
Transporters at the Interface between Cytosolic and Mitochondrial Amino Acid Metabolism

Abstract
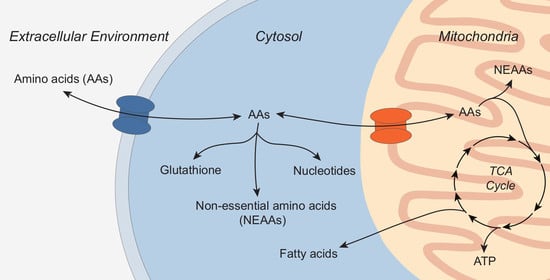
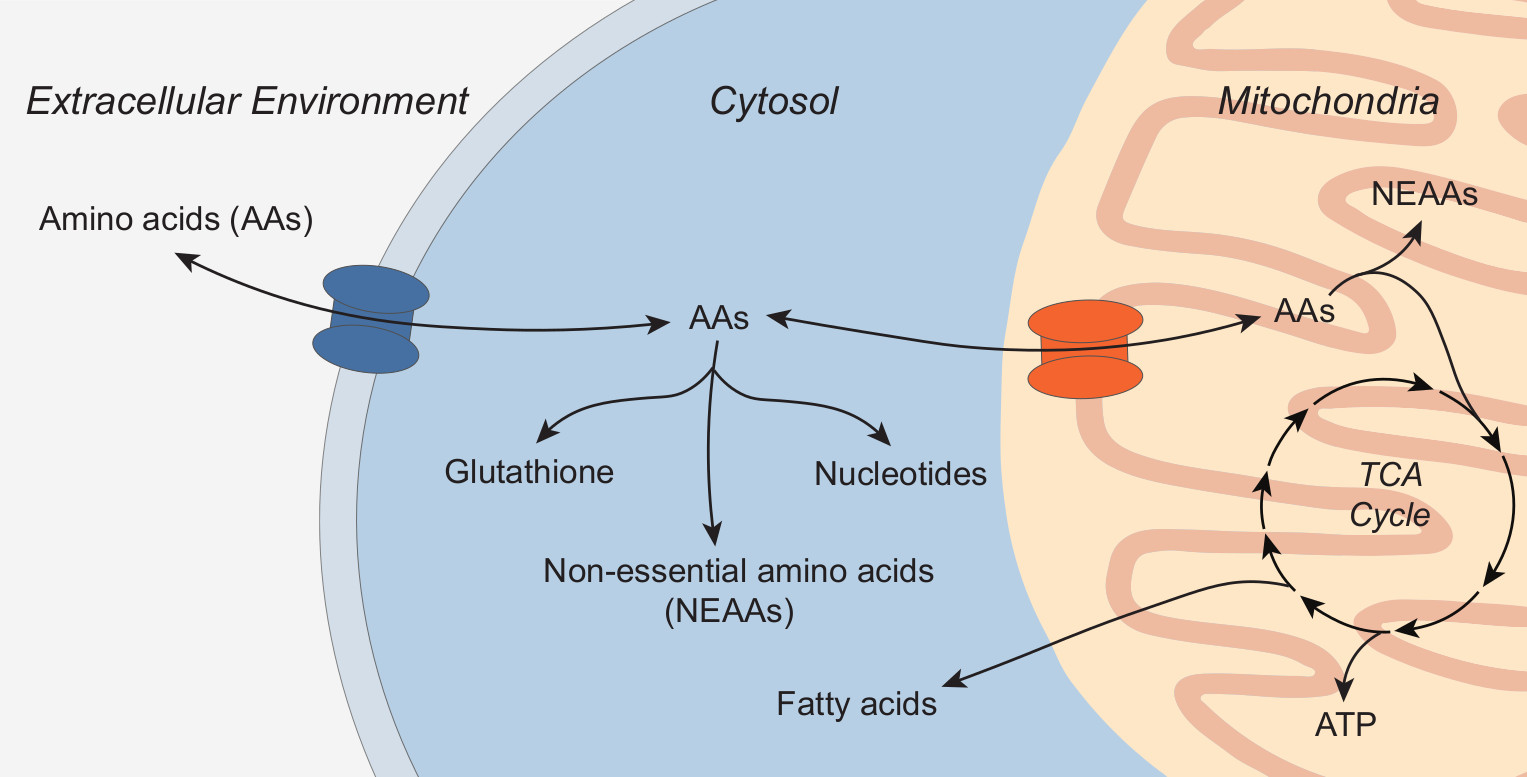
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Compartmentalized Amino Acid Metabolism
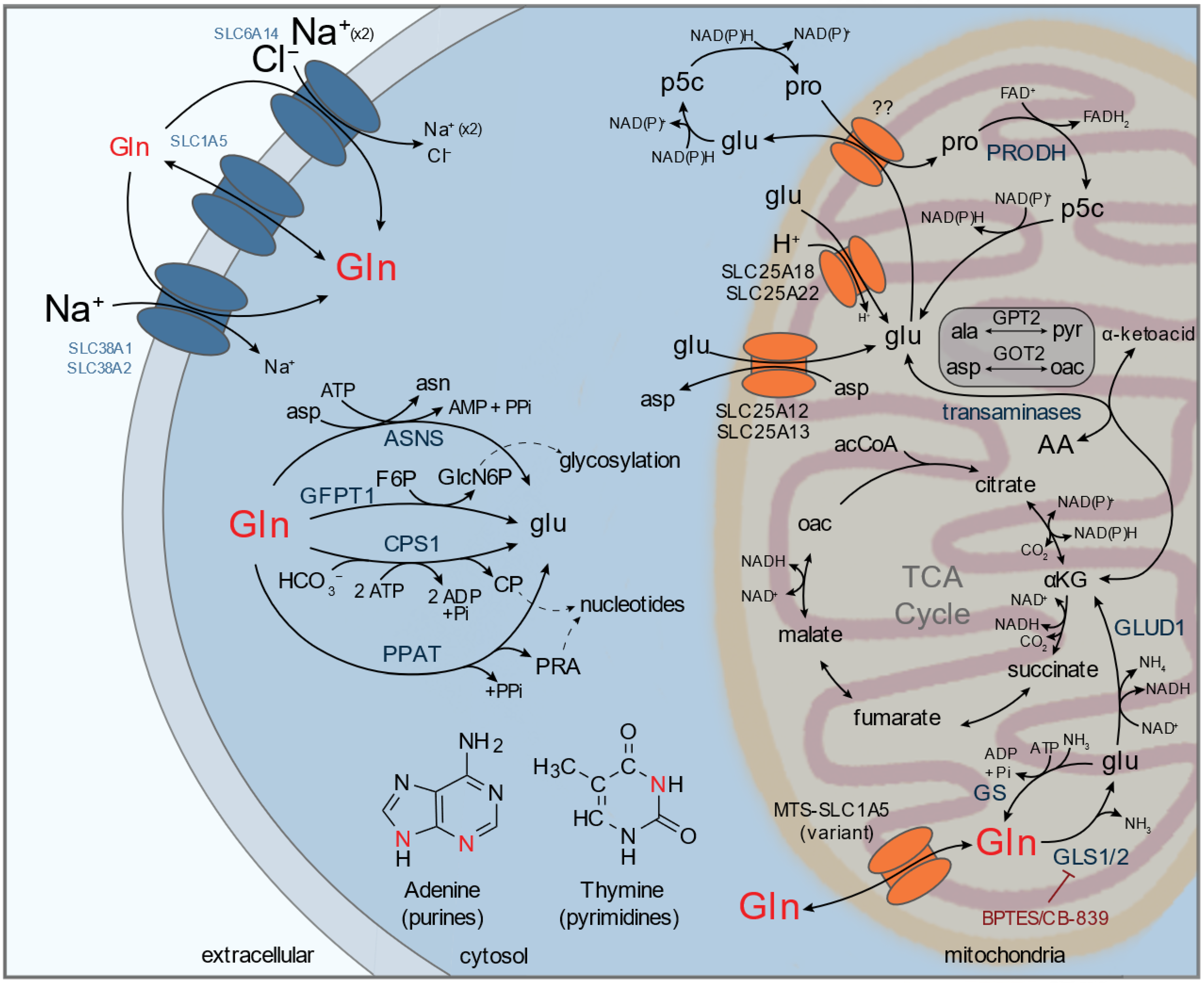
2.1. Glutamine
2.2. Aspartate
2.3. Serine, Glycine and Alanine
2.4. Branched-Chain Amino Acids
3. Mitochondrial Amino Acid Carriers
4. Approaches to Quantify Mitochondrial Metabolism and Transport
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCB | ATP-binding cassette transporter |
| AGC1/2 | Aspartate-glutamate carrier 1 or 2 |
| AML | Acute myeloid leukemia |
| ARG2 | Arginase 2 |
| ASNS | Asparagine synthetase |
| ASS1 | Argininosuccinate synthase 1 |
| BAT | Brown adipose tissue |
| BCAA | Branched-chain amino acid |
| BCAT1/2 | Branched-chain amino acid transaminase 1 or 2 |
| BCKA | Branched-chain ketoacid |
| BCKDH | Branched-chain ketoacid dehydrogenase |
| BCKDK | Branched-chain ketoacid kinase |
| CAF | Cancer-associated fibroblasts |
| CP | Carbamoyl phosphate |
| CPS | Carbamoyl phosphate synthase |
| CRAT | Carnitine acetyltransferase |
| CRC | Colorectal cancer |
| DMEM | Dulbecco’s modified eagle’s medium |
| DON | 6-diazo-5-oxo-L-norleucine |
| ECM | Extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| ETC | Electron transport chain |
| FASN | Fatty acid synthase |
| FH | Fumarate hydratase |
| FOCM | Folate-mediated one carbon metabolism |
| GCN2 | General control nonderepressible 2 |
| GCS | Glycine cleavage system |
| GFPT1 | Glutamine-fructose 6-phosphate aminotransferase 1 |
| GlcN6P | Glucosamine 6-phosphate |
| GlcNAc | N-acetyl-glucosamine |
| GLO1 | Glyoxalase 1 |
| GLS1/2 | Glutaminase 1 or 2 |
| GLUD1 | Glutamate dehydrogenase 1 |
| GOT1/2 | Glutamic-oxaloacetic transaminase 1 or 2 |
| gpASNase1 | Guinea pig asparaginase 1 |
| GPT1/2 | Glutamic-pyruvic transaminase 1 or 2 |
| GS | Glutamine synthetase |
| GSH | Reduced glutathione |
| HCC | Hepatocellular carcinoma |
| IDH1/2 | Isocitrate dehydrogenase 1 or 2 |
| IMM | Inner mitochondrial membrane |
| iNOS | Induced nitric oxide synthase |
| KIC | α-ketoisocaproic |
| KIV | α-ketoisovaleric |
| KMV | α-keto-β-methylvaleric |
| LAT1 | Large amino acid transporter 1 |
| LDH | Lactate dehydrogenase |
| MAS | Malate-aspartate-shuttle |
| MCT1/4 | Monocarboxylate transporter 1 or 4 |
| MDH | Malate dehydrogenase |
| ME1 | Malic enzyme 1 |
| MMA | Methylmalonic acid |
| MPC1/2 | Mitochondrial pyruvate carrier 1 or 2 |
| MSUD | Maple syrup urine disease |
| MTHFD1/1L/2 | Methylenetetrahydrofolate dehydrogenase 1, 1L, or 2 |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NMDA | N-methyl D-aspartate |
| NMR | Nuclear magnetic resonance |
| NO | Nitric oxide |
| NSCLC | Non-small cell lung cancer |
| OAA | Oxaloacetate |
| OMM | Outer mitochondrial membrane |
| PC | Pyruvate carboxylase |
| PDAC | Pancreatic ductal adenocarcinoma |
| PHGDH | Phosphoserine dehydrogenase |
| PPAT | Pyrophosphate amidotransferase |
| PPM1K | Mg2+/Mn2+-dependent 1K protein phosphatase |
| PRA | 5-phospho-β-D-ribosylamine |
| PRODH | Proline dehydrogenase |
| PSAT1 | Phosphoserine aminotransferase 1 |
| PSPH | Phosphoserine phosphatase |
| PYCR1 | Pyrroline 5-carboxylate reductase 1 |
| SDH | Succinate dehydrogenase |
| SFXN1/3 | Sideroflexin 1 or 3 |
| TCA | Tricarboxylic acid |
| UCP2 | Uncoupling protein 2 |
| δ-ALA | δ-aminolevulinic acid |
References
- Gray, M.W. Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V. The origin and early evolution of eukaryotes in the light of phylogenomics. Genome Biol. 2010, 11, 209. [Google Scholar] [CrossRef]
- Roger, A.J.; Munoz-Gomez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayers, J.R.; Heiden, M.G.V. Famine versus feast: Understanding the metabolism of tumors in vivo. Trends Biochem. Sci. 2015, 40, 130–140. [Google Scholar] [CrossRef]
- Stein, W.H.; Moore, S. The free amino acids of human blood plasma. J. Biol. Chem. 1954, 211, 915–926. [Google Scholar] [CrossRef]
- Fries, A.W.; Dadsetan, S.; Keiding, S.; Bak, L.K.; Schousboe, A.; Waagepetersen, H.S.; Simonsen, M.; Ott, P.; Vilstrup, H.; Sorensen, M. Effect of glutamine synthetase inhibition on brain and interorgan ammonia metabolism in bile duct ligated rats. J. Cereb. Blood Flow Metab. 2014, 34, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, J.; Dolinska, M. Glutamine as a pathogenic factor in hepatic encephalopathy. J. Neurosci. Res. 2001, 65, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 2018, 175, 1780–1795.e19. [Google Scholar] [CrossRef] [Green Version]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.O.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [Green Version]
- Francescone, R.; Vendramini-Costa, D.B.; Franco-Barraza, J.; Wagner, J.; Muir, A.; Lau, A.N.; Gabitova, L.; Pazina, T.; Gupta, S.; Luong, T.; et al. Netrin G1 promotes pancreatic tumorigenesis through cancer associated fibroblast driven nutritional support and immunosuppression. Cancer Discov. 2021, 11, 446–479. [Google Scholar] [CrossRef] [PubMed]
- Thai, M.; Thaker, S.K.; Feng, J.; Du, Y.; Hu, H.; Wu, T.T.; Graeber, T.G.; Braas, D.; Christofk, H.R. MYC-induced reprogramming of glutamine catabolism supports optimal virus replication. Nat. Commun. 2015, 6, 8873. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.I.; Huang, W.R.; Chiu, H.C.; Li, J.Y.; Nielsen, B.L.; Liu, H.J. Avian reovirus sigmaA-modulated suppression of lactate dehydrogenase and upregulation of glutaminolysis and the mTOC1/eIF4E/HIF-1alpha pathway to enhance glycolysis and the TCA cycle for virus replication. Cell Microbiol. 2018, 20, e12946. [Google Scholar] [CrossRef] [PubMed]
- Kedia-Mehta, N.; Finlay, D.K. Competition for nutrients and its role in controlling immune responses. Nat. Commun. 2019, 10, 2123. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gomez, C.; Campos-Sandoval, J.A.; Alonso, F.J.; Segura, J.A.; Manzanares, E.; Ruiz-Sanchez, P.; Gonzalez, M.E.; Marquez, J.; Mates, J.M. Co-expression of glutaminase K and L isoenzymes in human tumour cells. Biochem. J. 2005, 386, 535–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.N.; Berthezene, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.; Grunewald, T.G.P.; Fendt, S.M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef]
- Hollinshead, K.E.R.; Munford, H.; Eales, K.L.; Bardella, C.; Li, C.; Escribano-Gonzalez, C.; Thakker, A.; Nonnenmacher, Y.; Kluckova, K.; Jeeves, M.; et al. Oncogenic IDH1 Mutations Promote Enhanced Proline Synthesis through PYCR1 to Support the Maintenance of Mitochondrial Redox Homeostasis. Cell Rep. 2018, 22, 3107–3114. [Google Scholar] [CrossRef] [Green Version]
- Cruzat, V.; Rogero, M.M.; Keane, K.N.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, C.; Rafii, M.; Sgro, M.; Ball, R.O.; Pencharz, P. Arginine is synthesized from proline, not glutamate, in enterally fed human preterm neonates. Pediatr. Res. 2011, 69, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Song, P.; Jiang, T.; Dai, D.; Wang, H.; Sun, J.; Zhu, L.; Xu, W.; Feng, L.; Shin, V.Y.; et al. Heat Shock Factor 1 Epigenetically Stimulates Glutaminase-1-Dependent mTOR Activation to Promote Colorectal Carcinogenesis. Mol. Ther. 2018, 26, 1828–1839. [Google Scholar] [CrossRef] [Green Version]
- Rathore, M.G.; Saumet, A.; Rossi, J.F.; de Bettignies, C.; Tempe, D.; Lecellier, C.H.; Villalba, M. The NF-kappaB member p65 controls glutamine metabolism through miR-23a. Int. J. Biochem. Cell Biol. 2012, 44, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Lukey, M.J.; Greene, K.S.; Erickson, J.W.; Wilson, K.F.; Cerione, R.A. The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat. Commun. 2016, 7, 11321. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrego, J.; Gunda, V.; Vernucci, E.; Shukla, S.K.; King, R.J.; Dasgupta, A.; Goode, G.; Murthy, D.; Yu, F.; Singh, P.K. GOT1-mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett. 2017, 400, 37–46. [Google Scholar] [CrossRef]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Oosterveer, M.H.; Stein, S.; Demagny, H.; Ryu, D.; Moullan, N.; Wang, X.; Can, E.; Zamboni, N.; Comment, A.; et al. LRH-1-dependent programming of mitochondrial glutamine processing drives liver cancer. Genes Dev. 2016, 30, 1255–1260. [Google Scholar] [CrossRef]
- Robinson, M.M.; McBryant, S.J.; Tsukamoto, T.; Rojas, C.; Ferraris, D.V.; Hamilton, S.K.; Hansen, J.C.; Curthoys, N.P. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 2007, 406, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Stine, Z.E.; Xia, J.; Lu, Y.; O’Connor, R.S.; Altman, B.J.; Hsieh, A.L.; Gouw, A.M.; Thomas, A.G.; Gao, P.; et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Invest. 2015, 125, 2293–2306. [Google Scholar] [CrossRef] [Green Version]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, K.; Hartmann, R.; Tsiampali, J.; Uhlmann, C.; Nickel, A.C.; He, X.; Kamp, M.A.; Sabel, M.; Barker, R.A.; Steiger, H.J.; et al. A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov. 2020, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Biancur, D.E.; Paulo, J.A.; Malachowska, B.; Quiles Del Rey, M.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 2017, 8, 15965. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Kodama, M.; Oshikawa, K.; Shimizu, H.; Yoshioka, S.; Takahashi, M.; Izumi, Y.; Bamba, T.; Tateishi, C.; Tomonaga, T.; Matsumoto, M.; et al. A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nat. Commun. 2020, 11, 1320. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Morcillo, M.; Grande-Garcia, A.; Ruiz-Ramos, A.; Del Cano-Ochoa, F.; Boskovic, J.; Ramon-Maiques, S. Structural Insight into the Core of CAD, the Multifunctional Protein Leading De Novo Pyrimidine Biosynthesis. Structure 2017, 25, 912–923.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, A.; Haanstra, J.R.; Engqvist, M.; Gerding, A.; Bakker, B.M.; Klingmuller, U.; Teusink, B.; Nielsen, J. Quantitative analysis of amino acid metabolism in liver cancer links glutamate excretion to nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2020, 117, 10294–10304. [Google Scholar] [CrossRef]
- Chen, Q.; Muller, J.S.; Pang, P.C.; Laval, S.H.; Haslam, S.M.; Lochmuller, H.; Dell, A. Global N-linked Glycosylation is Not Significantly Impaired in Myoblasts in Congenital Myasthenic Syndromes Caused by Defective Glutamine-Fructose-6-Phosphate Transaminase 1 (GFPT1). Biomolecules 2015, 5, 2758–2781. [Google Scholar] [CrossRef] [Green Version]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 2018, 17, 1824–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magill, G.B.; Myers, W.P.; Reilly, H.C.; Putnam, R.C.; Magill, J.W.; Sykes, M.P.; Escher, G.C.; Karnofsky, D.A.; Burchenal, J.H. Pharmacological and initial therapeutic observations on 6-diazo-5-oxo-1-norleucine (DON) in human neoplastic disease. Cancer 1957, 10, 1138–1150. [Google Scholar] [CrossRef]
- Nedelcovych, M.T.; Tenora, L.; Kim, B.H.; Kelschenbach, J.; Chao, W.; Hadas, E.; Jancarik, A.; Prchalova, E.; Zimmermann, S.C.; Dash, R.P.; et al. N-(Pivaloyloxy)alkoxy-carbonyl Prodrugs of the Glutamine Antagonist 6-Diazo-5-oxo-l-norleucine (DON) as a Potential Treatment for HIV Associated Neurocognitive Disorders. J. Med. Chem. 2017, 60, 7186–7198. [Google Scholar] [CrossRef]
- Ueki, N.; Lee, S.; Sampson, N.S.; Hayman, M.J. Selective cancer targeting with prodrugs activated by histone deacetylases and a tumour-associated protease. Nat. Commun. 2013, 4, 2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rais, R.; Jancarik, A.; Tenora, L.; Nedelcovych, M.; Alt, J.; Englert, J.; Rojas, C.; Le, A.; Elgogary, A.; Tan, J.; et al. Discovery of 6-Diazo-5-oxo-l-norleucine (DON) Prodrugs with Enhanced CSF Delivery in Monkeys: A Potential Treatment for Glioblastoma. J. Med. Chem. 2016, 59, 8621–8633. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Zhao, L.; Wu, Y.; Veeravalli, V.; Alt, J.; Aguilar, J.M.H.; Dash, R.P.; Lam, J.; Tenora, L.; Rodriguez, C.; et al. Novel lymphoid targeted prodrugs of the glutamine antagonist DON for the treatment of hematological malignancies. FASEB J. 2016, 30, lb472. [Google Scholar] [CrossRef]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. Membrane transporters for the special amino acid glutamine: Structure/function relationships and relevance to human health. Front. Chem. 2014, 2, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhao, T.; Li, Z.; Wang, L.; Yuan, S.; Sun, L. The role of ASCT2 in cancer: A review. Eur. J. Pharmacol. 2018, 837, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Nalecz, K.A. Amino Acid Transporter SLC6A14 (ATB(0,+))—A Target in Combined Anti-cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 594464. [Google Scholar] [CrossRef]
- Broer, A.; Rahimi, F.; Broer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J. Biol. Chem. 2016, 291, 13194–13205. [Google Scholar] [CrossRef] [Green Version]
- Broer, A.; Fairweather, S.; Broer, S. Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Front. Pharmacol. 2018, 9, 785. [Google Scholar] [CrossRef] [PubMed]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef]
- Van Geldermalsen, M.; Quek, L.E.; Turner, N.; Freidman, N.; Pang, A.; Guan, Y.F.; Krycer, J.R.; Ryan, R.; Wang, Q.; Holst, J. Benzylserine inhibits breast cancer cell growth by disrupting intracellular amino acid homeostasis and triggering amino acid response pathways. BMC Cancer 2018, 18, 689. [Google Scholar] [CrossRef] [Green Version]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.J.; Amendola, C.R.; Hollinshead, K.E.R.; Yu, Q.; Yamamoto, K.; Encarnacion-Rosado, J.; Rose, R.E.; LaRue, M.M.; Sohn, A.S.W.; Biancur, D.E.; et al. Selective Alanine Transporter Utilization Creates a Targetable Metabolic Niche in Pancreatic Cancer. Cancer Discov. 2020, 10, 1018–1037. [Google Scholar] [CrossRef]
- Coothankandaswamy, V.; Cao, S.; Xu, Y.; Prasad, P.D.; Singh, P.K.; Reynolds, C.P.; Yang, S.; Ogura, J.; Ganapathy, V.; Bhutia, Y.D. Amino acid transporter SLC6A14 is a novel and effective drug target for pancreatic cancer. Br. J. Pharmacol. 2016, 173, 3292–3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luongo, T.S.; Eller, J.M.; Lu, M.J.; Niere, M.; Raith, F.; Perry, C.; Bornstein, M.R.; Oliphint, P.; Wang, L.; McReynolds, M.R.; et al. SLC25A51 is a mammalian mitochondrial NAD(+) transporter. Nature 2020, 588, 174–179. [Google Scholar] [CrossRef]
- Girardi, E.; Agrimi, G.; Goldmann, U.; Fiume, G.; Lindinger, S.; Sedlyarov, V.; Srndic, I.; Gurtl, B.; Agerer, B.; Kartnig, F.; et al. Epistasis-driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nat. Commun. 2020, 11, 6145. [Google Scholar] [CrossRef]
- Kory, N.; de Bos, J.U.; van der Rijt, S.; Jankovic, N.; Gura, M.; Arp, N.; Pena, I.A.; Prakash, G.; Chan, S.H.; Kunchok, T.; et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Raho, S.; Capobianco, L.; Malivindi, R.; Vozza, A.; Piazzolla, C.; De Leonardis, F.; Gorgoglione, R.; Scarcia, P.; Pezzuto, F.; Agrimi, G.; et al. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat. Metab. 2020, 2, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Heiden, M.G.V. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Hwang, S.; Kim, M.; Seo, S.B.; Lee, J.H.; Jeong, S.M. Mitochondrial glutamine metabolism via GOT2 supports pancreatic cancer growth through senescence inhibition. Cell Death Dis. 2018, 9, 55. [Google Scholar] [CrossRef]
- Hong, R.; Zhang, W.; Xia, X.; Zhang, K.; Wang, Y.; Wu, M.; Fan, J.; Li, J.; Xia, W.; Xu, F.; et al. Preventing BRCA1/ZBRK1 repressor complex binding to the GOT2 promoter results in accelerated aspartate biosynthesis and promotion of cell proliferation. Mol. Oncol. 2019, 13, 959–977. [Google Scholar] [CrossRef] [Green Version]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Heiden, M.G.V. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Alkan, H.F.; Walter, K.E.; Luengo, A.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Lau, A.N.; Al-Zoughbi, W.; Lewis, C.A.; Thomas, C.J.; Hoefler, G.; et al. Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab. 2018, 28, 706–720.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, L.B.; Luengo, A.; Danai, L.V.; Bush, L.N.; Diehl, F.F.; Hosios, A.M.; Lau, A.N.; Elmiligy, S.; Malstrom, S.; Lewis, C.A.; et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 2018, 20, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Tajan, M.; Hock, A.K.; Blagih, J.; Robertson, N.A.; Labuschagne, C.F.; Kruiswijk, F.; Humpton, T.J.; Adams, P.D.; Vousden, K.H. A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab. 2018, 28, 721–736.e6. [Google Scholar] [CrossRef] [Green Version]
- Melendez-Rodriguez, F.; Urrutia, A.A.; Lorendeau, D.; Rinaldi, G.; Roche, O.; Bogurcu-Seidel, N.; Muelas, M.O.; Mesa-Ciller, C.; Turiel, G.; Bouthelier, A.; et al. HIF1alpha Suppresses Tumor Cell Proliferation through Inhibition of Aspartate Biosynthesis. Cell Rep. 2019, 26, 2257–2265.e4. [Google Scholar] [CrossRef] [Green Version]
- Hollinshead, K.E.R.; Parker, S.J.; Eapen, V.V.; Encarnacion-Rosado, J.; Sohn, A.; Oncu, T.; Cammer, M.; Mancias, J.D.; Kimmelman, A.C. Respiratory Supercomplexes Promote Mitochondrial Efficiency and Growth in Severely Hypoxic Pancreatic Cancer. Cell Rep. 2020, 33, 108231. [Google Scholar] [CrossRef]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [Green Version]
- Folger, O.; Jerby, L.; Frezza, C.; Gottlieb, E.; Ruppin, E.; Shlomi, T. Predicting selective drug targets in cancer through metabolic networks. Mol. Syst. Biol. 2011, 7, 501. [Google Scholar] [CrossRef]
- Yang, Y.; Lane, A.N.; Ricketts, C.J.; Sourbier, C.; Wei, M.H.; Shuch, B.; Pike, L.; Wu, M.; Rouault, T.A.; Boros, L.G.; et al. Metabolic reprogramming for producing energy and reducing power in fumarate hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PLoS ONE 2013, 8, e72179. [Google Scholar] [CrossRef]
- Mullen, A.R.; Hu, Z.; Shi, X.; Jiang, L.; Boroughs, L.K.; Kovacs, Z.; Boriack, R.; Rakheja, D.; Sullivan, L.B.; Linehan, W.M.; et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014, 7, 1679–1690. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Mates, J.M.; DeBerardinis, R.J. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef] [Green Version]
- Cardaci, S.; Zheng, L.; MacKay, G.; van den Broek, N.J.; MacKenzie, E.D.; Nixon, C.; Stevenson, D.; Tumanov, S.; Bulusu, V.; Kamphorst, J.J.; et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol. 2015, 17, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Lussey-Lepoutre, C.; Hollinshead, K.E.; Ludwig, C.; Menara, M.; Morin, A.; Castro-Vega, L.J.; Parker, S.J.; Janin, M.; Martinelli, C.; Ottolenghi, C.; et al. Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat. Commun. 2015, 6, 8784. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.N.; Li, Z.; Danai, L.V.; Westermark, A.M.; Darnell, A.M.; Ferreira, R.; Gocheva, V.; Sivanand, S.; Lien, E.C.; Sapp, K.M.; et al. Dissecting cell-type-specific metabolism in pancreatic ductal adenocarcinoma. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcelli, V.; Fiermonte, G.; Longo, A.; Palmieri, F. The human gene SLC25A29, of solute carrier family 25, encodes a mitochondrial transporter of basic amino acids. J. Biol. Chem. 2014, 289, 13374–13384. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, Q.; Gu, J.; Yin, L.; Liang, S.; Wu, L.; Xu, H.; Zhao, C.; Gu, Y. Elevated mitochondrial SLC25A29 in cancer modulates metabolic status by increasing mitochondria-derived nitric oxide. Oncogene 2018, 37, 2545–2558. [Google Scholar] [CrossRef]
- Lim, H.K.; Lim, H.K.; Ryoo, S.; Benjo, A.; Shuleri, K.; Miriel, V.; Baraban, E.; Camara, A.; Soucy, K.; Nyhan, D.; et al. Mitochondrial arginase II constrains endothelial NOS-3 activity. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3317–H3324. [Google Scholar] [CrossRef]
- Wu, G.; Morris, S.M., Jr. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336 Pt 1, 1–17. [Google Scholar] [CrossRef]
- Lorenzi, P.L.; Reinhold, W.C.; Rudelius, M.; Gunsior, M.; Shankavaram, U.; Bussey, K.J.; Scherf, U.; Eichler, G.S.; Martin, S.E.; Chin, K.; et al. Asparagine synthetase as a causal, predictive biomarker for L-asparaginase activity in ovarian cancer cells. Mol. Cancer Ther. 2006, 5, 2613–2623. [Google Scholar] [CrossRef] [Green Version]
- Touzart, A.; Lengline, E.; Latiri, M.; Belhocine, M.; Smith, C.; Thomas, X.; Spicuglia, S.; Puthier, D.; Pflumio, F.; Leguay, T.; et al. Epigenetic Silencing Affects l-Asparaginase Sensitivity and Predicts Outcome in T-ALL. Clin. Cancer Res. 2019, 25, 2483–2493. [Google Scholar] [CrossRef] [Green Version]
- Sahu, D.; Gupta, S.; Hau, A.M.; Nakashima, K.; Leivo, M.Z.; Searles, S.C.; Elson, P.; Bomalaski, J.S.; Casteel, D.E.; Boss, G.R.; et al. Argininosuccinate Synthetase 1 Loss in Invasive Bladder Cancer Regulates Survival through General Control Nonderepressible 2 Kinase-Mediated Eukaryotic Initiation Factor 2alpha Activity and Is Targetable by Pegylated Arginine Deiminase. Am. J. Pathol. 2017, 187, 200–213. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, L.J.; Smith, P.R.; Hiller, L.; Szlosarek, P.W.; Kimberley, C.; Sehouli, J.; Koensgen, D.; Mustea, A.; Schmid, P.; Crook, T. Epigenetic silencing of argininosuccinate synthetase confers resistance to platinum-induced cell death but collateral sensitivity to arginine auxotrophy in ovarian cancer. Int. J. Cancer 2009, 125, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Furuya, S.; Osuka, S.; Mitoma, J.; Shinoda, Y.; Watanabe, M.; Azuma, N.; Tanaka, H.; Hashikawa, T.; Itohara, S.; et al. Targeted disruption of the mouse 3-phosphoglycerate dehydrogenase gene causes severe neurodevelopmental defects and results in embryonic lethality. J. Biol. Chem. 2004, 279, 3573–3577. [Google Scholar] [CrossRef] [Green Version]
- Mothet, J.P.; Parent, A.T.; Wolosker, H.; Brady, R.O., Jr.; Linden, D.J.; Ferris, C.D.; Rogawski, M.A.; Snyder, S.H. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 4926–4931. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Li, S.; Yang, C.; Jia, H.; Gu, Z.; Tu, X.; Tian, S.; Liu, J.; Li, G.; Ma, Y. D-serine Ameliorates Motor and Cognitive Impairments in beta-amyloid 1-42 Injected Mice by Inhibiting JNK Signaling Pathway. J. Chem. Neuroanat. 2020, 109, 101852. [Google Scholar] [CrossRef] [PubMed]
- Heresco-Levy, U.; Javitt, D.C.; Ebstein, R.; Vass, A.; Lichtenberg, P.; Bar, G.; Catinari, S.; Ermilov, M. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol. Psychiatry 2005, 57, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Tsai, G.; Yang, P.; Chung, L.C.; Lange, N.; Coyle, J.T. D-serine added to antipsychotics for the treatment of schizophrenia. Biol. Psychiatry 1998, 44, 1081–1089. [Google Scholar] [CrossRef]
- Beltran-Castillo, S.; Eugenin, J.; von Bernhardi, R. Impact of Aging in Microglia-Mediated D-Serine Balance in the CNS. Mediat. Inflamm. 2018, 2018, 7219732. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M. Role of the transcription factor ATF4 in the anabolic actions of insulin and the anti-anabolic actions of glucocorticoids. J. Biol. Chem. 2007, 282, 16744–16753. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Song, L.; Wan, Q.; Wu, G.; Li, X.; Wang, Y.; Wang, J.; Liu, Z.; Zhong, X.; He, X.; et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015, 25, 429–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, D.; Park, Y.; Andrabi, S.A.; Shelton, L.M.; Gilkes, D.M.; Semenza, G.L. PHGDH Expression Is Required for Mitochondrial Redox Homeostasis, Breast Cancer Stem Cell Maintenance, and Lung Metastasis. Cancer Res. 2016, 76, 4430–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Gravel, S.P.; Hulea, L.; Toban, N.; Birman, E.; Blouin, M.J.; Zakikhani, M.; Zhao, Y.; Topisirovic, I.; St-Pierre, J.; Pollak, M. Serine deprivation enhances antineoplastic activity of biguanides. Cancer Res. 2014, 74, 7521–7533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddocks, O.D.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef]
- Ngo, B.; Kim, E.; Osorio-Vasquez, V.; Doll, S.; Bustraan, S.; Liang, R.J.; Luengo, A.; Davidson, S.M.; Ali, A.; Ferraro, G.B.; et al. Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH Inhibition. Cancer Discov. 2020, 10, 1352–1373. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Mattaini, K.R.; Dennstedt, E.A.; Nguyen, A.A.; Sivanand, S.; Reilly, M.F.; Meeth, K.; Muir, A.; Darnell, A.M.; Bosenberg, M.W.; et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. 2019, 29, 1410–1421.e4. [Google Scholar] [CrossRef]
- Banh, R.S.; Biancur, D.E.; Yamamoto, K.; Sohn, A.S.W.; Walters, B.; Kuljanin, M.; Gikandi, A.; Wang, H.; Mancias, J.D.; Schneider, R.J.; et al. Neurons Release Serine to Support mRNA Translation in Pancreatic Cancer. Cell 2020, 183, 1202–1218.e25. [Google Scholar] [CrossRef]
- Weinstabl, H.; Treu, M.; Rinnenthal, J.; Zahn, S.K.; Ettmayer, P.; Bader, G.; Dahmann, G.; Kessler, D.; Rumpel, K.; Mischerikow, N.; et al. Intracellular Trapping of the Selective Phosphoglycerate Dehydrogenase (PHGDH) Inhibitor BI-4924 Disrupts Serine Biosynthesis. J. Med. Chem. 2019, 62, 7976–7997. [Google Scholar] [CrossRef] [PubMed]
- Pacold, M.E.; Brimacombe, K.R.; Chan, S.H.; Rohde, J.M.; Lewis, C.A.; Swier, L.J.; Possemato, R.; Chen, W.W.; Sullivan, L.B.; Fiske, B.P.; et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 2016, 12, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Mullarky, E.; Lucki, N.C.; Zavareh, R.B.; Anglin, J.L.; Gomes, A.P.; Nicolay, B.N.; Wong, J.C.; Christen, S.; Takahashi, H.; Singh, P.K.; et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl. Acad. Sci. USA 2016, 113, 1778–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Ducker, G.S.; Chen, L.; Morscher, R.J.; Ghergurovich, J.M.; Esposito, M.; Teng, X.; Kang, Y.; Rabinowitz, J.D. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016, 24, 640–641. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Heiden, M.G.V.; Metallo, C.M. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducker, G.S.; Ghergurovich, J.M.; Mainolfi, N.; Suri, V.; Jeong, S.K.; Hsin-Jung Li, S.; Friedman, A.; Manfredi, M.G.; Gitai, Z.; Kim, H.; et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11404–11409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [Green Version]
- Meiser, J.; Tumanov, S.; Maddocks, O.; Labuschagne, C.F.; Athineos, D.; Van Den Broek, N.; Mackay, G.M.; Gottlieb, E.; Blyth, K.; Vousden, K.; et al. Serine one-carbon catabolism with formate overflow. Sci. Adv. 2016, 2, e1601273. [Google Scholar] [CrossRef] [Green Version]
- Meiser, J.; Schuster, A.; Pietzke, M.; Voorde, J.V.; Athineos, D.; Oizel, K.; Burgos-Barragan, G.; Wit, N.; Dhayade, S.; Morton, J.P.; et al. Increased formate overflow is a hallmark of oxidative cancer. Nat. Commun. 2018, 9, 1368. [Google Scholar] [CrossRef]
- Yang, L.; Canaveras, J.C.G.; Chen, Z.; Wang, L.; Liang, L.; Jang, C.; Mayr, J.A.; Zhang, Z.; Ghergurovich, J.M.; Zhan, L.; et al. Serine Catabolism Feeds NADH when Respiration Is Impaired. Cell Metab. 2020, 31, 809–821.e6. [Google Scholar] [CrossRef] [PubMed]
- Reina-Campos, M.; Diaz-Meco, M.T.; Moscat, J. The complexity of the serine glycine one-carbon pathway in cancer. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Fiske, B.P.; Birsoy, K.; Freinkman, E.; Kami, K.; Possemato, R.L.; Chudnovsky, Y.; Pacold, M.E.; Chen, W.W.; Cantor, J.R.; et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 2015, 520, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luengo, A.; Abbott, K.L.; Davidson, S.M.; Hosios, A.M.; Faubert, B.; Chan, S.H.; Freinkman, E.; Zacharias, L.G.; Mathews, T.P.; Clish, C.B.; et al. Reactive metabolite production is a targetable liability of glycolytic metabolism in lung cancer. Nat. Commun. 2019, 10, 5604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.; Feng, J.; Cao, Y.; Shen, S.; Cai, Y.; Yang, D.; Yan, R.; Wang, L.; Zhang, H.; Zhong, X.; et al. Glycine cleavage system determines the fate of pluripotent stem cells via the regulation of senescence and epigenetic modifications. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [Green Version]
- Mari, M.; Morales, A.; Colell, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, R.; Nagata, A.; Murray, B.W. Protein redox chemistry: Post-translational cysteine modifications that regulate signal transduction and drug pharmacology. Front. Pharmacol. 2014, 5, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, Z.D.; Gates, K.S. Thiol-dependent recovery of catalytic activity from oxidized protein tyrosine phosphatases. Biochemistry 2013, 52, 6412–6423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, C.; Battaglia, E.; Burkholz, T.; Peng, D.; Bagrel, D.; Montenarh, M. Control of oxidative posttranslational cysteine modifications: From intricate chemistry to widespread biological and medical applications. Chem. Res. Toxicol. 2012, 25, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Mattaini, K.R.; Sullivan, M.R.; Heiden, M.G.V. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felig, P.; Pozefsky, T.; Marliss, E.; Cahill, G.F., Jr. Alanine: Key role in gluconeogenesis. Science 1970, 167, 1003–1004. [Google Scholar] [CrossRef]
- Mallet, L.E.; Exton, J.H.; Park, C.R. Control of gluconeogenesis from amino acids in the perfused rat liver. J. Biol. Chem. 1969, 244, 5713–5723. [Google Scholar] [CrossRef]
- Ross, B.D.; Hems, R.; Krebs, H.A. The rate of gluconeogenesis from various precursors in the perfused rat liver. Biochem. J. 1967, 102, 942–951. [Google Scholar] [CrossRef] [Green Version]
- Felig, P.; Wahren, J. Amino acid metabolism in exercising man. J. Clin. Invest. 1971, 50, 2703–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, K.F.; Dufour, S.; Cline, G.W.; Shulman, G.I. Regulation of hepatic mitochondrial oxidation by glucose-alanine cycling during starvation in humans. J. Clin. Invest. 2019, 129, 4671–4675. [Google Scholar] [CrossRef]
- Mayers, J.R.; Wu, C.; Clish, C.B.; Kraft, P.; Torrence, M.E.; Fiske, B.P.; Yuan, C.; Bao, Y.; Townsend, M.K.; Tworoger, S.S.; et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 2014, 20, 1193–1198. [Google Scholar] [CrossRef]
- Ni, J.; Zhang, L. Cancer Cachexia: Definition, Staging, and Emerging Treatments. Cancer Manag. Res. 2020, 12, 5597–5605. [Google Scholar] [CrossRef]
- Melville, S.; McNurlan, M.A.; Calder, A.G.; Garlick, P.J. Increased protein turnover despite normal energy metabolism and responses to feeding in patients with lung cancer. Cancer Res. 1990, 50, 1125–1131. [Google Scholar]
- Leij-Halfwerk, S.; van den Berg, J.W.; Sijens, P.E.; Wilson, J.H.; Oudkerk, M.; Dagnelie, P.C. Altered hepatic gluconeogenesis during L-alanine infusion in weight-losing lung cancer patients as observed by phosphorus magnetic resonance spectroscopy and turnover measurements. Cancer Res. 2000, 60, 618–623. [Google Scholar]
- Leij-Halfwerk, S.; Dagnelie, P.C.; van Den Berg, J.W.; Wattimena, J.D.; Hordijk-Luijk, C.H.; Wilson, J.P. Weight loss and elevated gluconeogenesis from alanine in lung cancer patients. Am. J. Clin. Nutr. 2000, 71, 583–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burt, M.E.; Gorschboth, C.M.; Brennan, M.F. A controlled, prospective, randomized trial evaluating the metabolic effects of enteral and parenteral nutrition in the cancer patient. Cancer 1982, 49, 1092–1105. [Google Scholar] [CrossRef]
- Waterhouse, C.; Jeanpretre, N.; Keilson, J. Gluconeogenesis from alanine in patients with progressive malignant disease. Cancer Res. 1979, 39, 1968–1972. [Google Scholar] [PubMed]
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New aspects of amino acid metabolism in cancer. Br. J. Cancer 2020, 122, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Groen, A.K.; Sips, H.J.; Vervoorn, R.C.; Tager, J.M. Intracellular compartmentation and control of alanine metabolism in rat liver parenchymal cells. Eur. J. Biochem. 1982, 122, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Dieterle, P.; Brawand, F.; Moser, U.K.; Walter, P. Alanine metabolism in rat liver mitochondria. Eur. J. Biochem. 1978, 88, 467–473. [Google Scholar] [CrossRef]
- Mendes-Mourao, J.; Halestrap, A.P.; Crisp, D.M.; Pogson, C.I. The involvement of mitochondrial pyruvate transport in the pathways of gluconeogenesis from serine and alanine in isolated rat and mouse liver cells. FEBS Lett. 1975, 53, 29–32. [Google Scholar] [CrossRef] [Green Version]
- DeRosa, G.; Swick, R.W. Metabolic implications of the distribution of the alanine aminotransferase isoenzymes. J. Biol. Chem. 1975, 250, 7961–7967. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron-Harel, N.; Ghergurovich, J.M.; Notarangelo, G.; LaFleur, M.W.; Tsubosaka, Y.; Sharpe, A.H.; Rabinowitz, J.D.; Haigis, M.C. T Cell Activation Depends on Extracellular Alanine. Cell Rep. 2019, 28, 3011–3021.e4. [Google Scholar] [CrossRef] [Green Version]
- Elia, I.; Rossi, M.; Stegen, S.; Broekaert, D.; Doglioni, G.; van Gorsel, M.; Boon, R.; Escalona-Noguero, C.; Torrekens, S.; Verfaillie, C.; et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 2019, 568, 117–121. [Google Scholar] [CrossRef]
- Sips, H.J.; Groen, A.K.; Tager, J.M. Plasma-membrane transport of alanine is rate-limiting for its metabolism in rat-liver parenchymal cells. FEBS Lett. 1980, 119, 271–274. [Google Scholar] [CrossRef] [Green Version]
- McGivan, J.D.; Ramsell, J.C.; Lacey, J.H. Stimulation of alanine transport and metabolism by dibutyryl cyclic AMP in the hepatocytes from fed rats. Assessment of transport as a potential rate-limiting step for alanine metabolism. Biochim. Biophys. Acta 1981, 644, 295–304. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Heiden, M.G.V.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Heiden, M.G.V.; Muir, A. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Segawa, H.; Miyamoto, K.; Uchino, H.; Takeda, E.; Endou, H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem. 1998, 273, 23629–23632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, H.N. Role of amino acid transport and countertransport in nutrition and metabolism. Physiol. Rev. 1990, 70, 43–77. [Google Scholar] [CrossRef]
- Sivanand, S.; Heiden, M.G.V. Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell 2020, 37, 147–156. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efeyan, A.; Zoncu, R.; Sabatini, D.M. Amino acids and mTORC1: From lysosomes to disease. Trends Mol. Med. 2012, 18, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Broer, S. Amino Acid Transporters as Targets for Cancer Therapy: Why, Where, When, and How. Int. J. Mol. Sci. 2020, 21, 6156. [Google Scholar] [CrossRef]
- Broer, A.; Gauthier-Coles, G.; Rahimi, F.; van Geldermalsen, M.; Dorsch, D.; Wegener, A.; Holst, J.; Broer, S. Ablation of the ASCT2 (SLC1A5) gene encoding a neutral amino acid transporter reveals transporter plasticity and redundancy in cancer cells. J. Biol. Chem. 2019, 294, 4012–4026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bothwell, P.J.; Kron, C.D.; Wittke, E.F.; Czerniak, B.N.; Bode, B.P. Targeted Suppression and Knockout of ASCT2 or LAT1 in Epithelial and Mesenchymal Human Liver Cancer Cells Fail to Inhibit Growth. Int. J. Mol. Sci. 2018, 19, 2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormerais, Y.; Giuliano, S.; LeFloch, R.; Front, B.; Durivault, J.; Tambutte, E.; Massard, P.A.; de la Ballina, L.R.; Endou, H.; Wempe, M.F.; et al. Genetic Disruption of the Multifunctional CD98/LAT1 Complex Demonstrates the Key Role of Essential Amino Acid Transport in the Control of mTORC1 and Tumor Growth. Cancer Res. 2016, 76, 4481–4492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najumudeen, A.K.; Ceteci, F.; Fey, S.K.; Hamm, G.; Steven, R.T.; Hall, H.; Nikula, C.J.; Dexter, A.; Murta, T.; Race, A.M.; et al. The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat. Genet. 2021, 53, 16–26. [Google Scholar] [CrossRef]
- Okano, N.; Naruge, D.; Kawai, K.; Kobayashi, T.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phase I study of JPH203, an L-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Invest. New Drugs 2020, 38, 1495–1506. [Google Scholar] [CrossRef]
- Rosilio, C.; Nebout, M.; Imbert, V.; Griessinger, E.; Neffati, Z.; Benadiba, J.; Hagenbeek, T.; Spits, H.; Reverso, J.; Ambrosetti, D.; et al. L-type amino-acid transporter 1 (LAT1): A therapeutic target supporting growth and survival of T-cell lymphoblastic lymphoma/T-cell acute lymphoblastic leukemia. Leukemia 2015, 29, 1253–1266. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Hosoda, N.; Endo, H.; Saito, K.; Tsujihara, K.; Yamamura, M.; Sakata, T.; Anzai, N.; Wempe, M.F.; Kanai, Y.; et al. L-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci. 2010, 101, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Bohmer, C.; Broer, A.; Munzinger, M.; Kowalczuk, S.; Rasko, J.E.; Lang, F.; Broer, S. Characterization of mouse amino acid transporter B0AT1 (slc6a19). Biochem. J. 2005, 389, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broer, A.; Klingel, K.; Kowalczuk, S.; Rasko, J.E.; Cavanaugh, J.; Broer, S. Molecular cloning of mouse amino acid transport system B0, a neutral amino acid transporter related to Hartnup disorder. J. Biol. Chem. 2004, 279, 24467–24476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Shah, N.; Broer, A.; Fairweather, S.; Jiang, Y.; Schmoll, D.; Corry, B.; Broer, S. Identification of novel inhibitors of the amino acid transporter B(0) AT1 (SLC6A19), a potential target to induce protein restriction and to treat type 2 diabetes. Br. J. Pharmacol. 2017, 174, 468–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, A.; Shah, N.; Tiwari, P.K.; Javed, K.; Cheng, Q.; Aidhen, I.S.; Broer, S. Novel Chemical Scaffolds to Inhibit the Neutral Amino Acid Transporter B(0)AT1 (SLC6A19), a Potential Target to Treat Metabolic Diseases. Front. Pharmacol. 2020, 11, 140. [Google Scholar] [CrossRef]
- Peng, H.; Wang, Y.; Luo, W. Multifaceted role of branched-chain amino acid metabolism in cancer. Oncogene 2020, 39, 6747–6756. [Google Scholar] [CrossRef] [PubMed]
- Holecek, M. Branched-chain amino acids in health and disease: Metabolism, alterations in blood plasma, and as supplements. Nutr. Metab. (Lond.) 2018, 15, 33. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.R.; Wallin, R.; Reinhart, G.D.; Hutson, S.M. Branched chain aminotransferase isoenzymes. Purification and characterization of the rat brain isoenzyme. J. Biol. Chem. 1993, 268, 3092–3098. [Google Scholar] [CrossRef]
- Li, J.T.; Yin, M.; Wang, D.; Wang, J.; Lei, M.Z.; Zhang, Y.; Liu, Y.; Zhang, L.; Zou, S.W.; Hu, L.P.; et al. BCAT2-mediated BCAA catabolism is critical for development of pancreatic ductal adenocarcinoma. Nat. Cell Biol. 2020, 22, 167–174. [Google Scholar] [CrossRef]
- Zhang, L.; Han, J. Branched-chain amino acid transaminase 1 (BCAT1) promotes the growth of breast cancer cells through improving mTOR-mediated mitochondrial biogenesis and function. Biochem. Biophys. Res. Commun. 2017, 486, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Canela, M.; Toledo, E.; Clish, C.B.; Hruby, A.; Liang, L.; Salas-Salvado, J.; Razquin, C.; Corella, D.; Estruch, R.; Ros, E.; et al. Plasma Branched-Chain Amino Acids and Incident Cardiovascular Disease in the PREDIMED Trial. Clin. Chem. 2016, 62, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, F.; Zhang, Y.; Li, X.; Chen, C.; Zhou, M.; Yu, Z.; Liu, Y.; Zhao, Y.; Hao, X.; et al. PPM1K Regulates Hematopoiesis and Leukemogenesis through CDC20-Mediated Ubiquitination of MEIS1 and p21. Cell Rep. 2018, 23, 1461–1475. [Google Scholar] [CrossRef]
- Goldberg, E.J.; Buddo, K.A.; McLaughlin, K.L.; Fernandez, R.F.; Pereyra, A.S.; Psaltis, C.E.; Lin, C.T.; Hagen, J.T.; Boykov, I.N.; Nguyen, T.K.; et al. Tissue-specific characterization of mitochondrial branched-chain keto acid oxidation using a multiplexed assay platform. Biochem. J. 2019, 476, 1521–1537. [Google Scholar] [CrossRef]
- Son, S.M.; Park, S.J.; Stamatakou, E.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Leucine regulates autophagy via acetylation of the mTORC1 component raptor. Nat. Commun. 2020, 11, 3148. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Park, S.J.; Lee, H.; Siddiqi, F.; Lee, J.E.; Menzies, F.M.; Rubinsztein, D.C. Leucine Signals to mTORC1 via Its Metabolite Acetyl-Coenzyme A. Cell Metab. 2019, 29, 192–201.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef]
- Dey, P.; Baddour, J.; Muller, F.; Wu, C.C.; Wang, H.; Liao, W.T.; Lan, Z.; Chen, A.; Gutschner, T.; Kang, Y.; et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 2017, 542, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Grankvist, N.; Watrous, J.D.; Lagerborg, K.A.; Lyutvinskiy, Y.; Jain, M.; Nilsson, R. Profiling the Metabolism of Human Cells by Deep (13)C Labeling. Cell Chem. Biol. 2018, 25, 1419–1427.e4. [Google Scholar] [CrossRef]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Achreja, A.; Meurs, N.; Animasahun, O.; Owen, S.; Mittal, A.; Parikh, P.; Lo, T.W.; Franco-Barraza, J.; Shi, J.; et al. Tumour-reprogrammed stromal BCAT1 fuels branched-chain ketoacid dependency in stromal-rich PDAC tumours. Nat. Metab. 2020, 2, 775–792. [Google Scholar] [CrossRef] [PubMed]
- Suh, E.H.; Hackett, E.P.; Wynn, R.M.; Chuang, D.T.; Zhang, B.; Luo, W.; Sherry, A.D.; Park, J.M. In vivo assessment of increased oxidation of branched-chain amino acids in glioblastoma. Sci. Rep. 2019, 9, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mac, M.; Nehlig, A.; Nalecz, M.J.; Nalecz, K.A. Transport of alpha-ketoisocaproate in rat cerebral cortical neurons. Arch. Biochem. Biophys. 2000, 376, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Broer, S.; Schneider, H.P.; Broer, A.; Rahman, B.; Hamprecht, B.; Deitmer, J.W. Characterization of the monocarboxylate transporter 1 expressed in Xenopus laevis oocytes by changes in cytosolic pH. Biochem. J. 1998, 333 Pt 1, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Dimmer, K.S.; Friedrich, B.; Lang, F.; Deitmer, J.W.; Broer, S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem. J. 2000, 350 Pt 1, 219–227. [Google Scholar] [CrossRef]
- Silva, L.S.; Poschet, G.; Nonnenmacher, Y.; Becker, H.M.; Sapcariu, S.; Gaupel, A.C.; Schlotter, M.; Wu, Y.; Kneisel, N.; Seiffert, M.; et al. Branched-chain ketoacids secreted by glioblastoma cells via MCT1 modulate macrophage phenotype. EMBO Rep. 2017, 18, 2172–2185. [Google Scholar] [CrossRef]
- Crown, S.B.; Marze, N.; Antoniewicz, M.R. Catabolism of Branched Chain Amino Acids Contributes Significantly to Synthesis of Odd-Chain and Even-Chain Fatty Acids in 3T3-L1 Adipocytes. PLoS ONE 2015, 10, e0145850. [Google Scholar] [CrossRef] [Green Version]
- Oku, H.; Yagi, N.; Nagata, J.; Chinen, I. Precursor role of branched-chain amino acids in the biosynthesis of iso and anteiso fatty acids in rat skin. Biochim. Biophys. Acta 1994, 1214, 279–287. [Google Scholar]
- Green, C.R.; Wallace, M.; Divakaruni, A.S.; Phillips, S.A.; Murphy, A.N.; Ciaraldi, T.P.; Metallo, C.M. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 2016, 12, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Wallace, M.; Green, C.R.; Roberts, L.S.; Lee, Y.M.; McCarville, J.L.; Sanchez-Gurmaches, J.; Meurs, N.; Gengatharan, J.M.; Hover, J.D.; Phillips, S.A.; et al. Enzyme promiscuity drives branched-chain fatty acid synthesis in adipose tissues. Nat. Chem. Biol. 2018, 14, 1021–1031. [Google Scholar] [CrossRef]
- Gomes, A.P.; Ilter, D.; Low, V.; Endress, J.E.; Fernandez-Garcia, J.; Rosenzweig, A.; Schild, T.; Broekaert, D.; Ahmed, A.; Planque, M.; et al. Age-induced accumulation of methylmalonic acid promotes tumour progression. Nature 2020, 585, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Reichert, A.S.; Neupert, W. Contact sites between the outer and inner membrane of mitochondria-role in protein transport. Biochim. Biophys. Acta 2002, 1592, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lackner, L.L. The Expanding and Unexpected Functions of Mitochondria Contact Sites. Trends Cell Biol. 2019, 29, 580–590. [Google Scholar] [CrossRef]
- Palmieri, F.; Monne, M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. Biophys. Acta 2016, 1863, 2362–2378. [Google Scholar] [CrossRef]
- Kunji, E.R.S.; King, M.S.; Ruprecht, J.J.; Thangaratnarajah, C. The SLC25 Carrier Family: Important Transport Proteins in Mitochondrial Physiology and Pathology. Physiology (Bethesda) 2020, 35, 302–327. [Google Scholar] [CrossRef]
- Cunningham, C.N.; Rutter, J. 20,000 picometers under the OMM: Diving into the vastness of mitochondrial metabolite transport. EMBO Rep. 2020, 21, e50071. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef]
- Kory, N.; Wyant, G.A.; Prakash, G.; Uit de Bos, J.; Bottanelli, F.; Pacold, M.E.; Chan, S.H.; Lewis, C.A.; Wang, T.; Keys, H.R.; et al. SFXN1 is a mitochondrial serine transporter required for one-carbon metabolism. Science 2018, 362. [Google Scholar] [CrossRef] [Green Version]
- Yoneshiro, T.; Wang, Q.; Tajima, K.; Matsushita, M.; Maki, H.; Igarashi, K.; Dai, Z.; White, P.J.; McGarrah, R.W.; Ilkayeva, O.R.; et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature 2019, 572, 614–619. [Google Scholar] [CrossRef]
- Fernandez-Murray, J.P.; Prykhozhij, S.V.; Dufay, J.N.; Steele, S.L.; Gaston, D.; Nasrallah, G.K.; Coombs, A.J.; Liwski, R.S.; Fernandez, C.V.; Berman, J.N.; et al. Glycine and Folate Ameliorate Models of Congenital Sideroblastic Anemia. PLoS Genet. 2016, 12, e1005783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guernsey, D.L.; Jiang, H.; Campagna, D.R.; Evans, S.C.; Ferguson, M.; Kellogg, M.D.; Lachance, M.; Matsuoka, M.; Nightingale, M.; Rideout, A.; et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 2009, 41, 651–653. [Google Scholar] [CrossRef]
- Molina, M.; Segura, J.A.; Aledo, J.C.; Medina, M.A.; de Castro, I.N.; Marquez, J. Glutamine transport by vesicles isolated from tumour-cell mitochondrial inner membrane. Biochem. J. 1995, 308 Pt 2, 629–633. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fendt, S.M.; Bell, E.L.; Keibler, M.A.; Olenchock, B.A.; Mayers, J.R.; Wasylenko, T.M.; Vokes, N.I.; Guarente, L.; Heiden, M.G.V.; Stephanopoulos, G. Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013, 4, 2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative metabolic flux profiling of melanoma cell lines: Beyond the Warburg effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef] [Green Version]
- Hussien, R.; Brooks, G.A. Mitochondrial and plasma membrane lactate transporter and lactate dehydrogenase isoform expression in breast cancer cell lines. Physiol. Genom. 2011, 43, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Hussien, R.; Brooks, G.A. Colocalization of MCT1, CD147, and LDH in mitochondrial inner membrane of L6 muscle cells: Evidence of a mitochondrial lactate oxidation complex. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1237–E1244. [Google Scholar] [CrossRef]
- Hoek, J.B.; Njogu, R.M. The role of glutamate transport in the regulation of the pathway of proline oxidation in rat liver mitochondria. J. Biol. Chem. 1980, 255, 8711–8718. [Google Scholar] [CrossRef]
- Meyer, J. Proline transport in rat liver mitochondria. Arch. Biochem. Biophys. 1977, 178, 387–395. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2020, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Smith, A.C.; Robinson, A.J. MitoMiner v4.0: An updated database of mitochondrial localization evidence, phenotypes and diseases. Nucleic Acids Res. 2019, 47, D1225–D1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.W.; Freinkman, E.; Wang, T.; Birsoy, K.; Sabatini, D.M. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 2016, 166, 1324–1337.e11. [Google Scholar] [CrossRef] [PubMed]
- Roede, J.R.; Park, Y.; Li, S.; Strobel, F.H.; Jones, D.P. Detailed mitochondrial phenotyping by high resolution metabolomics. PLoS ONE 2012, 7, e33020. [Google Scholar] [CrossRef] [Green Version]
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813. [Google Scholar] [CrossRef] [Green Version]
- Ray, G.J.; Boydston, E.A.; Shortt, E.; Wyant, G.A.; Lourido, S.; Chen, W.W.; Sabatini, D.M. A PEROXO-Tag Enables Rapid Isolation of Peroxisomes from Human Cells. iScience 2020, 23, 101109. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Saulnier, J.L.; Wang, W.; Jones, D.R.; Pacold, M.E.; Sabatini, B.L. Rapid purification and metabolomic profiling of synaptic vesicles from mammalian brain. Elife 2020, 9. [Google Scholar] [CrossRef]
- Adelmann, C.H.; Traunbauer, A.K.; Chen, B.; Condon, K.J.; Chan, S.H.; Kunchok, T.; Lewis, C.A.; Sabatini, D.M. MFSD12 mediates the import of cysteine into melanosomes and lysosomes. Nature 2020, 588, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, E.C.; Baudrier, L.; Ozerdem, C.; Lewis, C.A.; Chan, S.H.; Kunchok, T.; Abu-Remaileh, M.; Cangelosi, A.L.; Sabatini, D.M.; Birsoy, K.; et al. MITO-Tag Mice enable rapid isolation and multimodal profiling of mitochondria from specific cell types in vivo. Proc. Natl. Acad. Sci. USA 2019, 116, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Salabei, J.K.; Gibb, A.A.; Hill, B.G. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 2014, 9, 421–438. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.Y.; Heerklotz, H. Digitonin does not flip across cholesterol-poor membranes. J. Colloid Interface Sci. 2017, 504, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Zuurendonk, P.F.; Tager, J.M. Rapid separation of particulate components and soluble cytoplasm of isolated rat-liver cells. Biochim. Biophys. Acta 1974, 333, 393–399. [Google Scholar] [CrossRef]
- Nonnenmacher, Y.; Palorini, R.; d’Herouel, A.F.; Kramer, L.; Neumann-Schaal, M.; Chiaradonna, F.; Skupin, A.; Wegner, A.; Hiller, K. Analysis of mitochondrial metabolism in situ: Combining stable isotope labeling with selective permeabilization. Metab. Eng. 2017, 43, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Gravel, S.P.; Andrzejewski, S.; Avizonis, D.; St-Pierre, J. Stable isotope tracer analysis in isolated mitochondria from mammalian systems. Metabolites 2014, 4, 166–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.D.; Mukha, D.; Aizenshtein, E.; Shlomi, T. Spatial-fluxomics provides a subcellular-compartmentalized view of reductive glutamine metabolism in cancer cells. Nat. Commun. 2019, 10, 1351. [Google Scholar] [CrossRef] [Green Version]
- Saez, M.J.; Lagunas, R. Determination of intermediary metabolites in yeast. Critical examination of the effect of sampling conditions and recommendations for obtaining true levels. Mol. Cell. Biochem. 1976, 13, 73–78. [Google Scholar] [CrossRef]
- Pinu, F.R.; Villas-Boas, S.G.; Aggio, R. Analysis of Intracellular Metabolites from Microorganisms: Quenching and Extraction Protocols. Metabolites 2017, 7, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, G.A.; Dubouchaud, H.; Brown, M.; Sicurello, J.P.; Butz, C.E. Role of mitochondrial lactate dehydrogenase and lactate oxidation in the intracellular lactate shuttle. Proc. Natl. Acad. Sci. USA 1999, 96, 1129–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.J.; Mahieu, N.G.; Huang, X.; Singh, M.; Crawford, P.A.; Johnson, S.L.; Gross, R.W.; Schaefer, J.; Patti, G.J. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 2016, 12, 937–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indiveri, C.; Tonazzi, A.; De Palma, A.; Palmieri, F. Kinetic mechanism of antiports catalyzed by reconstituted ornithine/citrulline carrier from rat liver mitochondria. Biochim. Biophys. Acta 2001, 1503, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Giangregorio, N.; Tonazzi, A.; Console, L.; Indiveri, C. Post-translational modification by acetylation regulates the mitochondrial carnitine/acylcarnitine transport protein. Mol. Cell. Biochem. 2017, 426, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Console, L.; Giangregorio, N.; Indiveri, C.; Tonazzi, A. Carnitine/acylcarnitine translocase and carnitine palmitoyltransferase 2 form a complex in the inner mitochondrial membrane. Mol. Cell. Biochem. 2014, 394, 307–314. [Google Scholar] [CrossRef]
- Console, L.; Tolomeo, M.; Colella, M.; Barile, M.; Indiveri, C. Reconstitution in Proteoliposomes of the Recombinant Human Riboflavin Transporter 2 (SLC52A2) Overexpressed in E. coli. Int. J. Mol. Sci. 2019, 20, 4416. [Google Scholar] [CrossRef] [Green Version]
- Scalise, M.; Pochini, L.; Giangregorio, N.; Tonazzi, A.; Indiveri, C. Proteoliposomes as tool for assaying membrane transporter functions and interactions with xenobiotics. Pharmaceutics 2013, 5, 472–497. [Google Scholar] [CrossRef] [Green Version]
- Krumpochova, P.; Sapthu, S.; Brouwers, J.F.; de Haas, M.; de Vos, R.; Borst, P.; van de Wetering, K. Transportomics: Screening for substrates of ABC transporters in body fluids using vesicular transport assays. FASEB J. 2012, 26, 738–747. [Google Scholar] [CrossRef]
- Saliba, A.E.; Vonkova, I.; Ceschia, S.; Findlay, G.M.; Maeda, K.; Tischer, C.; Deghou, S.; van Noort, V.; Bork, P.; Pawson, T.; et al. A quantitative liposome microarray to systematically characterize protein-lipid interactions. Nat. Methods 2014, 11, 47–50. [Google Scholar] [CrossRef]
- Cesar-Razquin, A.; Snijder, B.; Frappier-Brinton, T.; Isserlin, R.; Gyimesi, G.; Bai, X.; Reithmeier, R.A.; Hepworth, D.; Hediger, M.A.; Edwards, A.M.; et al. A Call for Systematic Research on Solute Carriers. Cell 2015, 162, 478–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hewton, K.G.; Johal, A.S.; Parker, S.J. Transporters at the Interface between Cytosolic and Mitochondrial Amino Acid Metabolism. Metabolites 2021, 11, 112. https://doi.org/10.3390/metabo11020112
Hewton KG, Johal AS, Parker SJ. Transporters at the Interface between Cytosolic and Mitochondrial Amino Acid Metabolism. Metabolites. 2021; 11(2):112. https://doi.org/10.3390/metabo11020112
Chicago/Turabian StyleHewton, Keeley G., Amritpal S. Johal, and Seth J. Parker. 2021. "Transporters at the Interface between Cytosolic and Mitochondrial Amino Acid Metabolism" Metabolites 11, no. 2: 112. https://doi.org/10.3390/metabo11020112
APA StyleHewton, K. G., Johal, A. S., & Parker, S. J. (2021). Transporters at the Interface between Cytosolic and Mitochondrial Amino Acid Metabolism. Metabolites, 11(2), 112. https://doi.org/10.3390/metabo11020112