
Early Noninvasive Metabolic Biomarkers of Mutant IDH Inhibition in Glioma

 ,
,
Abstract
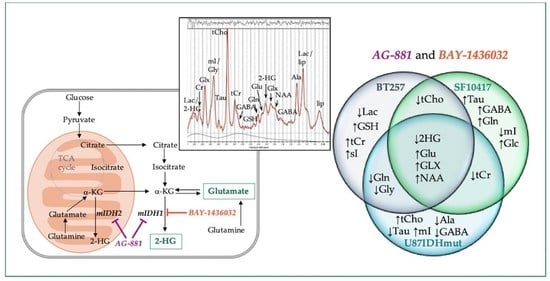
:
1. Introduction
2. Results
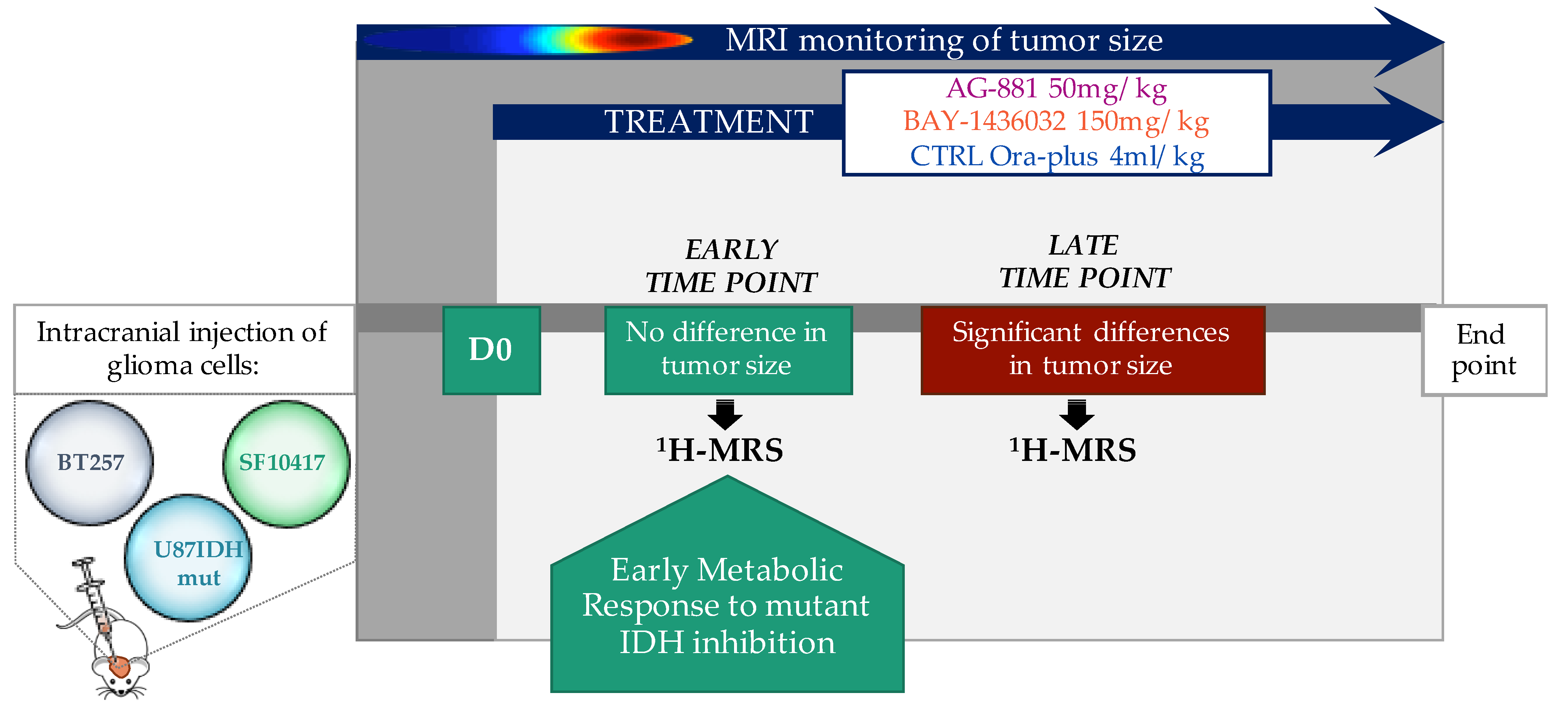
2.1. In Vivo 1H-MRS Studies Using U87IDHmut Genetically Engineered Model
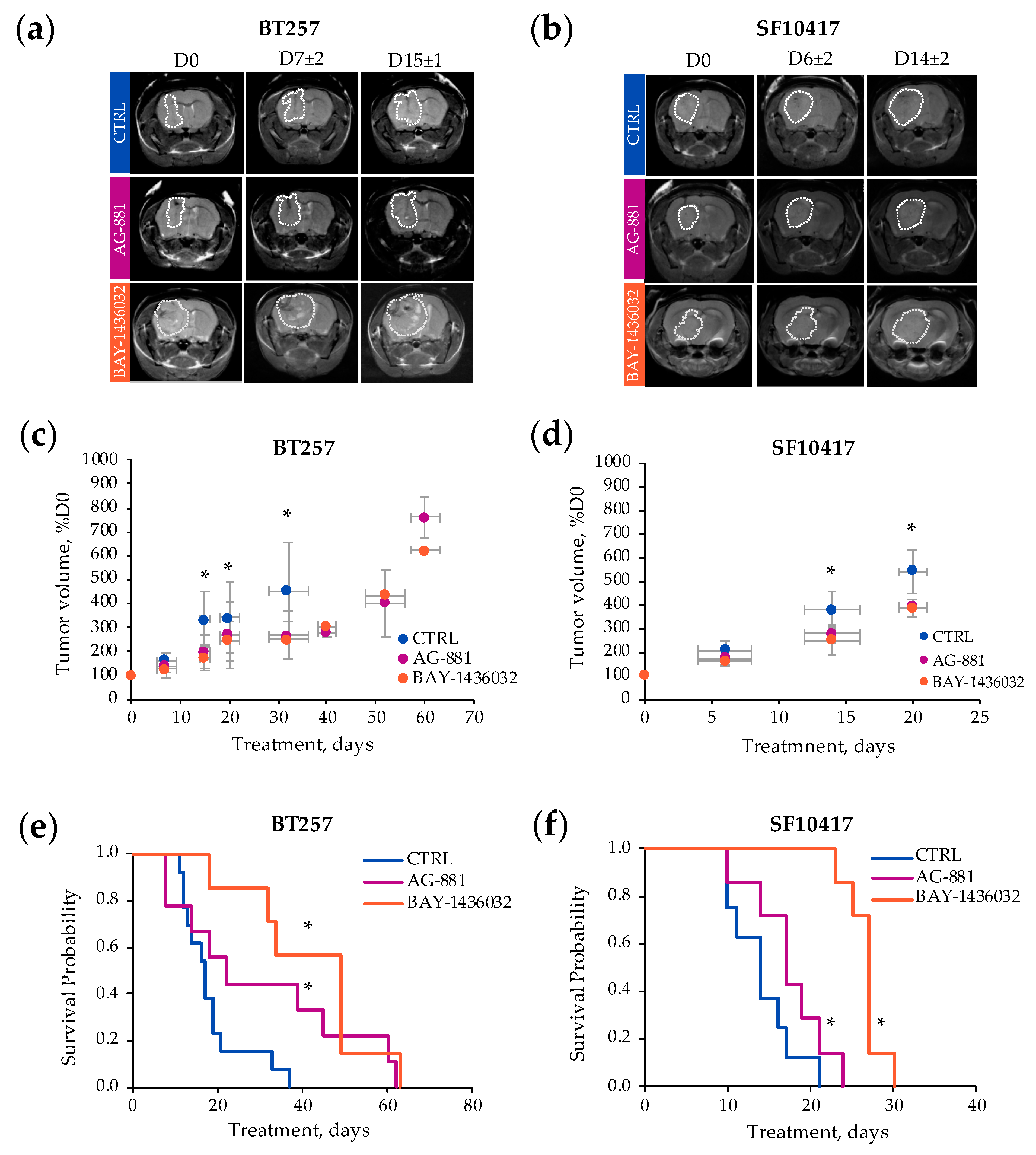
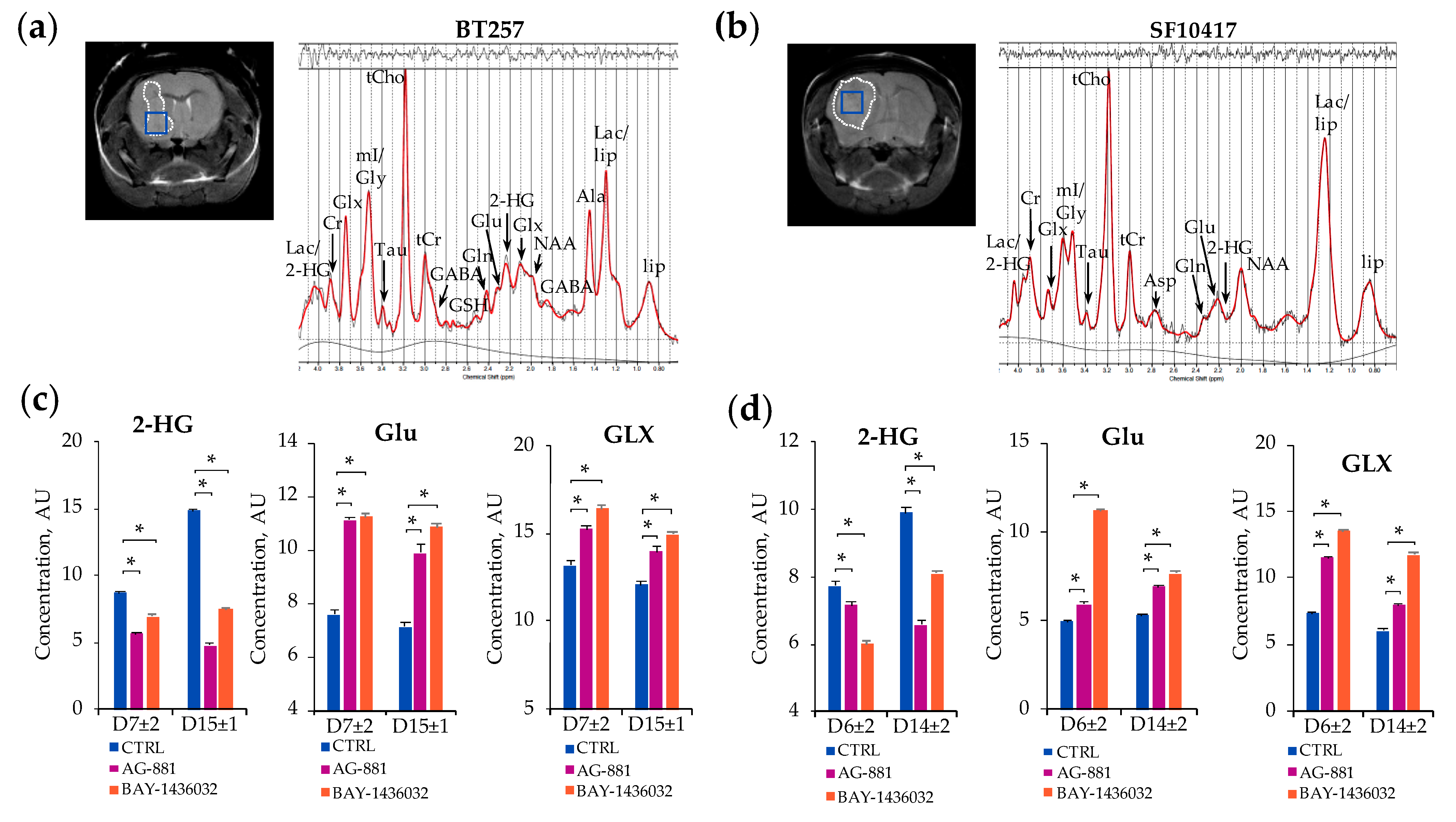
2.2. In Vivo 1H-MRS Studies Using Patient-Derived BT257 and SF10417 Glioma Models
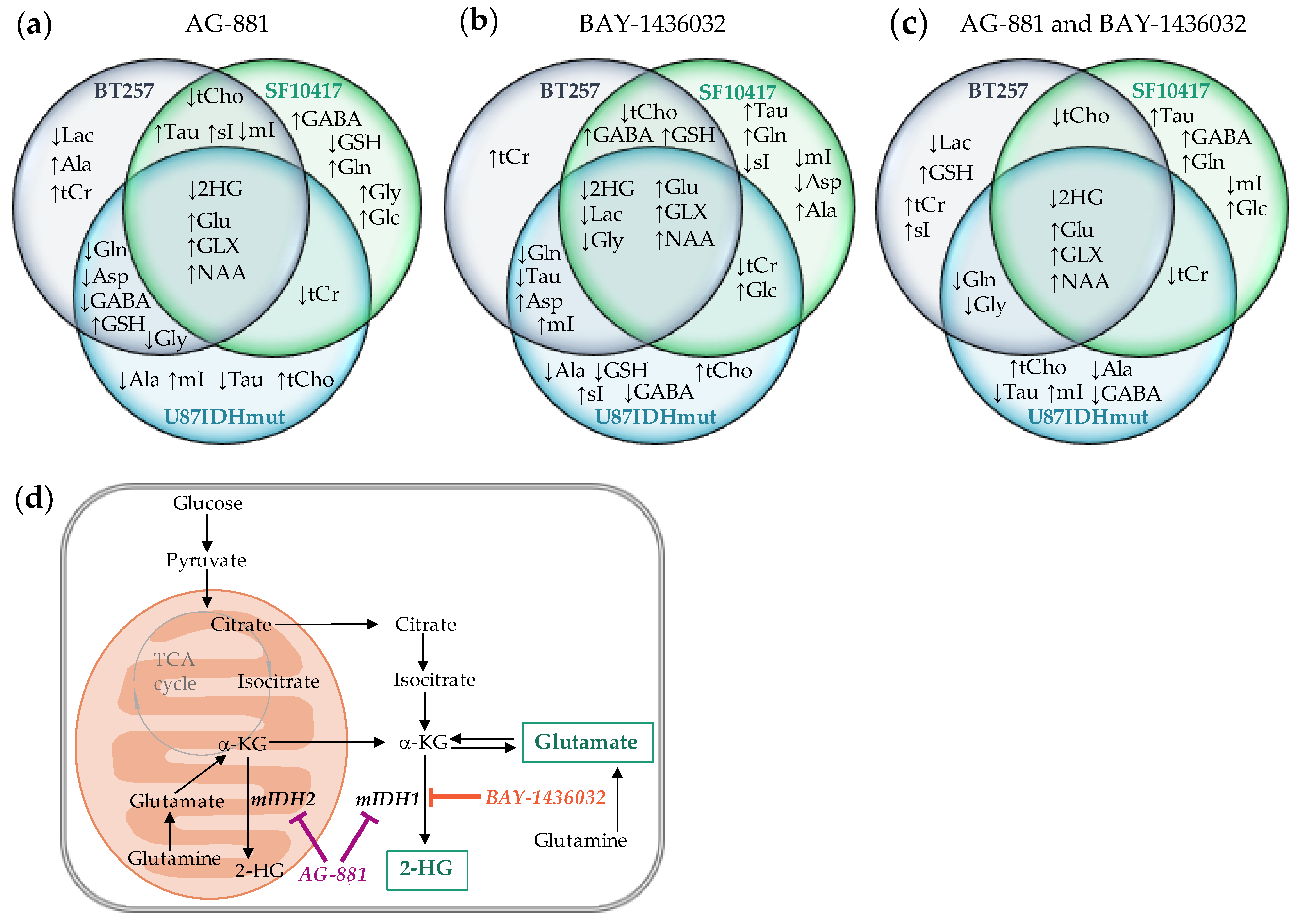
2.3. Metabolic Profile Commonly Associated with Mutant IDH1 Inhibition
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Mutant IDH Inhibitors
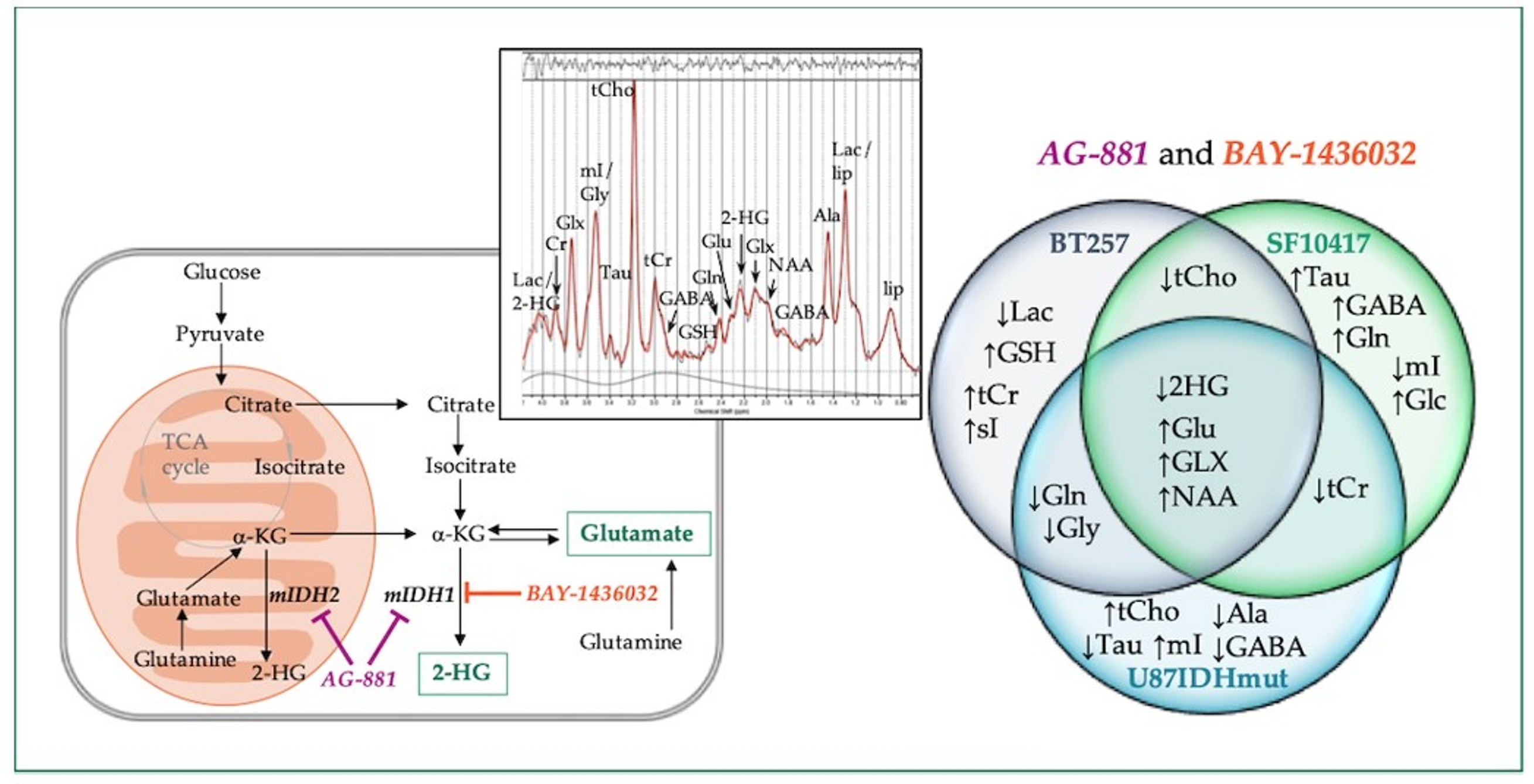
4.3. Animal Models and Study Design
4.4. In Vivo MR Studies
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalinina, J.; Carroll, A.; Wang, L.; Yu, Q.; Mancheno, D.E.; Wu, S.; Liu, F.; Ahn, J.; He, M.; Mao, H.; et al. Detection of “oncometabolite” 2-hydroxyglutarate by magnetic resonance analysis as a biomarker of IDH1/2 mutations in glioma. J. Mol. Med. 2012, 90, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pope, W.B.; Prins, R.M.; Thomas, M.A.; Nagarajan, R.; Yen, K.E.; Bittinger, M.A.; Salamon, N.; Chou, A.P.; Yong, W.H.; Soto, H.; et al. Non-invasive detection of 2-hydroxyglutarate and other metabolites in IDH1 mutant glioma patients using magnetic resonance spectroscopy. J. Neurooncol. 2012, 107, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Smits, A.; Jakola, A.S. Clinical Presentation, Natural History, and Prognosis of Diffuse Low-Grade Gliomas. Neurosurg. Clin.N. Am. 2019, 30, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Bush, N.A.O.; Chang, S. Treatment Strategies for Low-Grade Glioma in Adults. J. Clin. Oncol. 2016, 12, 1235–1241. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol. 2020, 22, iv1–iv96. [Google Scholar] [CrossRef]
- Jakola, A.S.; Skjulsvik, A.J.; Myrmel, K.S.; Sjåvik, K.; Unsgård, G.; Torp, S.H.; Aaberg, K.; Berg, T.; Dai, H.Y.; Johnsen, K.; et al. Surgical resection versus watchful waiting in low-grade gliomas. Ann. Oncol. 2017, 28, 1942–1948. [Google Scholar] [CrossRef]
- Delgado-López, P.D.; Corrales-García, E.M.; Martino, J.; Lastra-Aras, E.; Dueñas-Polo, M.T. Diffuse low-grade glioma: A review on the new molecular classification, natural history and current management strategies. Clin. Transl. Oncol. 2017, 19, 931–944. [Google Scholar] [CrossRef]
- Leao, D.J.; Craig, P.G.; Godoy, L.F.; da La Leite, C.; Policeni, B. Response Assessment in Neuro-Oncology Criteria for Gliomas: Practical Approach Using Conventional and Advanced Techniques. Am. J. Neuroradiol. 2019, 41, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Claus, E.B.; Walsh, K.M.; Wiencke, J.K.; Molinaro, A.M.; Wiemels, J.L.; Schildkraut, J.M.; Bondy, M.L.; Berger, M.; Jenkins, R.; Wrensch, M. Survival and low-grade glioma: The emergence of genetic information. Neurosurg. Focus 2015, 38. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Ye, D.; Guan, K.-L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1,010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 2019, 9, 417–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, T.; Khawaja, M.R.; DiNardo, C.D.; Atkins, J.T.; Janku, F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov. Med. 2016, 21, 373–380. [Google Scholar]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and Deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascades Which Alter Therapy Response. Oncotarget 2012, 3, 954–987. [Google Scholar] [CrossRef] [Green Version]
- Kaminska, B.; Czapski, B.; Guzik, R.; Król, S.K.; Gielniewski, B. Consequences of IDH1/2 Mutations in Gliomas and an Assessment of Inhibitors Targeting Mutated IDH Proteins. Molecules 2019, 24, 968. [Google Scholar] [CrossRef] [Green Version]
- Villanueva-Meyer, J.E.; Mabray, M.C.; Cha, S. Current Clinical Brain Tumor Imaging. Neurosurgery 2017, 81, 397–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Park, I.; Nelson, S.J. Imaging tumor metabolism using in vivo magnetic resonance spectroscopy. Cancer J. 2015, 21, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Horská, A.; Barker, P.B. Imaging of Brain Tumors: MR Spectroscopy and Metabolic Imaging. Neuroimaging Clin. N. Am. 2010, 20, 293–310. [Google Scholar] [CrossRef] [Green Version]
- van der Graaf, M. In vivo magnetic resonance spectroscopy: Basic methodology and clinical applications. Eur. Biophys. J. 2010, 39, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Verma, G.; Mohan, S.; Nasrallah, M.P.; Brem, S.; Lee, J.Y.K.; Chawla, S.; Wang, S.; Nagarajan, R.; Thomas, M.A.; Poptani, H. Non-invasive detection of 2-hydroxyglutarate in IDH-mutated gliomas using two-dimensional localized correlation spectroscopy (2D L-COSY) at 7 Tesla. J. Transl. Med. 2016, 14, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.; Andronesi, O.; Barker, P.B.; Bartha, R.; Bizzi, A.; Bolan, P.J.; Brindle, K.M.; Choi, I.-Y.; Cudalbu, C.; Dydak, U.; et al. A Methodological Consensus on Clinical Proton MR Spectroscopy of the Brain: Review and Recommendations. Magn. Reson. Med. 2019, 82, 527–550. [Google Scholar] [CrossRef] [Green Version]
- Andronesi, O.C.; Kim, G.; Gerstner, E.; Batchelor, T.; Tzika, A.A.; Fantin, V.R.; Heiden, M.G.V.; Sorensen, A.G. Detection of 2-Hydroxyglutarate in IDH-mutated Glioma Patients by Spectral-editing and 2D Correlation Magnetic Resonance Spectroscopy. Sci. Trans. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andronesi, O.C.; Loebel, F.; Bogner, W.; Marjańska, M.; Heiden, M.G.V.; Iafrate, A.J.; Dietrich, J.; Batchelor, T.T.; Gerstner, E.R.; Kaelin, W.G.; et al. Treatment Response Assessment in IDH-Mutant Glioma Patients by Noninvasive 3D Functional Spectroscopic Mapping of 2-Hydroxyglutarate. Clin. Cancer. Res. 2016, 22, 1632–1641. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.; Ganji, S.K.; DeBerardinis, R.J.; Hatanpaa, K.J.; Rakheja, D.; Kovacs, Z.; Yang, X.-L.; Mashimo, T.; Raisane, J.M.; Marin-Valencia, I.; et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat. Med. 2012, 18, 624–629. [Google Scholar] [CrossRef] [Green Version]
- Emir, U.E.; Larkin, S.J.; Pennington, N.d.; Voets, N.; Plaha, P.; Stacey, R.; Al-Qahtani, K.; Mccullagh, J.; Schofield, C.J.; Clare, S.; et al. Noninvasive Quantification of 2-Hydroxyglutarate in Human Gliomas with IDH1 and IDH2 Mutations. Cancer Res. 2016, 76, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Branzoli, F.; Stefano, A.L.D.; Capelle, L.; Ottolenghi, C.; Valabrègue, R.; Deelchand, D.K.; Bielle, F.; Villa, C.; Baussart, B.; Lehéricy, S.; et al. Highly specific determination of IDH status using edited in vivo magnetic resonance spectroscopy. Neuro Oncol. 2018, 20, 907–916. [Google Scholar] [CrossRef]
- Chaumeil, M.M.; Larson, P.E.Z.; Yoshihara, H.A.I.; Danforth, O.M.; Vigneron, D.B.; Nelson, S.J.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Non-invasive in vivo assessment of IDH1 mutational status in glioma. Nat. Commun. 2013, 4, 2429–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkhaled, A.; Jalbert, L.E.; Phillips, J.J.; Yoshihara, H.A.I.; Parvataneni, R.; Srinivasan, R.; Bourne, G.; Berger, M.S.; Chang, S.M.; Cha, S.; et al. Magnetic resonance of 2-hydroxyglutarate in IDH1-mutated low-grade gliomas. Sci. Trans. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leather, T.; Jenkinson, M.D.; Das, K.; Poptani, H. Magnetic Resonance Spectroscopy for Detection of 2-Hydroxyglutarate as a Biomarker for IDH Mutation in Gliomas. Metabolites 2017, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Andronesi, O.C.; Arrillaga-Romany, I.C.; Ly, K.I.; Bogner, W.; Ratai, E.M.; Reitz, K.; Iafrate, A.J.; Dietrich, J.; Gerstner, E.R.; Chi, A.S.; et al. Pharmacodynamics of mutant-IDH1 inhibitors in glioma patients probed by in vivo 3D MRS imaging of 2-hydroxyglutarate. Nat. Commun. 2018, 9, 1474–1483. [Google Scholar] [CrossRef]
- Molloy, A.R.; Najac, C.; Viswanath, P.; Lakhani, A.; Subramani, E.; Batsios, G.; Radoul, M.; Gillespie, A.M.; Pieper, R.O.; Ronen, S.M. MR-detectable metabolic biomarkers of response to mutant IDH inhibition in low-grade glioma. Theranostics 2020, 10, 8757–8770. [Google Scholar] [CrossRef] [PubMed]
- Karpel-Massler, G.; Nguyen, T.T.T.; Shang, E.; Siegelin, M.D. Novel IDH1-Targeted Glioma Therapies. CNS Drugs 2019, 33, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Cloughesy, T.F.; Wen, P.Y.; Taylor, J.W.; Maher, E.A.; Arrillaga, I.; Peters, K.B.; Choi, C.; Ellingson, B.M.; Lin, A.P.; et al. A phase 1, open-label, perioperative study of ivosidenib (AG-120) and vorasidenib (AG-881) in recurrent IDH1 mutant, low-grade glioma: Updated results. J. Clin. Oncol. 2019, 37, 2003. [Google Scholar] [CrossRef]
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Chaumeil, M.M.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Metabolic reprogramming in mutant IDH1 glioma cells. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Konteatis, Z.; Artin, E.; Nicolay, B.; Straley, K.; Padyana, A.K.; Jin, L.; Chen, Y.; Narayaraswamy, R.; Ton, S.; Wang, F.; et al. Vorasidenib (AG-881): A First-in-Class, Brain-Penetrant Dual Inhibitor of Mutant IDH1 and 2 for Treatment of Glioma. ACS Med. Chem. Lett. 2020, 11, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Palmisiano, N.; Mantzaris, I.; Mims, A.; DiNardo, C.; Silverman, L.R.; Wang, E.S.; Fiedler, W.; Baldus, C.; Schwind, S.; et al. Safety and efficacy of BAY1436032 in IDH1-mutant AML: Phase I study results. Leukemia 2020, 34, 2903–2913. [Google Scholar] [CrossRef]
- Pusch, S.; Krausert, S.; Fischer, V.; Balss, J.; Ott, M.; Schrimpf, D.; Capper, D.; Sahm, F.; Eisel, J.; Beck, A.-C.; et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017, 133, 629–644. [Google Scholar] [CrossRef]
- Choi, C.; Ganji, S.; Hulsey, K.; Madan, A.; Kovacs, Z.; Dimitrov, I.; Zhang, S.; Pichumani, K.; Mendelsohn, D.; Mickey, B.; et al. A comparative study of short- and long-TE 1H MRS at 3 T for in vivo detection of 2-hydroxyglutarate in brain tumors. NMR Biomed. 2013, 26, 1242–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenger, K.J.; Hattingen, E.; Harter, P.N.; Richter, C.; Franz, K.; Steinbach, J.P.; Bähr, O.; Pilatus, U. Fitting algorithms and baseline correction influence the results of non-invasive in vivo quantitation of 2-hydroxyglutarate with 1H-MRS. NMR Biomed. 2019, 32, e4027. [Google Scholar] [CrossRef] [Green Version]
- Wenger, K.J.; Richter, C.; Burger, M.C.; Urban, H.; Kaulfuss, S.; Harter, P.N.; Sreeramulu, S.; Schwalbe, H.; Steinbach, J.P.; Hattingen, E.; et al. Non-Invasive Measurement of Drug and 2-HG Signals Using 19F and 1H MR Spectroscopy in Brain Tumors Treated with the Mutant IDH1 Inhibitor BAY1436032. Cancers 2020, 12, 3175. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalbert, L.E.; Elkhaled, A.; Phillips, J.J.; Neill, E.; Williams, A.; Crane, J.C.; Olson, M.P.; Molinaro, A.M.; Berger, M.S.; Kurhanewicz, J.; et al. Metabolic Profiling of IDH Mutation and Malignant Progression in Infiltrating Glioma. Sci. Rep. 2017, 7, 44792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannessen, T.C.A.; Mukherjee, J.; Viswanath, P.; Ohba, S.; Ronen, S.M.; Bjerkvig, R.; Pieper, R.O. Rapid Conversion of Mutant IDH1 from Driver to Passenger in a Model of Human Gliomagenesis. Mol. Cancer Res. 2016, 14, 976–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batsios, G.; Viswanath, P.; Subramani, E.; Najac, C.; Gillespie, A.M.; Santos, R.D.; Molloy, A.R.; Pieper, R.O.; Ronen, S.M. PI3K/mTOR inhibition of IDH1 mutant glioma leads to reduced 2HG production that is associated with increased survival. Sci. Rep. 2019, 9, 10521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, L.E.; Hilz, S.; Grimmer, M.R.; Mazor, T.; Najac, C.; Mukherjee, J.; McKinney, A.; Chow, T.; Pieper, R.O.; Ronen, S.M.; et al. Patient-derived cells from recurrent tumors that model the evolution of IDH-mutant glioma. Neuro Oncol. Adv. 2020, 2. [Google Scholar] [CrossRef]
- Mazor, T.; Chesnelong, C.; Pankov, A.; Jalbert, L.E.; Hong, C.; Hayes, J.; Smirnov, I.V.; Marshall, R.; Souza, C.F.; Shen, Y.; et al. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc. Natl. Acad. Sci. USA 2017, 114, 10743–10748. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.J. Analysis of volume MRI and MR spectroscopic imaging data for the evaluation of patients with brain tumors. Magn. Reson. Med. 2001, 46, 228–239. [Google Scholar] [CrossRef]
- Subramani, E.; Radoul, M.; Najac, C.; Batsios, G.; Molloy, A.R.; Hong, D.; Gillespie, A.M.; Santos, R.D.; Viswanath, P.; Costello, J.F.; et al. Glutamate Is a Noninvasive Metabolic Biomarker of IDH1-Mutant Glioma Response to Temozolomide Treatment. Cancer Res. 2020, 80, 5098–5108. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.W. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn. Reson. Med. 1993, 30, 6. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.W. LCModel and LCMgui User’s Manual. 2020. Available online: http://s-provencher.com/pub/LCModel/manual/manual.pdf (accessed on 1 December 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Cramer-Rao Lower Bounds (%) | Concentration (AU) | ||||
|---|---|---|---|---|---|---|
| Control | AG-881 | BAY-1436032 | Control | AG-881 | BAY-1436032 | |
| 2HG | 8 | 22 | 10 | 4.10 ± 0.17 | 1.73 ± 0.12 | 3.09 ± 0.12 |
| GSH | 4 | 5 | 4 | 4.72 ± 0.07 | 5.12 ± 0.08 | 4.26 ± 0.06 |
| NAA | 7 | 8 | 7 | 3.36 ± 0.13 | 4.97 ± 0.15 | 4.62 ± 0.13 |
| Lac | 5 | 7 | 6 | 15.04 ± 0.27 | - | 8.67 ± 0.25 |
| Glc | 30 | 28 | 22 | 1.12 ± 0.22 | - | 2.25 ± 0.12 |
| mI | 14 | 6 | 7 | 2.17 ± 0.07 | 2.97 ± 0.09 | 2.49 ± 0.07 |
| Asp | 7 | 13 | 5 | 6.53 ± 0.27 | 1.19 ± 0.12 | 14.35 ± 0.29 |
| Tau | 4 | 4 | 6 | 9.30 ± 0.15 | 8.04 ± 0.19 | 6.45 ± 0.14 |
| GABA | 6 | 9 | 7 | 7.30 ± 0.20 | 2.57 ± 0.14 | 3.87 ± 0.14 |
| Ala | 13 | 15 | 18 | 3.03 ± 0.10 | 1.09 ± 0.05 | 0.81 ± 0.04 |
| Gln | 6 | 21 | 8 | 3.15 ± 0.11 | 1.54 ± 0.09 | 1.74 ± 0.06 |
| Glu | 3 | 3 | 2 | 12.17 ± 0.17 | 17.35 ± 0.24 | 16.98 ± 0.17 |
| Gly | 7 | 13 | 7 | 7.20 ± 0.21 | 0.30 ± 0.04 | - |
| sI | 44 | 13 | 17 | 0.15 ± 0.03 | - | - |
| tCho | 1 | 1 | 1 | 9.78 ± 0.05 | 14.51 ± 0.07 | 10.31 ± 0.05 |
| tCr | 7 | 12 | 8 | 2.03 ± 0.06 | 1.53 ± 0.07 | 1.36 ± 0.05 |
| GLX | 3 | 3 | 2 | 16.47 ± 0.22 | 18.74 ± 0.25 | 19.08 ± 0.20 |
| D7 ± 2 | ||||||
|---|---|---|---|---|---|---|
| Metabolite | Cramer-Rao Lower Bounds (%) | Concentration (AU) | ||||
| Control | AG-881 | BAY-1436032 | Control | AG-881 | BAY-1436032 | |
| 2HG | 6 | 7 | 5 | 8.63 ± 0.16 | 5.57 ± 0.13 | 6.89 ± 0.13 |
| GSH | 6 | 6 | 5 | 2.19 ± 0.04 | 2.34 ± 0.05 | 2.53 ± 0.05 |
| NAA | 4 | 4 | 3 | 8.54 ± 0.10 | 11.38 ± 0.10 | 13.44 ± 0.11 |
| Lac | 14 | 8 | 6 | 5.73 ± 0.22 | 4.48 ± 0.10 | 4.29 ± 0.11 |
| mI | 4 | 3 | 2 | 19.83 ± 0.14 | 15.27 ± 0.12 | 20.38 ± 0.13 |
| Asp | 22 | 28 | 7 | 6.30 ± 0.19 | 2.26 ± 0.13 | 7.75 ± 0.23 |
| Tau | 5 | 3 | 3 | 10.74 ± 0.13 | 14.16 ± 0.16 | 10.42 ± 0.13 |
| GABA | 5 | 12 | 4 | 12.64 ± 0.17 | 6.62 ± 0.14 | 13.28 ± 0.19 |
| Ala | 22 | 10 | 10 | 0.98 ± 0.05 | 1.66 ± 0.07 | - |
| Gln | 6 | 5 | 5 | 4.93 ± 0.08 | 4.55 ± 0.09 | 4.48 ± 0.08 |
| Glu | 7 | 4 | 4 | 7.59 ± 0.14 | 11.10 ± 0.14 | 11.22 ± 0.16 |
| Gly | 5 | 4 | 4 | 27.39 ± 0.35 | 21.03 ± 0.19 | 16.23 ± 0.15 |
| sI | 14 | 8 | 13 | 0.46 ± 0.02 | 0.66 ± 0.02 | 0.88 ± 0.03 |
| tCho | 1 | 1 | 1 | 12.70 ± 0.04 | 8.70 ± 0.04 | 9.31 ± 0.04 |
| tCr | 4 | 2 | 2 | 7.87 ± 0.06 | 12.13 ± 0.09 | 8.75 ± 0.07 |
| GLX | 5 | 4 | 3 | 13.19 ± 0.18 | 15.21 ± 0.20 | 16.39 ± 0.19 |
| D15 ± 1 | ||||||
| 2HG | 4 | 7 | 4 | 14.79 ± 0.20 | 4.73 ± 0.17 | 7.42 ± 0.17 |
| GSH | 11 | 7 | 4 | 3.41 ± 0.06 | 2.20 ± 0.07 | 1.89 ± 0.04 |
| NAA | 4 | 4 | 4 | 11.45 ± 0.10 | - | 8.73 ± 0.08 |
| Lac | 8 | 13 | 10 | 7.28 ± 0.19 | 5.94 ± 0.38 | 3.18 ± 0.17 |
| mI | 3 | 2 | 2 | 19.45 ± 0.11 | 22.92 ± 0.25 | 22.49 ± 0.17 |
| Asp | 9 | 10 | 6 | 8.44 ± 0.31 | 4.75 ± 0.24 | 6.49 ± 0.18 |
| Tau | 4 | 5 | 4 | 11.04 ± 0.12 | 15.38 ± 0.38 | 11.68 ± 0.17 |
| GABA | 4 | 6 | 5 | 20.74 ± 0.18 | 13.82 ± 0.37 | 8.48 ± 0.14 |
| Ala | 10 | 11 | 15 | 2.29 ± 0.06 | - | 1.00 ± 0.03 |
| Gln | 7 | 6 | 6 | 5.47 ± 0.10 | 3.90 ± 0.12 | 2.48 ± 0.07 |
| Glu | 5 | 7 | 4 | 7.12 ± 0.13 | 9.89 ± 0.31 | 10.83 ± 0.15 |
| Gly | 4 | 6 | 4 | 19.87 ± 0.15 | 13.32 ± 0.38 | 19.19 ± 0.17 |
| sI | 13 | 14 | 11 | 0.57 ± 0.03 | 0.81 ± 0.06 | 0.34 ± 0.02 |
| tCho | 1 | 1 | 1 | 12.37 ± 0.05 | 10.60 ± 0.06 | 11.45 ± 0.06 |
| tCr | 3 | 3 | 3 | 7.89 ± 0.06 | 10.79 ± 0.16 | 13.31 ± 0.11 |
| GLX | 4 | 5 | 4 | 12.03 ± 0.18 | 13.90 ± 0.34 | 14.91 ± 0.19 |
| D6 ± 2 | ||||||
|---|---|---|---|---|---|---|
| Metabolite | Cramer-Rao Lower Bounds (%) | Concentration (AU) | ||||
| Control | AG-881 | BAY-1436032 | Control | AG-881 | BAY-1436032 | |
| 2-HG | 4 | 5 | 5 | 7.73 ± 0.13 | 7.17 ± 0.11 | 5.98 ± 0.11 |
| GSH | 5 | 5 | 3 | 2.71 ± 0.05 | 1.73 ± 0.03 | 3.08 ± 0.04 |
| NAA | 4 | 3 | 3 | 6.37 ± 0.10 | 8.10 ± 0.06 | 8.57 ± 0.06 |
| Lac | 30 | 13 | 7 | 3.62 ± 0.19 | - | 3.13 ± 0.06 |
| Glc | 29 | 14 | 5 | 3.17 ± 0.20 | 3.98 ± 0.12 | 5.39 ± 0.12 |
| mI | 3 | 2 | 2 | 8.99 ± 0.07 | 7.00 ± 0.07 | 8.13 ± 0.04 |
| Asp | 6 | 5 | 5 | 11.37 ± 0.26 | - | 5.34 ± 0.12 |
| Tau | 5 | 4 | 3 | 5.27 ± 0.10 | 8.66 ± 0.08 | 13.07 ± 0.09 |
| GABA | 7 | 4 | 5 | 4.34 ± 0.11 | 6.75 ± 0.09 | 5.93 ± 0.09 |
| Ala | 36 | 11 | 10 | 0.47 ± 0.04 | - | 0.83 ± 0.03 |
| Gln | 16 | 6 | 4 | 1.71 ± 0.09 | 3.28 ± 0.07 | 2.67 ± 0.05 |
| Glu | 6 | 5 | 3 | 4.93 ± 0.10 | 5.92 ± 0.08 | 11.24 ± 0.09 |
| Gly | 5 | 3 | 5 | 12.68 ± 0.16 | 13.79 ± 0.11 | 10.25 ± 0.10 |
| sI | 30 | 13 | 10 | 0.29 ± 0.02 | 0.38 ± 0.03 | 0.18 ± 0.01 |
| tCho | 1 | 1 | 1 | 9.75 ± 0.04 | 7.99 ± 0.03 | 6.18 ± 0.03 |
| tCr | 3 | 2 | 2 | 9.77 ± 0.07 | 7.20 ± 0.05 | 8.03 ± 0.04 |
| GLX | 5 | 4 | 3 | 7.31 ± 0.15 | 11.48 ± 0.11 | 13.54 ± 0.11 |
| D14 ± 2 | ||||||
| 2-HG | 3 | 4 | 4 | 9.91 ± 0.16 | 6.56 ± 0.13 | 8.09 ± 0.12 |
| GSH | 5 | 5 | 5 | 1.71 ± 0.05 | 3.31 ± 0.06 | 2.62 ± 0.04 |
| NAA | 4 | 3 | 3 | 9.42 ± 0.17 | 6.01 ± 0.10 | 9.04 ± 0.08 |
| Lac | 7 | 15 | 7 | 2.43 ± 0.17 | - | - |
| Glc | 10 | 8 | 12 | 3.19 ± 0.18 | - | - |
| mI | 2 | 3 | 3 | 10.21 ± 0.09 | 9.72 ± 0.09 | 8.34 ± 0.07 |
| Asp | 6 | 7 | 7 | 15.89 ± 0.42 | 9.05 ± 0.32 | 5.50 ± 0.14 |
| Tau | 4 | 3 | 4 | 5.02 ± 0.12 | 7.46 ± 0.12 | 13.98 ± 0.13 |
| GABA | 8 | 5 | 5 | 4.71 ± 0.15 | - | 7.71 ± 0.12 |
| Ala | 16 | 42 | 17 | 0.96 ± 0.15 | - | - |
| Gln | 25 | 9 | 5 | 0.88 ± 0.09 | 1.74 ± 0.10 | 3.20 ± 0.06 |
| Glu | 6 | 3 | 6 | 5.26 ± 0.14 | 6.93 ± 0.10 | 7.63 ± 0.12 |
| Gly | 4 | 4 | 5 | 7.88 ± 0.19 | 14.64 ± 0.14 | 10.58 ± 0.17 |
| sI | 20 | 19 | 16 | 0.22 ± 0.03 | - | 0.32 ± 0.01 |
| tCho | 1 | 1 | 1 | 9.28 ± 0.05 | 8.89 ± 0.05 | 6.06 ± 0.03 |
| tCr | 3 | 2 | 2 | 7.41 ± 0.11 | 10.62 ± 0.10 | - |
| GLX | 6 | 3 | 4 | 5.98 ± 0.18 | 7.90 ± 0.12 | 11.69 ± 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radoul, M.; Hong, D.; Gillespie, A.M.; Najac, C.; Viswanath, P.; Pieper, R.O.; Costello, J.F.; Luchman, H.A.; Ronen, S.M. Early Noninvasive Metabolic Biomarkers of Mutant IDH Inhibition in Glioma. Metabolites 2021, 11, 109. https://doi.org/10.3390/metabo11020109
Radoul M, Hong D, Gillespie AM, Najac C, Viswanath P, Pieper RO, Costello JF, Luchman HA, Ronen SM. Early Noninvasive Metabolic Biomarkers of Mutant IDH Inhibition in Glioma. Metabolites. 2021; 11(2):109. https://doi.org/10.3390/metabo11020109
Chicago/Turabian StyleRadoul, Marina, Donghyun Hong, Anne Marie Gillespie, Chloé Najac, Pavithra Viswanath, Russell O. Pieper, Joseph F. Costello, Hema Artee Luchman, and Sabrina M. Ronen. 2021. "Early Noninvasive Metabolic Biomarkers of Mutant IDH Inhibition in Glioma" Metabolites 11, no. 2: 109. https://doi.org/10.3390/metabo11020109
APA StyleRadoul, M., Hong, D., Gillespie, A. M., Najac, C., Viswanath, P., Pieper, R. O., Costello, J. F., Luchman, H. A., & Ronen, S. M. (2021). Early Noninvasive Metabolic Biomarkers of Mutant IDH Inhibition in Glioma. Metabolites, 11(2), 109. https://doi.org/10.3390/metabo11020109