
Resistance to Androgen Deprivation Leads to Altered Metabolism in Human and Murine Prostate Cancer Cell and Tumor Models

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
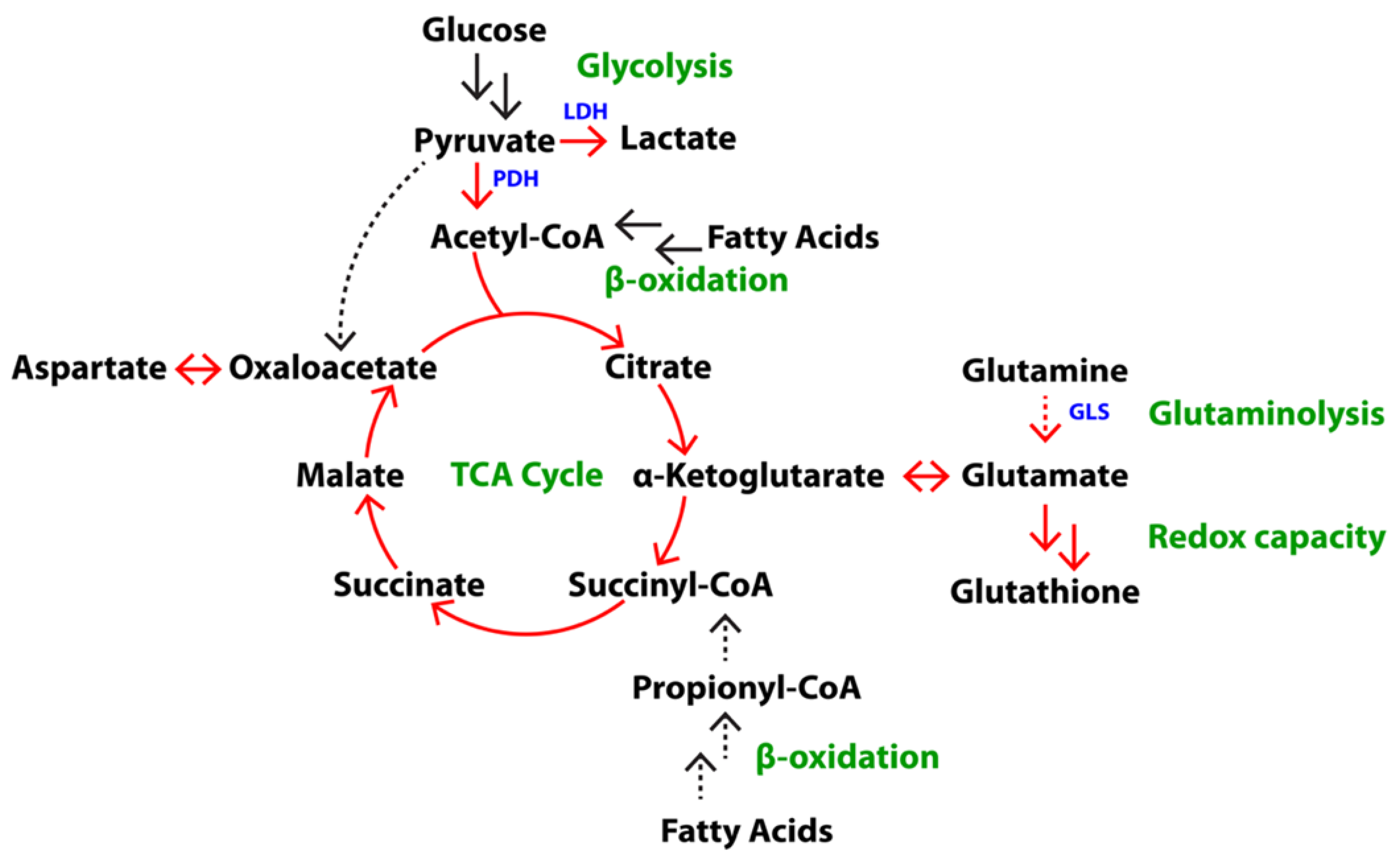
2.1. Differences in Steady State Levels of Metabolites between Androgen-Dependent and Castration-Resistant PCa
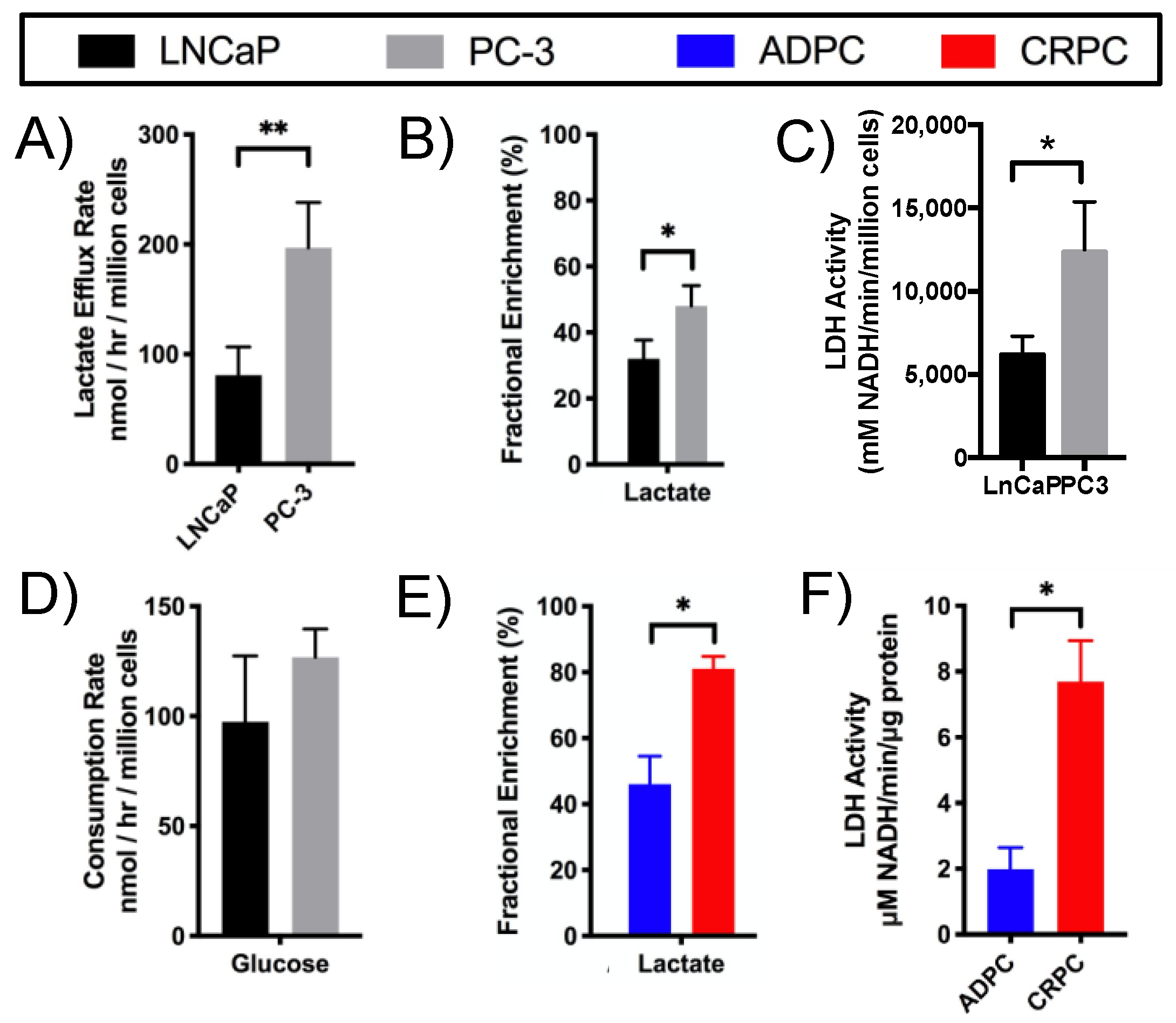
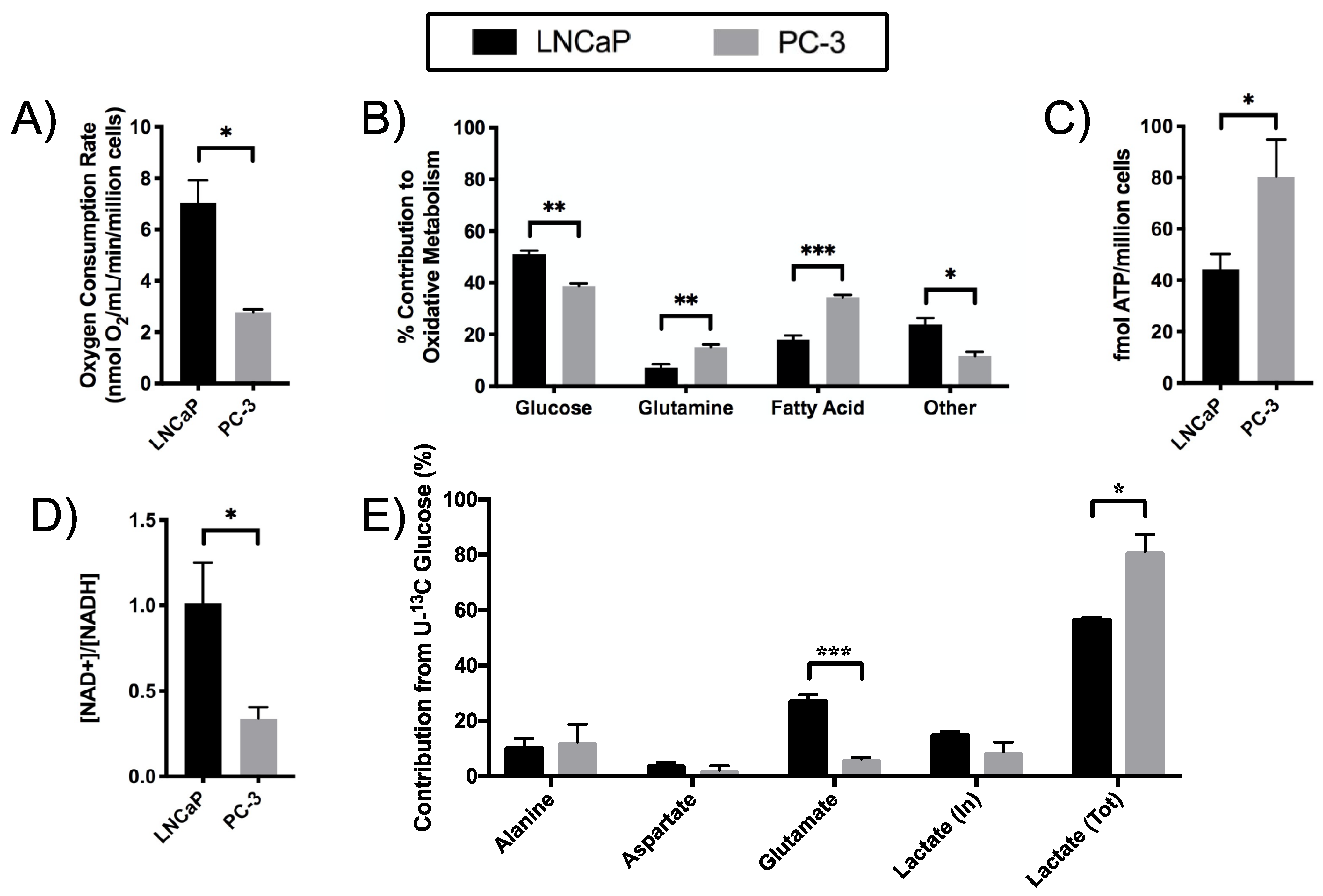
2.2. Elevated Glycolysis in CRPC versus ADPC
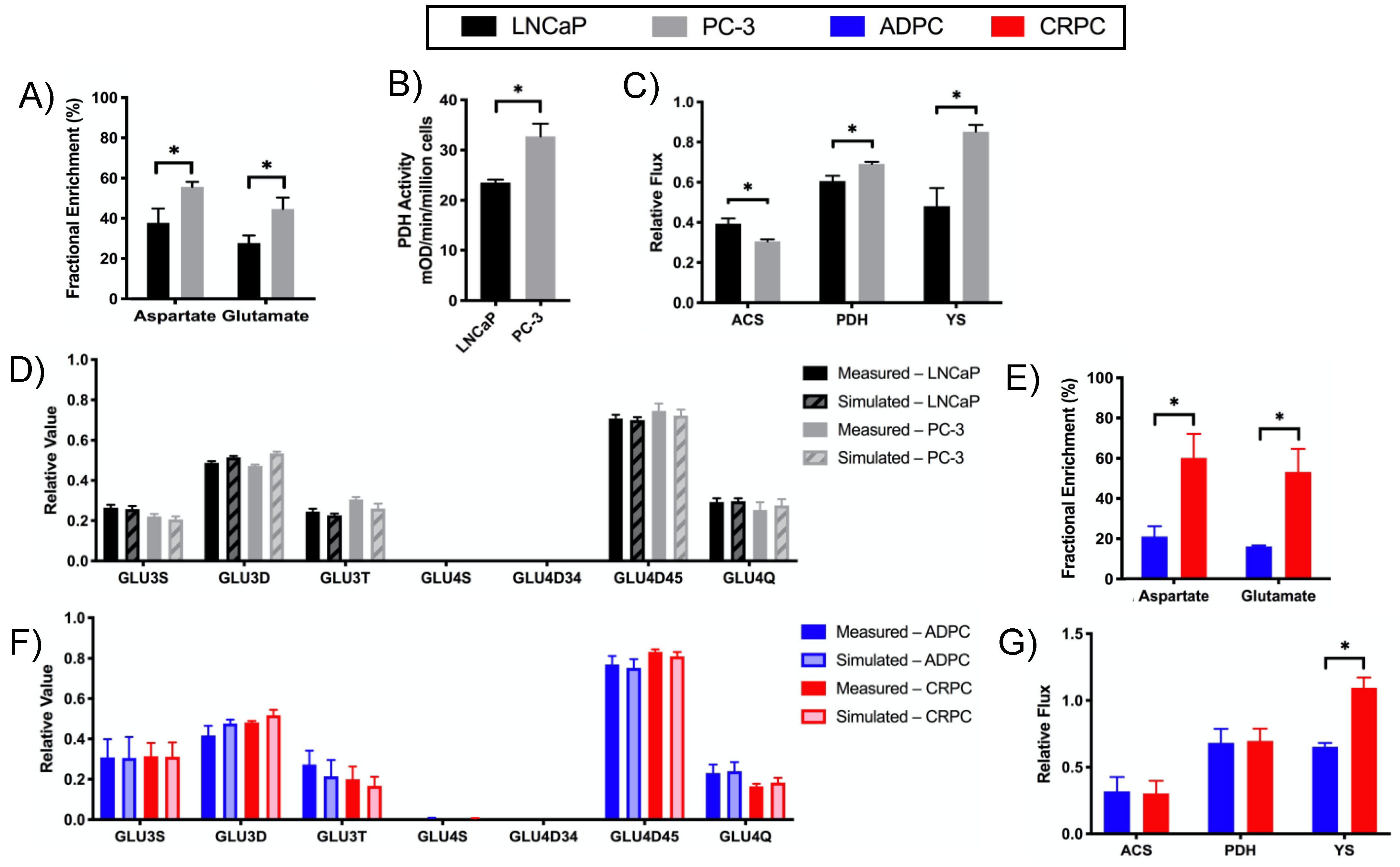
2.3. Increased Utilization of Pyruvate in the TCA Cycle in CRPC versus ADPC
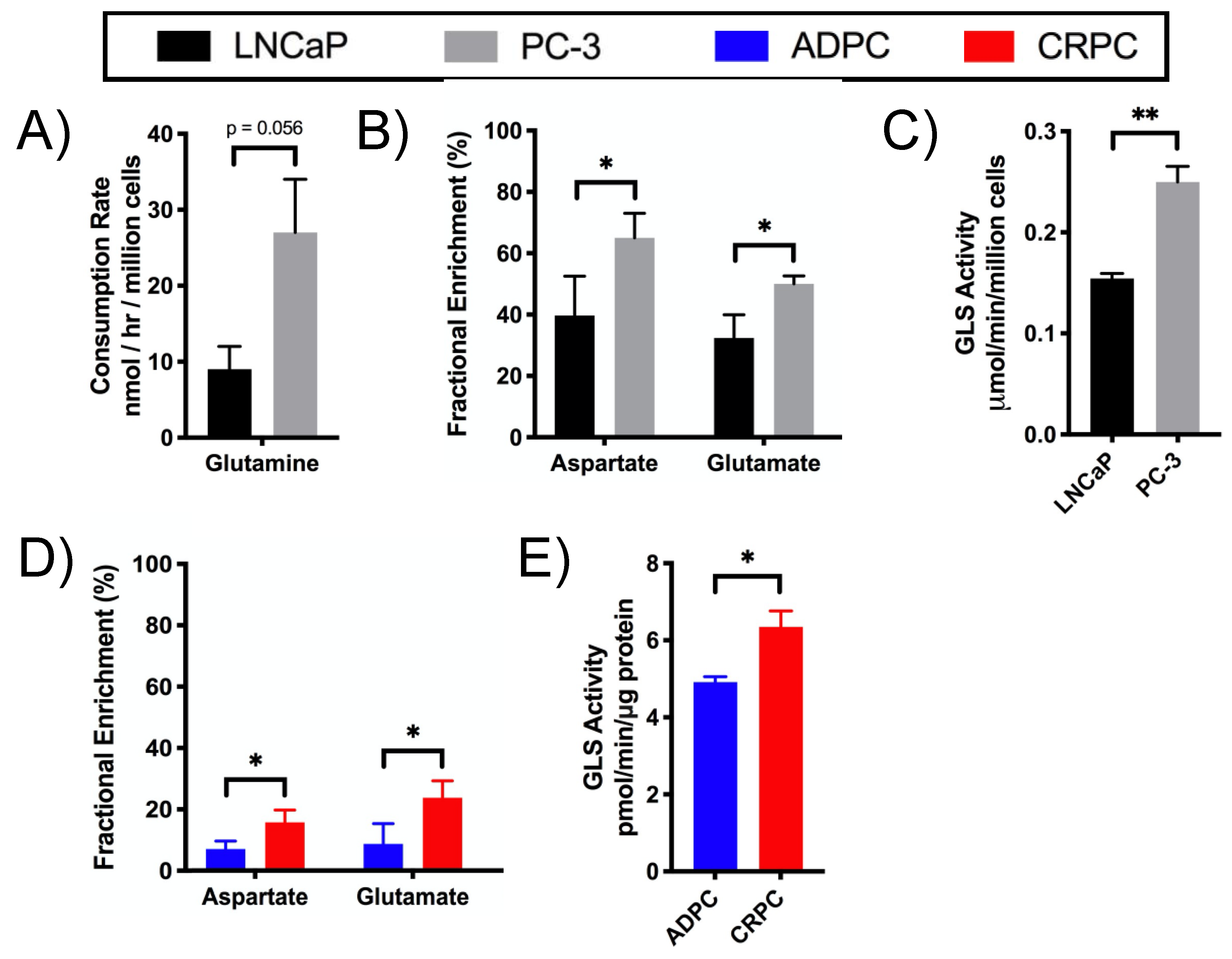
2.4. Glutamine Utilization in the TCA Cycle and Glutaminolysis in CRPC versus ADPC
2.5. Differential Substrate Utilization into Oxidative Phosphorylation (OXPHOS) by LNCaP and PC-3 Cells
2.6. Increased Redox Capacity in CRPC versus ADPC
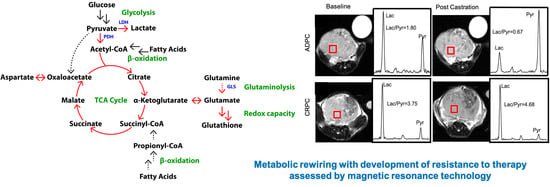
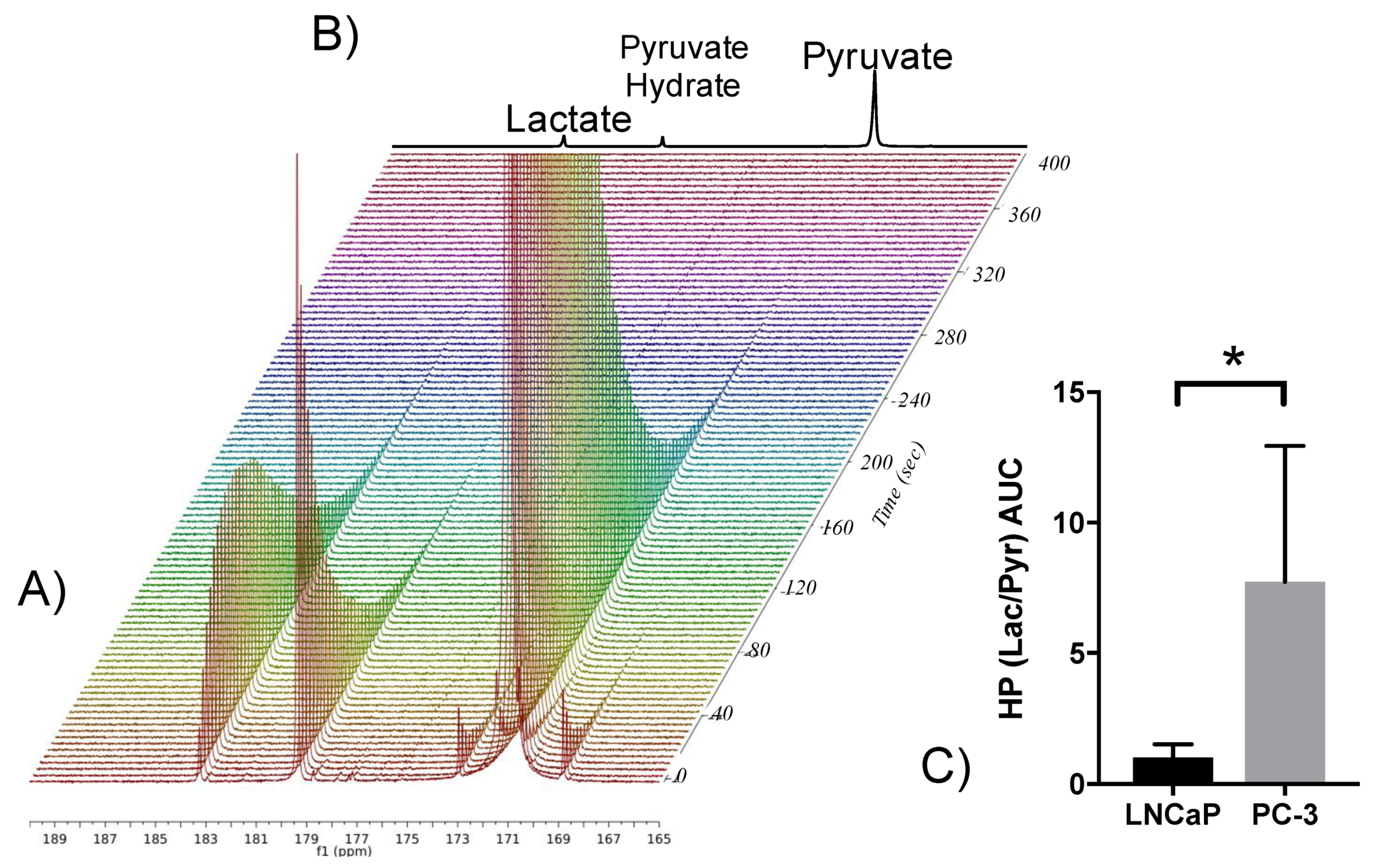
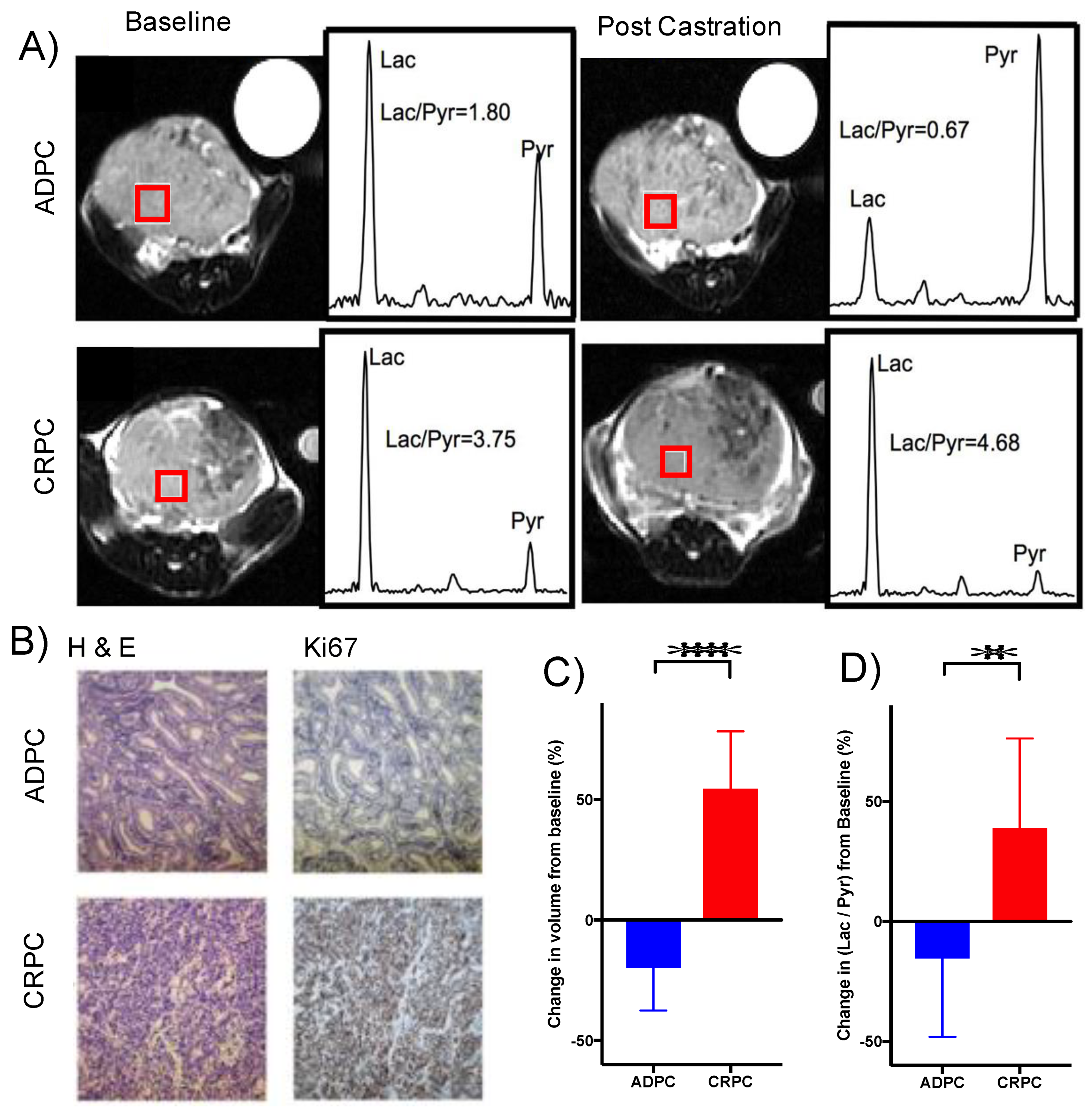
2.7. Elevated Glycolysis Observed in CRPC vs. ADPC Using HP [1-13C]pyruvate
3. Discussion
4. Materials and Methods
4.1. Cell Culture, 13C-Labeling, and Extraction of Metabolites
4.2. Generation of Androgen-Dependent and CRPC TRAMP Tumors, 13C-Labeling, and Extraction of Metabolites
4.3. NMR Acquisition and Quantification
4.4. 13C Isotopomer Modeling
4.5. Enzyme Activity Assays
4.6. ATP Quantification
4.7. NAD/NADH and NADP/NADPH Assays
4.8. Glutathione Assay
4.9. Oxygen Consumption
4.10. Seahorse Analysis
4.11. HP 13C NMR Spectroscopy of Cell Slurries
4.12. TRAMP Hyperpolarized 13C MRI
4.13. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ryan, C.J.; Small, E.J. Role of secondary hormonal therapy in the management of recurrent prostate cancer. Urology 2003, 62 (Suppl. S1), 87–94. [Google Scholar] [CrossRef]
- Kirby, M.; Hirst, C.; Crawford, E.D. Characterising the castration-resistant prostate cancer population: A systematic review. Int. J. Clin. Pract. 2011, 65, 1180–1192. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S. Androgen receptor signaling and mutations in prostate cancer. Asian J. Androl. 2010, 12, 639–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jernberg, E.; Bergh, A.; Wikström, P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect. 2017, 6, R146–R161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 2013, 23, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [Green Version]
- Coller, H.A. Is cancer a metabolic disease? Am. J. Pathol. 2014, 184, 4–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell 2017, 32, 474–489.e6. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.J.; Kurhanewicz, J.; Vigneron, D.B.; Larson, P.E.Z.; Harzstark, A.L.; Ferrone, M.; Van Criekinge, M.; Chang, J.W.; Bok, R.; Park, I.; et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]Pyruvate. Sci. Transl. Med. 2013, 5, 198. [Google Scholar] [CrossRef] [Green Version]
- Albers, M.J.; Bok, R.; Chen, A.P.; Cunningham, C.H.; Zierhut, M.L.; Zhang, V.Y.; Kohler, S.J.; Tropp, J.; Hurd, R.E.; Yen, Y.-F.; et al. Hyperpolarized 13C lactate, pyruvate, and alanine: Noninvasive biomarkers for prostate cancer detection and grading. Cancer Res. 2008, 68, 8607–8615. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-Y.; Larson, P.E.Z.; Bok, R.A.; von Morze, C.; Sriram, R.; Delos Santos, R.; DeLos Santos, J.; Gordon, J.W.; Bahrami, N.; Ferrone, M.; et al. Assessing prostate cancer aggressiveness with hyperpolarized dual-agent 3D dynamic imaging of metabolism and perfusion. Cancer Res. 2017, 77, 3207–3216. [Google Scholar] [CrossRef] [Green Version]
- Sriram, R.; Van Criekinge, M.; DeLos Santos, J.; Ahamed, F.; Qin, H.; Nolley, R.; Delos Santos, R.; Tabatabai, Z.L.; Bok, R.A.; Keshari, K.R.; et al. Elevated tumor lactate and efflux in high-grade prostate cancer demonstrated by hyperpolarized 13C magnetic resonance spectroscopy of prostate tissue slice cultures. Cancers 2020, 12, 537. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.; Vigneron, D.B.; Kurhanewicz, J. Hyperpolarized 1-[13C]-Pyruvate magnetic resonance imaging detects an early metabolic response to androgen ablation therapy in prostate cancer. Eur. Urol. 2017, 72, 1028–1029. [Google Scholar] [CrossRef]
- Gingrich, J.R.; Barrios, R.J.; Kattan, M.W.; Nahm, H.S.; Finegold, M.J.; Greenberg, N.M. Androgen-Independent prostate cancer progression in the TRAMP model. Cancer Res. 1997, 57, 4687–4691. [Google Scholar]
- Crawford, E.D. Hormonal therapy in prostate cancer: Historical approaches. Rev. Urol. 2004, 6 (Suppl. S7), S3–S11. [Google Scholar]
- Gomella, L.G. Effective testosterone suppression for prostate cancer: Is there a best castration therapy? Rev. Urol. 2009, 11, 52–60. [Google Scholar]
- Sherry, A.D.; Malloy, C.R. 13C isotopomer analysis of glutamate a NMR method to probe metabolic pathways intersecting in the citric acid cycle. In In Vivo Carbon-13 NMR; Berliner, L.J., Robitaille, P.M., Eds.; Springer: Boston, MA, USA, 2002; Volume 15, pp. 59–97. [Google Scholar]
- Costello, L.C.; Franklin, R.B. “Why do tumour cells glycolyse?”: From glycolysis through citrate to lipogenesis. Mol. Cell. Biochem. 2005, 280, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Mulder, H.; Zhao, P.; Burgess, S.C.; Jensen, M.V.; Kamzolova, S.; Newgard, C.B.; Sherry, A.D. 13C NMR isotopomer analysis reveals a connection between pyruvate cycling and glucose-stimulated insulin secretion (GSIS). Proc. Natl. Acad. Sci. USA 2002, 99, 2708–2713. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Wu, C.-Y.; Wynn, R.M.; Gui, W.; Malloy, C.R.; Sherry, A.D.; Chuang, D.T.; Khemtong, C. Real-Time hyperpolarized 13C magnetic resonance detects increased pyruvate oxidation in pyruvate dehydrogenase kinase 2/4-double knockout mouse livers. Sci. Rep. 2019, 9, 16480. [Google Scholar] [CrossRef]
- Malloy, C.R.; Jones, J.G.; Jeffrey, F.M.; Jessen, M.E.; Sherry, A.D. Contribution of various substrates to total citric acid cycle flux and ]anaplerosis as determined by 13C isotopomer analysis and O2 consumption in the heart. Magn. Reson. Mater. Phy. 1996, 4, 35–46. [Google Scholar] [CrossRef]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Cohen, A.; Sun, Y.; Squires, J.; Braas, D.; Graeber, T.G.; Du, L.; Li, G.; Li, Z.; Xu, X.; et al. The role of CD44 in glucose metabolism in prostatic small cell neuroendocrine carcinoma. Mol. Cancer Res. 2016, 14, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Vaz, C.V.; Alves, M.G.; Marques, R.; Moreira, P.I.; Oliveira, P.F.; Maia, C.J.; Socorro, S. Androgen-Responsive and nonresponsive prostate cancer cells present a distinct glycolytic metabolism profile. Int. J. Biochem. Cell Biol. 2012, 44, 2077–2084. [Google Scholar] [CrossRef]
- Zacharias, N.; Lee, J.; Ramachandran, S.; Shanmugavelandy, S.; McHenry, J.; Dutta, P.; Millward, S.; Gammon, S.; Efstathiou, E.; Troncoso, P.; et al. Androgen receptor signaling in castration-resistant prostate cancer alters hyperpolarized pyruvate to lactate conversion and lactate levels in vivo. Mol. Imaging Biol. 2019, 21, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.C.; Xue, H.; Wu, R.; Fazli, L.; Lin, D.; Collins, C.C.; Gleave, M.E.; Gout, P.W.; Wang, Y. The MCT4 gene: A novel, potential target for therapy of advanced prostate cancer. Clin. Cancer Res. 2016, 22, 2721–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ros, S.; Santos, C.R.; Moco, S.; Baenke, F.; Kelly, G.; Howell, M.; Zamboni, N.; Schulze, A. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2012, 2, 328–343. [Google Scholar] [CrossRef] [Green Version]
- Higgins, L.H.; Withers, H.G.; Garbens, A.; Love, H.D.; Magnoni, L.; Hayward, S.W.; Moyes, C.D. Hypoxia and the metabolic phenotype of prostate cancer cells. Biochim. Biophys. Acta 2009, 1787, 1433–1443. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, A.K.; Vareed, S.K.; Basu, S.; Putluri, V.; Putluri, N.; Panzitt, K.; Brennan, C.A.; Chinnaiyan, A.M.; Vergara, I.A.; Erho, N.; et al. Metabolomic profiling identifies biochemical pathways associated with castration-resistant prostate cancer. J. Proteome Res. 2013, 13, 1088–1100. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hardie, R.-A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Chaiswing, L.; Zhong, W.; Oberley, T.D. Distinct redox profiles of selected human prostate carcinoma cell lines: Implications for rational design of redox therapy. Cancers 2011, 3, 3557–3584. [Google Scholar] [CrossRef]
- Naiki, T.; Naiki-Ito, A.; Asamoto, M.; Kawai, N.; Tozawa, K.; Etani, T.; Sato, S.; Suzuki, S.; Shirai, T.; Kohri, K.; et al. GPX2 overexpression is involved in cell proliferation and prognosis of castration-resistant prostate cancer. Carcinogenesis 2014, 35, 1962–1967. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Kido, L.A.; de Almeida Lamas, C.; Maróstica, M.R.; Cagnon, V.H.A. Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) model: A good alternative to study PCa progression and chemoprevention approaches. Life Sci. 2019, 217, 141–147. [Google Scholar] [CrossRef]
- Bok, R.; Lee, J.; Sriram, R.; Keshari, K.; Sukumar, S.; Daneshmandi, S.; Korenchan, D.E.; Flavell, R.R.; Vigneron, D.B.; Kurhanewicz, J.; et al. The role of lactate metabolism in prostate cancer progression and metastases revealed by dual-agent hyperpolarized 13C MRSI. Cancers 2019, 11, 257. [Google Scholar] [CrossRef] [Green Version]
- Labrecque, M.P.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; da Costa, R.M.G.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 129, 4492–4505. [Google Scholar] [CrossRef] [Green Version]
- Connelly, Z.M.; Yang, S.; Chen, F.; Yeh, Y.; Khater, N.; Jin, R.; Matusik, R.; Yu, X. Foxa2 activates the transcription of androgen receptor target genes in castrate resistant prostatic tumors. Am. J. Clin. Exp. Urol. 2018, 6, 172–181. [Google Scholar]
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zong, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2019, 1, 70–85. [Google Scholar] [CrossRef]
- Zacharias, N.M.; McCullough, C.; Shanmugavelandy, S.; Lee, J.; Lee, Y.; Dutta, P.; McHenry, J.; Nguyen, L.; Norton, W.; Jones, L.W.; et al. Metabolic differences in glutamine utilization lead to metabolic vulnerabilities in prostate cancer. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Van Heijster, F.H.A.; Heskamp, S.; Breukels, V.; Veltien, A.; Franssen, G.M.; Jansen, K.C.F.J.; Boerman, O.C.; Schalken, J.A.; Scheenen, T.W.J.; Heerschap, A. Pyruvate-Lactate exchange and glucose uptake in human prostate cancer cell models. A study in xenografts and suspensions by hyperpolarized [1-13C]pyruvate MRS and [18F]FDG-PET. NMR Biomed. 2020, 33, e4362. [Google Scholar] [CrossRef]
- Kurhanewicz, J.; Vigneron, D.B.; Brindle, K.; Chekmenev, E.Y.; Comment, A.; Cunningham, C.H.; DeBerardinis, R.J.; Green, G.G.; Leach, M.O.; Rajan, S.S.; et al. Analysis of cancer metabolism by imaging hyperpolarized nuclei: Prospects for translation to clinical research. Neoplasia 2011, 13, 81–97. [Google Scholar] [CrossRef] [Green Version]
- Maharjan, R.P.; Ferenci, T. Global metabolite analysis: The influence of extraction methodology on metabolome profiles of Escherichia coli. Anal. Biochem. 2003, 313, 145–154. [Google Scholar] [CrossRef]
- Lane, A.N.; Yan, J.; Fan, T.W.M. 13C tracer studies of metabolism in mouse tumor xenografts. Bio-Protocol 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, R.; Nguyen, J.; Santos, J.D.; Nguyen, L.; Sun, J.; Vigneron, S.; Van Criekinge, M.; Kurhanewicz, J.; MacKenzie, J.D. Molecular detection of inflammation in cell models using hyperpolarized 13C-pyruvate. Theranostics 2018, 8, 3400–3407. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, S.; Toikka, M.M.; Karhunen, P.T.; Kilpeläinen, I.A. Quantitative 2D HSQC (Q-HSQC) via suppression of J-dependence of polarization transfer in NMR spectroscopy: Application to wood lignin. J. Am. Chem. Soc. 2003, 125, 4362–4367. [Google Scholar] [CrossRef]
- Malloy, C.R.; Sherry, A.D.; Jeffrey, F.M. Analysis of tricarboxylic acid cycle of the heart using 13C isotope isomers. Am. J. Physiol. 1990, 259, H987–H995. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.; Bok, R.A.; DeLos Santos, J.; Upadhyay, D.; DeLos Santos, R.; Agarwal, S.; Van Criekinge, M.; Vigneron, D.B.; Aggarwal, R.; Peehl, D.M.; et al. Resistance to Androgen Deprivation Leads to Altered Metabolism in Human and Murine Prostate Cancer Cell and Tumor Models. Metabolites 2021, 11, 139. https://doi.org/10.3390/metabo11030139
Sun J, Bok RA, DeLos Santos J, Upadhyay D, DeLos Santos R, Agarwal S, Van Criekinge M, Vigneron DB, Aggarwal R, Peehl DM, et al. Resistance to Androgen Deprivation Leads to Altered Metabolism in Human and Murine Prostate Cancer Cell and Tumor Models. Metabolites. 2021; 11(3):139. https://doi.org/10.3390/metabo11030139
Chicago/Turabian StyleSun, Jinny, Robert A. Bok, Justin DeLos Santos, Deepti Upadhyay, Romelyn DeLos Santos, Shubhangi Agarwal, Mark Van Criekinge, Daniel B. Vigneron, Rahul Aggarwal, Donna M. Peehl, and et al. 2021. "Resistance to Androgen Deprivation Leads to Altered Metabolism in Human and Murine Prostate Cancer Cell and Tumor Models" Metabolites 11, no. 3: 139. https://doi.org/10.3390/metabo11030139
APA StyleSun, J., Bok, R. A., DeLos Santos, J., Upadhyay, D., DeLos Santos, R., Agarwal, S., Van Criekinge, M., Vigneron, D. B., Aggarwal, R., Peehl, D. M., Kurhanewicz, J., & Sriram, R. (2021). Resistance to Androgen Deprivation Leads to Altered Metabolism in Human and Murine Prostate Cancer Cell and Tumor Models. Metabolites, 11(3), 139. https://doi.org/10.3390/metabo11030139