Romero, A.; Baker, D.; et al. The Stability Landscape of de novo TIM Barrels Explored by a Modular Design Approach. J. Mol. Biol. 2021, 433, 167153. [Google Scholar] [CrossRef] [PubMed]
- Steuber, H.; Heine, A.; Podjarny, A.; Klebe, G. Merging the binding sites of aldose and aldehyde reductase for detection of inhibitor selectivity-determining features. J. Mol. Biol. 2008, 379, 991–1016. [Google Scholar] [CrossRef] [PubMed]
- El-Kabbani, O.; Podjarny, A. Selectivity determinants of the aldose and aldehyde reductase inhibitor-binding sites. Cell Mol. Life Sci. 2007, 64, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Dejoz, J.; Kolar, M.H.; Ruiz, F.X.; Crespo, I.; Cousido-Siah, A.; Podjarny, A.; Barski, O.A.; Fanfrlik, J.; Parés, X.; Farrés, J.; et al. Substrate Specificity, Inhibitor Selectivity and Structure-Function Relationships of Aldo-Keto Reductase 1B15: A Novel Human Retinaldehyde Reductase. PLoS ONE 2015, 10, e0134506. [Google Scholar] [CrossRef]
- Rondeau, J.M.; Tete-Favier, F.; Podjarny, A.; Reymann, J.M.; Barth, P.; Biellmann, J.F.; Moras, D. Novel NADPH-binding domain revealed by the crystal structure of aldose reductase. Nature 1992, 355, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Urzhumtsev, A.; Tete-Favier, F.; Mitschler, A.; Barbanton, J.; Barth, P.; Urzhumtseva, L.; Biellmann, J.F.; Podjarny, A.; Moras, D. A ‘specificity’ pocket inferred from the crystal structures of the complexes of aldose reductase with the pharmaceutically important inhibitors tolrestat and sorbinil. Structure 1997, 5, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Gallego, O.; Ruiz, F.X.; Ardèvol, A.; Domínguez, M.; Álvarez, R.; de Lera, A.R.; Rovira, C.; Farrés, J.; Fita, I.; Parés, X. Structural basis for the high all-trans-retinaldehyde reductase activity of the tumor marker AKR1B10. Proc. Natl. Acad. Sci. USA 2007, 104, 20764–20769. [Google Scholar] [CrossRef] [Green Version]
- Cousido-Siah, A.; Ruiz, F.X.; Mitschler, A.; Porté, S.; de Lera, A.R.; Martin, M.J.; Manzanaro, S.; de la Fuente, J.A.; Terwesten, F.; Betz, M.; et al. Identification of a novel polyfluorinated compound as a lead to inhibit the human enzymes aldose reductase and AKR1B10: Structure determination of both ternary complexes and implications for drug design. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 889–903. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, F.X.; Porté, S.; Parés, X.; Farrés, J. Biological role of aldo-keto reductases in retinoic Acid biosynthesis and signaling. Front. Pharmacol. 2012, 3, 58. [Google Scholar] [CrossRef] [Green Version]
- Balestri, F.; Poli, G.; Pineschi, C.; Moschini, R.; Cappiello, M.; Mura, U.; Tuccinardi, T.; Del Corso, A. Aldose Reductase Differential Inhibitors in Green Tea. Biomolecules 2020, 10, 1003. [Google Scholar] [CrossRef] [PubMed]
- Cappiello, M.; Balestri, F.; Moschini, R.; Mura, U.; Del-Corso, A. Intra-site differential inhibition of multi-specific enzymes. J. Enzyme Inhib. Med. Chem. 2020, 35, 840–846. [Google Scholar] [CrossRef]
- Del-Corso, A.; Balestri, F.; Di Bugno, E.; Moschini, R.; Cappiello, M.; Sartini, S.; La-Motta, C.; Da-Settimo, F.; Mura, U. A new approach to control the enigmatic activity of aldose reductase. PLoS ONE 2013, 8, e74076. [Google Scholar] [CrossRef] [Green Version]
- Endo, S.; Matsunaga, T.; Nishinaka, T. The Role of AKR1B10 in Physiology and Pathophysiology. Metabolites 2021, 11, 332. [Google Scholar] [CrossRef]
- Huang, L.; He, R.; Luo, W.; Zhu, Y.S.; Li, J.; Tan, T.; Zhang, X.; Hu, Z.; Luo, D. Aldo-Keto Reductase Family 1 Member B10 Inhibitors: Potential Drugs for Cancer Treatment. Recent Pat. Anticancer Drug Discov. 2016, 11, 184–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, S.; Xia, S.; Suyama, M.; Morikawa, Y.; Oguri, H.; Hu, D.; Ao, Y.; Takahara, S.; Horino, Y.; Hayakawa, Y.; et al. Synthesis of Potent and Selective Inhibitors of Aldo-Keto Reductase 1B10 and Their Efficacy against Proliferation, Metastasis, and Cisplatin Resistance of Lung Cancer Cells. J. Med. Chem. 2017, 60, 8441–8455. [Google Scholar] [CrossRef] [PubMed]
- Crosas, B.; Hyndman, D.J.; Gallego, O.; Martras, S.; Parés, X.; Flynn, T.G.; Farrés, J. Human aldose reductase and human small intestine aldose reductase are efficient retinal reductases: Consequences for retinoid metabolism. Biochem. J. 2003, 373, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Gallego, O.; Belyaeva, O.V.; Porté, S.; Ruiz, F.X.; Stetsenko, A.V.; Shabrova, E.V.; Kostereva, N.V.; Farrés, J.; Parés, X.; Kedishvili, N.Y. Comparative functional analysis of human medium-chain dehydrogenases, short-chain dehydrogenases/reductases and aldo-keto reductases with retinoids. Biochem. J. 2006, 399, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, F.X.; Cousido-Siah, A.; Mitschler, A.; Farrés, J.; Parés, X.; Podjarny, A. X-ray structure of the V301L aldo-keto reductase 1B10 complexed with NADP(+) and the potent aldose reductase inhibitor fidarestat: Implications for inhibitor binding and selectivity. Chem. Biol. Interact. 2013, 202, 178–185. [Google Scholar] [CrossRef]
- Verma, M.; Martin, H.J.; Haq, W.; O’Connor, T.R.; Maser, E.; Balendiran, G.K. Inhibiting wild-type and C299S mutant AKR1B10; a homologue of aldose reductase upregulated in cancers. Eur. J. Pharmacol. 2008, 584, 213–221. [Google Scholar] [CrossRef]
- Endo, S.; Matsunaga, T.; Kuwata, K.; Zhao, H.T.; El-Kabbani, O.; Kitade, Y.; Hara, A. Chromene-3-carboxamide derivatives discovered from virtual screening as potent inhibitors of the tumour maker, AKR1B10. Bioorg. Med. Chem. 2010, 18, 2485–2490. [Google Scholar] [CrossRef]
- Zemanova, L.; Hofman, J.; Novotna, E.; Musilek, K.; Lundova, T.; Havrankova, J.; Hostalkova, A.; Chlebek, J.; Cahlikova, L.; Wsol, V. Flavones Inhibit the Activity of AKR1B10, a Promising Therapeutic Target for Cancer Treatment. J. Nat. Prod. 2015, 78, 2666–2674. [Google Scholar] [CrossRef]
- Bohren, K.M.; Grimshaw, C.E. The sorbinil trap: A predicted dead-end complex confirms the mechanism of aldose reductase inhibition. Biochemistry 2000, 39, 9967–9974. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Penning, T.M. Aldo-keto reductases and bioactivation/detoxication. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent Inhibition in Drug Discovery. ChemMedChem 2019, 14, 889–906. [Google Scholar] [CrossRef] [Green Version]
- Díez-Dacal, B.; Gayarre, J.; Gharbi, S.; Timms, J.F.; Coderch, C.; Gago, F.; Pérez-Sala, D. Identification of aldo-keto reductase AKR1B10 as a selective target for modification and inhibition by prostaglandin A(1): Implications for antitumoral activity. Cancer Res. 2011, 71, 4161–4171. [Google Scholar] [CrossRef] [Green Version]
- Balendiran, G.K.; Sawaya, M.R.; Schwarz, F.P.; Ponniah, G.; Cuckovich, R.; Verma, M.; Cascio, D. The role of Cys-298 in aldose reductase function. J. Biol. Chem. 2011, 286, 6336–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Gomez, F.J.; Diez-Dacal, B.; Garcia-Martin, E.; Agundez, J.A.; Pajares, M.A.; Perez-Sala, D. Detoxifying Enzymes at the Cross-Roads of Inflammation, Oxidative Stress, and Drug Hypersensitivity: Role of Glutathione Transferase P1-1 and Aldose Reductase. Front. Pharmacol. 2016, 7, 237. [Google Scholar] [CrossRef] [Green Version]
- Castellví, A.; Crespo, I.; Crosas, E.; Cámara-Artigas, A.; Gavira, J.A.; Aranda, M.A.G.; Parés, X.; Farrés, J.; Juanhuix, J. Efficacy of aldose reductase inhibitors is affected by oxidative stress induced under X-ray irradiation. Sci. Rep. 2019, 9, 3177. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, H.; Zhao, Y.; Li, Z.; Chen, S.; Zhai, J.; Chen, Y.; Xie, W.; Wang, Z.; Li, Q.; et al. Inhibitor selectivity between aldo-keto reductase superfamily members AKR1B10 and AKR1B1: Role of Trp112 (Trp111). FEBS Lett. 2013, 587, 3681–3686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.; Rajput, S.; Wang, C.; Ma, J.; Cao, D. Murine aldo-keto reductase family 1 subfamily B: Identification of AKR1B8 as an ortholog of human AKR1B10. Biol. Chem. 2010, 391, 1371–1378. [Google Scholar] [CrossRef]
- Ruiz, F.X.; Moro, A.; Gallego, O.; Ardèvol, A.; Rovira, C.; Petrash, J.M.; Parés, X.; Farrés, J. Human and rodent aldo-keto reductases from the AKR1B subfamily and their specificity with retinaldehyde. Chem. Biol. Interact. 2011, 191, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Diez-Dacal, B.; Sanchez-Gomez, F.J.; Sanchez-Murcia, P.A.; Milackova, I.; Zimmerman, T.; Ballekova, J.; Garcia-Martin, E.; Agundez, J.A.; Gharbi, S.; Gago, F.; et al. Molecular Interactions and Implications of Aldose Reductase Inhibition by PGA1 and Clinically Used Prostaglandins. Mol. Pharmacol. 2016, 89, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Hara, A.; Endo, S.; Matsunaga, T.; Soda, M.; El-Kabbani, O.; Yashiro, K. Inhibition of aldo-keto reductase family 1 member B10 by unsaturated fatty acids. Arch. Biochem. Biophys. 2016, 609, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Kemper, E.K.; Suciu, R.M.; Vinogradova, E.V.; Backus, K.M.; Horning, B.D.; Paul, T.A.; Ichu, T.A.; Svensson, R.U.; Olucha, J.; et al. Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 2017, 171, 696–709.e623. [Google Scholar] [CrossRef] [Green Version]
- Crowley, V.M.; Thielert, M.; Cravatt, B.F. Functionalized Scout Fragments for Site-Specific Covalent Ligand Discovery and Optimization. ACS Cent. Sci. 2021, 7, 613–623. [Google Scholar] [CrossRef]
- Ruiz, F.X.; Gallego, O.; Ardèvol, A.; Moro, A.; Domínguez, M.; Álvarez, S.; Álvarez, R.; de Lera, A.R.; Rovira, C.; Fita, I.; et al. Aldo-keto reductases from the AKR1B subfamily: Retinoid specificity and control of cellular retinoic acid levels. Chem. Biol. Interact. 2009, 178, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; Mruk, K.; Rahighi, S.; Raub, A.G.; Chen, C.H.; Dorn, L.E.; Horikoshi, N.; Wakatsuki, S.; Chen, J.K.; Mochly-Rosen, D. Correcting glucose-6-phosphate dehydrogenase deficiency with a small-molecule activator. Nat. Commun. 2018, 9, 4045. [Google Scholar] [CrossRef]
- Kok, B.P.; Ghimire, S.; Kim, W.; Chatterjee, S.; Johns, T.; Kitamura, S.; Eberhardt, J.; Ogasawara, D.; Xu, J.; Sukiasyan, A.; et al. Discovery of small-molecule enzyme activators by activity-based protein profiling. Nat. Chem. Biol. 2020, 16, 997–1005. [Google Scholar] [CrossRef]
- Endo, S.; Matsunaga, T.; Fujita, A.; Kuragano, T.; Soda, M.; Sundaram, K.; Dhagat, U.; Tajima, K.; El-Kabbani, O.; Hara, A. Activation of aldo-keto reductase family member 1B14 (AKR1B14) by bile acids: Activation mechanism and bile acid-binding site. Biochimie 2011, 93, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Endo, S.; Matsunaga, T.; Mamiya, H.; Ohta, C.; Soda, M.; Kitade, Y.; Tajima, K.; Zhao, H.T.; El-Kabbani, O.; Hara, A. Kinetic studies of AKR1B10, human aldose reductase-like protein: Endogenous substrates and inhibition by steroids. Arch. Biochem. Biophys. 2009, 487, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Steckelbroeck, S.; Oyesanmi, B.; Jin, Y.; Lee, S.H.; Kloosterboer, H.J.; Penning, T.M. Tibolone metabolism in human liver is catalyzed by 3α/3β-hydroxysteroid dehydrogenase activities of the four isoforms of the aldo-keto reductase (AKR)1C subfamily. J. Pharmacol. Exp. Ther. 2006, 316, 1300–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, E.I.; Sanishvili, R.; Cachau, R.E.; Mitschler, A.; Chevrier, B.; Barth, P.; Lamour, V.; Van Zandt, M.; Sibley, E.; Bon, C.; et al. Ultrahigh resolution drug design I: Details of interactions in human aldose reductase-inhibitor complex at 0.66 A. Proteins 2004, 55, 792–804. [Google Scholar] [CrossRef]
- Blakeley, M.P.; Ruiz, F.; Cachau, R.; Hazemann, I.; Meilleur, F.; Mitschler, A.; Ginell, S.; Afonine, P.; Ventura, O.N.; Cousido-Siah, A.; et al. Quantum model of catalysis based on a mobile proton revealed by subatomic X-ray and neutron diffraction studies of h-aldose reductase. Proc. Natl. Acad. Sci. USA 2008, 105, 1844–1848. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, F.X.; Crespo, I.; Alvarez, S.; Porté, S.; Giménez-Dejoz, J.; Cousido-Siah, A.; Mitschler, A.; de Lera, A.R.; Parés, X.; Podjarny, A.; et al. Structural basis for the inhibition of AKR1B10 by the C3 brominated TTNPB derivative UVI2008. Chem. Biol. Interact. 2017, 276, 174–181. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, H.; Zheng, X.; Zhao, Y.; Chen, S.; Chen, Y.; Zhang, R.; Li, Q.; Hu, X. Structural basis for the inhibition of AKR1B10 by caffeic acid phenethyl ester (CAPE). ChemMedChem 2014, 9, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, F.X.; Cousido-Siah, A.; Porté, S.; Domínguez, M.; Crespo, I.; Rechlin, C.; Mitschler, A.; de Lera, A.R.; Martin, M.J.; de la Fuente, J.A.; et al. Structural Determinants of the Selectivity of 3-Benzyluracil-1-acetic Acids toward Human Enzymes Aldose Reductase and AKR1B10. ChemMedChem 2015, 10, 1989–2003. [Google Scholar] [CrossRef]
- Cousido-Siah, A.; Ruiz, F.X.; Fanfrlik, J.; Giménez-Dejoz, J.; Mitschler, A.; Kamlar, M.; Vesely, J.; Ajani, H.; Parés, X.; Farrés, J.; et al. IDD388 Polyhalogenated Derivatives as Probes for an Improved Structure-Based Selectivity of AKR1B10 Inhibitors. ACS Chem. Biol. 2016, 11, 2693–2705. [Google Scholar] [CrossRef]
- Cousido-Siah, A.; Ruiz, F.X.; Crespo, I.; Porté, S.; Mitschler, A.; Parés, X.; Podjarny, A.; Farrés, J. Structural analysis of sulindac as an inhibitor of aldose reductase and AKR1B10. Chem. Biol. Interact. 2015, 234, 290–296. [Google Scholar] [CrossRef]
- Endo, S.; Matsunaga, T.; Soda, M.; Tajima, K.; Zhao, H.T.; El-Kabbani, O.; Hara, A. Selective inhibition of the tumor marker AKR1B10 by antiinflammatory N-phenylanthranilic acids and glycyrrhetic acid. Biol. Pharm. Bull. 2010, 33, 886–890. [Google Scholar] [CrossRef] [Green Version]
- Fanfrlik, J.; Kolar, M.; Kamlar, M.; Hurny, D.; Ruiz, F.X.; Cousido-Siah, A.; Mitschler, A.; Rezac, J.; Munusamy, E.; Lepsik, M.; et al. Modulation of aldose reductase inhibition by halogen bond tuning. ACS Chem. Biol. 2013, 8, 2484–2492. [Google Scholar] [CrossRef] [PubMed]
- Chatzopoulou, M.; Patsilinakos, A.; Vallianatou, T.; Prnova, M.S.; Zakelj, S.; Ragno, R.; Stefek, M.; Kristl, A.; Tsantili-Kakoulidou, A.; Demopoulos, V.J. Decreasing acidity in a series of aldose reductase inhibitors: 2-Fluoro-4-(1H-pyrrol-1-yl)phenol as a scaffold for improved membrane permeation. Bioorg. Med. Chem. 2014, 22, 2194–2207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, L.; Zhang, L.; Chen, W.; Chen, X.; Xie, M.; Yan, G.; Hu, X.; Xu, J.; Zhang, J. Synthesis and biological evaluation of steroidal derivatives as selective inhibitors of AKR1B10. Steroids 2014, 86, 39–44. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz, F.X.; Parés, X.; Farrés, J. Perspective on the Structural Basis for Human Aldo-Keto Reductase 1B10 Inhibition. Metabolites 2021, 11, 865. https://doi.org/10.3390/metabo11120865
Ruiz FX, Parés X, Farrés J. Perspective on the Structural Basis for Human Aldo-Keto Reductase 1B10 Inhibition. Metabolites. 2021; 11(12):865. https://doi.org/10.3390/metabo11120865
Chicago/Turabian StyleRuiz, Francesc Xavier, Xavier Parés, and Jaume Farrés. 2021. "Perspective on the Structural Basis for Human Aldo-Keto Reductase 1B10 Inhibition" Metabolites 11, no. 12: 865. https://doi.org/10.3390/metabo11120865
APA StyleRuiz, F. X., Parés, X., & Farrés, J. (2021). Perspective on the Structural Basis for Human Aldo-Keto Reductase 1B10 Inhibition. Metabolites, 11(12), 865. https://doi.org/10.3390/metabo11120865