L-Carnitine and Acylcarnitines: Mitochondrial Biomarkers for Precision Medicine

 , ,
, ,  and
and
Abstract
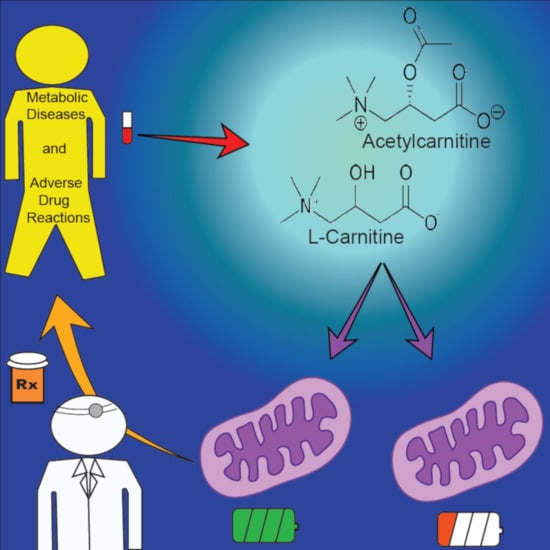
:
1. Introduction
2. Carnitine, Acylcarnitines, and Mitochondrial Bioenergetics
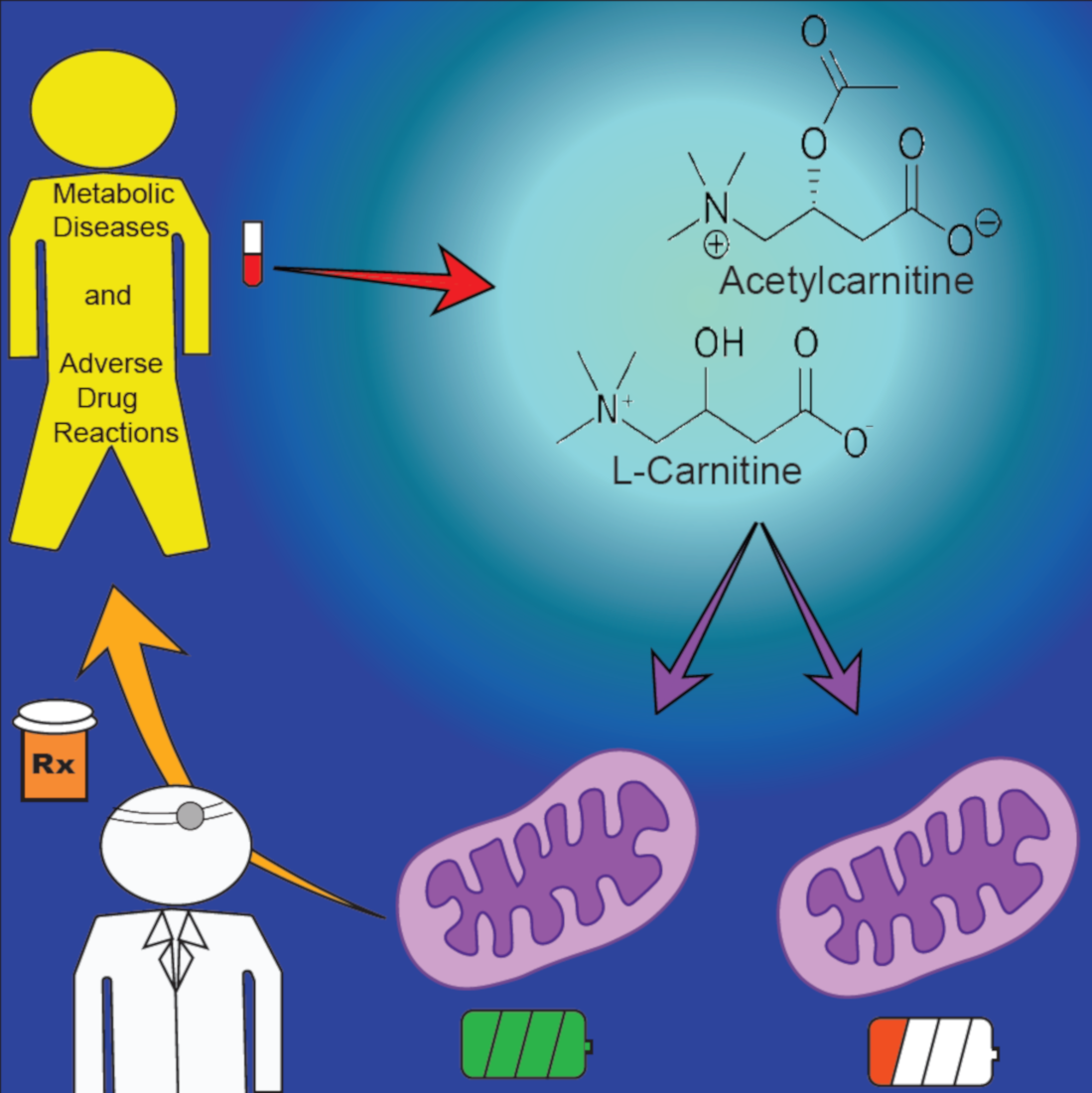
2.1. Primary Role: The Carnitine Shuttle
2.2. Endogenous Carnitine Homeostasis
2.3. Metabolic Pathways of Acylcarnitine Production
3. Disease-Induced Alterations to Carnitine Metabolism
3.1. Diabetes Mellitus
3.2. Sepsis and Septic Shock
3.3. Cancer
3.4. Heart Failure
4. Drug-Induced Alterations to Carnitine Metabolism
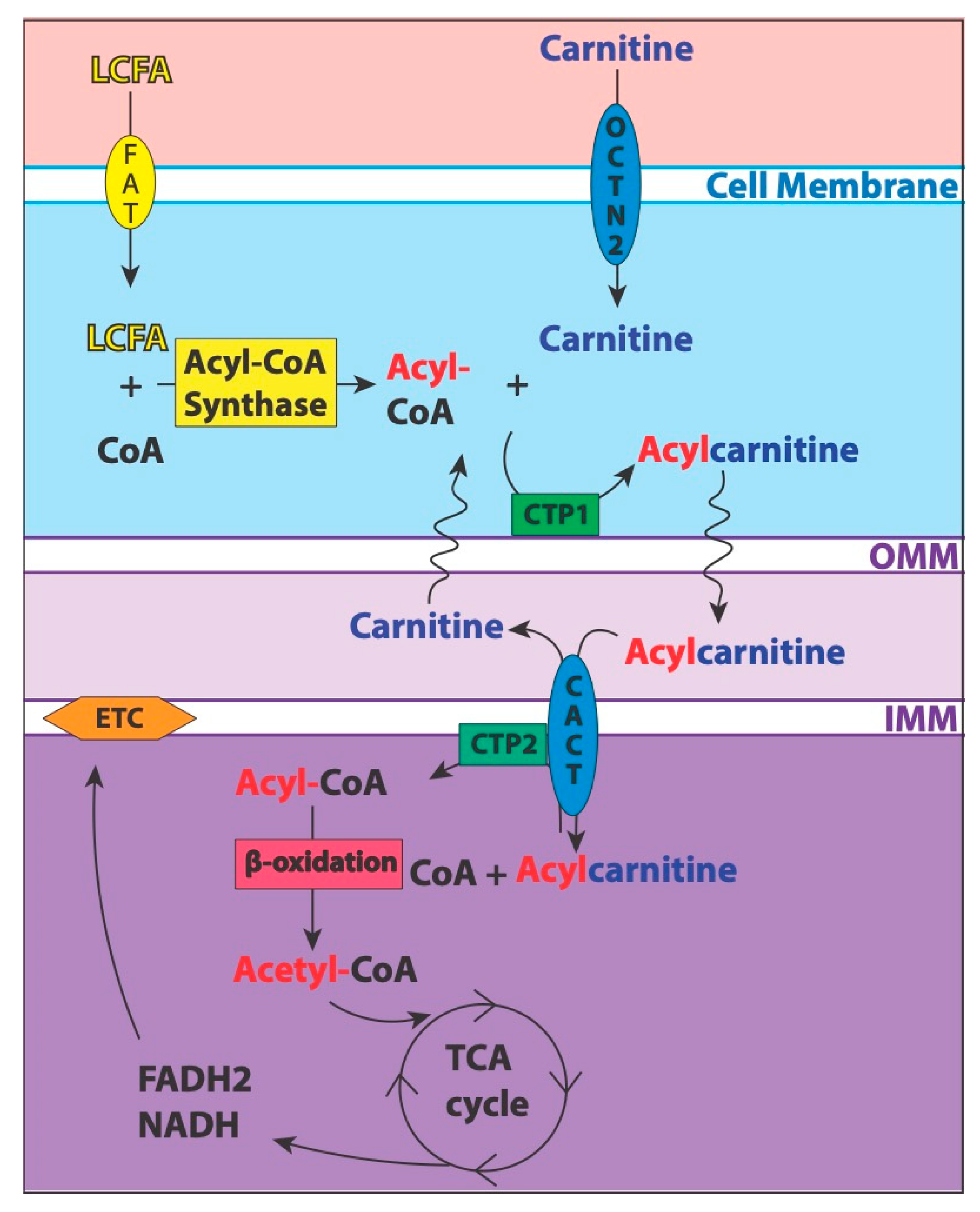
4.1. Valproic Acid
4.2. Clofazimine
4.3. Zidovudine
4.4. Cisplatin
4.5. Propofol
4.6. Cyclosporine
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Boyapati, R.K.; Dorward, D.A.; Tamborska, A.; Kalla, R.; Ventham, N.T.; Doherty, M.K.; Whitfield, P.D.; Gray, M.; Loane, J.; Rossi, A.G.; et al. Mitochondrial DNA Is a Pro-Inflammatory Damage-Associated Molecular Pattern Released During Active IBD. Inflamm. Bowel Dis. 2018, 24, 2113–2122. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-Derived Damage-Associated Molecular Patterns in Neurodegeneration. Front. Immunol. 2017, 8, 508. [Google Scholar] [CrossRef]
- Kraft, B.D.; Chen, L.; Suliman, H.B.; Piantadosi, C.A.; Welty-Wolf, K.E. Peripheral Blood Mononuclear Cells Demonstrate Mitochondrial Damage Clearance During Sepsis. Crit. Care Med. 2019, 47, 651–658. [Google Scholar] [CrossRef]
- Khatami, F.; Payab, M.; Sarvari, M.; Gilany, K.; Larijani, B.; Arjmand, B.; Tavangar, S.M. Oncometabolites as biomarkers in thyroid cancer: A systematic review. Cancer Manag. Res. 2019, 11, 1829–1841. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. Int. J. Mol. Sci. 2019, 20, 805. [Google Scholar] [CrossRef] [Green Version]
- Reuter, S.E.; Evans, A.M. Carnitine and acylcarnitines: Pharmacokinetic, pharmacological and clinical aspects. Clin. Pharmacokinet. 2012, 51, 553–572. [Google Scholar] [CrossRef]
- Evans, A.M.; Fornasini, G. Pharmacokinetics of L-carnitine. Clin. Pharmacokinet. 2003, 42, 941–967. [Google Scholar] [CrossRef]
- Knottnerus, S.J.G.; Bleeker, J.C.; Wüst, R.C.I.; Ferdinandusse, S.; IJlst, L.; Wijburg, F.A.; Wanders, R.J.A.; Visser, G.; Houtkooper, R.H. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev. Endocr. Metab. Disord. 2018, 19, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Wanders, R.J.A.; Ruiter, J.P.N.; IJlst, L.; Waterham, H.R.; Houten, S.M. The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J. Inherit. Metab. Dis. 2010, 33, 479–494. [Google Scholar] [CrossRef] [Green Version]
- Merritt, J.L.; Norris, M.; Kanungo, S. Fatty acid oxidation disorders. Ann. Transl. Med. 2018, 6. [Google Scholar] [CrossRef]
- American College of Medical Genetics. Newborn Screening: Towards a Uniform Screening Panel and System. Genet. Med. 2006, 117, S296–S307. [Google Scholar]
- Almannai, M.; Alfadhel, M.; El-Hattab, A.W. Carnitine Inborn Errors of Metabolism. Molecules 2019, 24, 3251. [Google Scholar] [CrossRef] [Green Version]
- Nasser, M.; Javaheri, H.; Fedorowicz, Z.; Noorani, Z. Carnitine supplementation for inborn errors of metabolism. Cochrane Database Syst. Rev. 2012, 2012, Cd006659. [Google Scholar] [CrossRef]
- Eckerle, M.; Ambroggio, L.; Puskarich, M.A.; Winston, B.; Jones, A.E.; Standiford, T.J.; Stringer, K.A. Metabolomics as a Driver in Advancing Precision Medicine in Sepsis. Pharmacotherapy 2017, 37, 1023–1032. [Google Scholar] [CrossRef]
- Kaddurah-Daouk, R.; Weinshilboum, R.M. Pharmacometabolomics: Implications for clinical pharmacology and systems pharmacology. Clin. Pharmacol. Ther. 2014, 95, 154–167. [Google Scholar] [CrossRef]
- Puskarich, M.A.; Finkel, M.A.; Karnovsky, A.; Jones, A.E.; Trexel, J.; Harris, B.N.; Stringer, K.A. Pharmacometabolomics of l-carnitine treatment response phenotypes in patients with septic shock. Ann. Am. Thorac. Soc. 2015, 12, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Kohoutova, M.; Dejmek, J.; Tuma, Z.; Kuncova, J. Variability of mitochondrial respiration in relation to sepsis-induced multiple organ dysfunction. Physiol. Res. 2018, 67, S577–S592. [Google Scholar] [CrossRef]
- El-Gharbawy, A.; Vockley, J. Defects of Fatty Acid Oxidation and the Carnitine Shuttle System. Pediatr. Clin. N. Am. 2018, 65, 317–335. [Google Scholar] [CrossRef]
- Saiki, S.; Hatano, T.; Fujimaki, M.; Ishikawa, K.I.; Mori, A.; Oji, Y.; Okuzumi, A.; Fukuhara, T.; Koinuma, T.; Imamichi, Y.; et al. Decreased long-chain acylcarnitines from insufficient beta-oxidation as potential early diagnostic markers for Parkinson’s disease. Sci. Rep. 2017, 7, 7328. [Google Scholar] [CrossRef]
- Bogusiewicz, A.; Horvath, T.D.; Stratton, S.L.; Mock, D.M.; Boysen, G. Measurement of acylcarnitine substrate to product ratios specific to biotin-dependent carboxylases offers a combination of indicators of biotin status in humans. J. Nutr. 2012, 142, 1621–1625. [Google Scholar] [CrossRef] [Green Version]
- Mihalik, S.J.; Goodpaster, B.H.; Kelley, D.E.; Chace, D.H.; Vockley, J.; Toledo, F.G.; DeLany, J.P. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity 2010, 18, 1695–1700. [Google Scholar] [CrossRef] [Green Version]
- Bruls, Y.M.; de Ligt, M.; Lindeboom, L.; Phielix, E.; Havekes, B.; Schaart, G.; Kornips, E.; Wildberger, J.E.; Hesselink, M.K.; Muoio, D.; et al. Carnitine supplementation improves metabolic flexibility and skeletal muscle acetylcarnitine formation in volunteers with impaired glucose tolerance: A randomised controlled trial. EBioMedicine 2019, 49, 318–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schooneman, M.G.; Vaz, F.M.; Houten, S.M.; Soeters, M.R. Acylcarnitines: Reflecting or Inflicting Insulin Resistance? Diabetes 2013, 62, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muoio, D.M. Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell 2014, 159, 1253–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, M.A.; Atherton, H.J.; Dodd, M.S.; Lee, P.; Cochlin, L.E.; Radda, G.K.; Clarke, K.; Tyler, D.J. The cycling of acetyl-coenzyme A through acetylcarnitine buffers cardiac substrate supply: A hyperpolarized 13C magnetic resonance study. Circ. Cardiovasc Imaging 2012, 5, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansford, R.G.; Cohen, L. Relative importance of pyruvate dehydrogenase interconversion and feed-back inhibition in the effect of fatty acids on pyruvate oxidation by rat heart mitochondria. Arch. Biochem. Biophys. 1978, 191, 65–81. [Google Scholar] [CrossRef]
- Arenas, J.; Huertas, R.; Campos, Y.; Diaz, A.E.; Villalon, J.M.; Vilas, E. Effects of L-carnitine on the pyruvate dehydrogenase complex and carnitine palmitoyl transferase activities in muscle of endurance athletes. FEBS Lett. 1994, 341, 91–93. [Google Scholar] [CrossRef] [Green Version]
- McAllister, A.; Allison, S.P.; Randle, P.J. Effects of dichloroacetate on the metabolism of glucose, pyruvate, acetate, 3-hydroxybutyrate and palmitate in rat diaphragm and heart muscle in vitro and on extraction of glucose, lactate, pyruvate and free fatty acids by dog heart in vivo. Biochem. J. 1973, 134, 1067–1081. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zeng, X.; Ren, M.; Mao, X.; Qiao, S. Novel metabolic and physiological functions of branched chain amino acids: A review. J. Anim. Sci. Biotechnol. 2017, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.G.; Reily, M.D.; Lehman-McKeeman, L.D.; Vaillancourt, R.R.; Cherrington, N.J. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino Acids 2015, 47, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R. The role of the carnitine system in peroxisomal fatty acid oxidation. Am. J. Med. Sci. 1999, 318, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Jakobs, B.S.; Wanders, R.J. Fatty acid beta-oxidation in peroxisomes and mitochondria: The first, unequivocal evidence for the involvement of carnitine in shuttling propionyl-CoA from peroxisomes to mitochondria. Biochem. Biophys. Res. Commun. 1995, 213, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Osmundsen, H.; Bremer, J.; Pedersen, J.I. Metabolic aspects of peroxisomal beta-oxidation. Biochim. Biophys. Acta 1991, 1085, 141–158. [Google Scholar] [CrossRef]
- Wanders, R.J.; Tager, J.M. Lipid metabolism in peroxisomes in relation to human disease. Mol. Asp. Med. 1998, 19, 69–154. [Google Scholar] [CrossRef]
- Demarquoy, J.; Le Borgne, F. Crosstalk between mitochondria and peroxisomes. World J. Biol. Chem. 2015, 6, 301–309. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2015, 3, 83. [Google Scholar] [CrossRef] [Green Version]
- National Center for Biotechnology Information. PubChem. Database. Palmitoylcarnitine, CID=461. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Palmitoylcarnitine (accessed on 19 February 2020).
- Bene, J.; Hadzsiev, K.; Melegh, B. Role of carnitine and its derivatives in the development and management of type 2 diabetes. Nutr. Diabetes 2018, 8. [Google Scholar] [CrossRef]
- Poorabbas, A.; Fallah, F.; Bagdadchi, J.; Mahdavi, R.; Aliasgarzadeh, A.; Asadi, Y.; Koushavar, H.; Vahed Jabbari, M. Determination of free L-carnitine levels in type II diabetic women with and without complications. Eur. J. Clin. Nutr. 2007, 61, 892–895. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.H.; Hoppel, C.L.; Lok, K.H.; Zhao, L.; Wong, S.W.; Minkler, P.E.; Hwang, D.H.; Newman, J.W.; Garvey, W.T. Plasma Acylcarnitine Profiles Suggest Incomplete Long-Chain Fatty Acid β-Oxidation and Altered Tricarboxylic Acid Cycle Activity in Type 2 Diabetic African-American Women. J. Nutr. 2009, 139, 1073–1081. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A Branched-Chain Amino Acid-Related Metabolic Signature that Differentiates Obese and Lean Humans and Contributes to Insulin Resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Liang, L.; Gao, X.; Zhang, H.; Yao, P.; Hu, Y.; Ma, Y.; Wang, F.; Jin, Q.; Li, H.; et al. Early Prediction of Developing Type 2 Diabetes by Plasma Acylcarnitines: A Population-Based Study. Diabetes Care 2016, 39, 1563–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batchuluun, B.; Al Rijjal, D.; Prentice, K.J.; Eversley, J.A.; Burdett, E.; Mohan, H.; Bhattacharjee, A.; Gunderson, E.P.; Liu, Y.; Wheeler, M.B. Elevated Medium-Chain Acylcarnitines Are Associated With Gestational Diabetes Mellitus and Early Progression to Type 2 Diabetes and Induce Pancreatic beta-Cell Dysfunction. Diabetes 2018, 67, 885–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langley, R.J.; Tsalik, E.L.; van Velkinburgh, J.C.; Glickman, S.W.; Rice, B.J.; Wang, C.; Chen, B.; Carin, L.; Suarez, A.; Mohney, R.P.; et al. An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci. Transl. Med. 2013, 5, 195ra95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, K.P.; Chen, G.Y.; Chuang, T.Y.; Huang, Y.T.; Chang, H.T.; Chen, Y.F.; Liu, W.L.; Chen, Y.J.; Hsu, C.L.; Huang, M.T.; et al. Increased Plasma Acetylcarnitine in Sepsis Is Associated With Multiple Organ Dysfunction and Mortality: A Multicenter Cohort Study. Crit. Care Med. 2019, 47, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Puskarich, M.A.; Evans, C.R.; Karnovsky, A.; Das, A.K.; Jones, A.E.; Stringer, K.A. Septic Shock Nonsurvivors Have Persistently Elevated Acylcarnitines Following Carnitine Supplementation. Shock 2018, 49, 412–419. [Google Scholar] [CrossRef]
- Ferrario, M.; Cambiaghi, A.; Brunelli, L.; Giordano, S.; Caironi, P.; Guatteri, L.; Raimondi, F.; Gattinoni, L.; Latini, R.; Masson, S.; et al. Mortality prediction in patients with severe septic shock: A pilot study using a target metabolomics approach. Sci. Rep. 2016, 6, 20391. [Google Scholar] [CrossRef]
- Rogers, A.J.; McGeachie, M.; Baron, R.M.; Gazourian, L.; Haspel, J.A.; Nakahira, K.; Fredenburgh, L.E.; Hunninghake, G.M.; Raby, B.A.; Matthay, M.A.; et al. Metabolomic derangements are associated with mortality in critically ill adult patients. PLoS ONE 2014, 9, e87538. [Google Scholar] [CrossRef] [Green Version]
- Enooku, K.; Nakagawa, H.; Fujiwara, N.; Kondo, M.; Minami, T.; Hoshida, Y.; Shibahara, J.; Tateishi, R.; Koike, K. Altered serum acylcarnitine profile is associated with the status of nonalcoholic fatty liver disease (NAFLD) and NAFLD-related hepatocellular carcinoma. Sci. Rep. 2019, 9, 10663. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Q.; Yin, P.; Xing, W.; Wu, Z.; Chen, S.; Lu, X.; Zhang, Y.; Lin, X.; Xu, G. Serum metabolomics reveals the deregulation of fatty acids metabolism in hepatocellular carcinoma and chronic liver diseases. Anal. Bioanal. Chem. 2012, 403, 203–213. [Google Scholar] [CrossRef]
- Chen, S.; Kong, H.; Lu, X.; Li, Y.; Yin, P.; Zeng, Z.; Xu, G. Pseudotargeted metabolomics method and its application in serum biomarker discovery for hepatocellular carcinoma based on ultra high-performance liquid chromatography/triple quadrupole mass spectrometry. Anal. Chem. 2013, 85, 8326–8333. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ding, L.; Yin, P.; Lu, X.; Wang, X.; Niu, J.; Gao, P.; Xu, G. Serum metabolic profiling study of hepatocellular carcinoma infected with hepatitis B or hepatitis C virus by using liquid chromatography-mass spectrometry. J. Proteome Res. 2012, 11, 5433–5442. [Google Scholar] [CrossRef] [PubMed]
- His, M.; Viallon, V.; Dossus, L.; Gicquiau, A.; Achaintre, D.; Scalbert, A.; Ferrari, P.; Romieu, I.; Onland-Moret, N.C.; Weiderpass, E.; et al. Prospective analysis of circulating metabolites and breast cancer in EPIC. BMC Med. 2019, 17, 178. [Google Scholar] [CrossRef]
- Farshidfar, F.; Kopciuk, K.A.; Hilsden, R.; McGregor, S.E.; Mazurak, V.C.; Buie, W.D.; MacLean, A.; Vogel, H.J.; Bathe, O.F. A quantitative multimodal metabolomic assay for colorectal cancer. BMC Cancer 2018, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Yin, P.; Su, D.; Peng, Z.; Zhou, L.; Ma, L.; Guo, W.; Xu, G.; Shi, J.; Jiao, B. Serum metabolic profiling and features of papillary thyroid carcinoma and nodular goiter. Mol. BioSyst. 2011, 7, 2608–2614. [Google Scholar] [CrossRef]
- Hunter, W.G.; Kelly, J.P.; McGarrah, R.W., 3rd; Khouri, M.G.; Craig, D.; Haynes, C.; Ilkayeva, O.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.J.; et al. Metabolomic Profiling Identifies Novel Circulating Biomarkers of Mitochondrial Dysfunction Differentially Elevated in Heart Failure With Preserved Versus Reduced Ejection Fraction: Evidence for Shared Metabolic Impairments in Clinical Heart Failure. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, T.; Kelly, J.P.; McGarrah, R.W.; Hellkamp, A.S.; Fiuzat, M.; Testani, J.M.; Wang, T.S.; Verma, A.; Samsky, M.D.; Donahue, M.P.; et al. Long-Chain Acylcarnitine Metabolites are Associated with Adverse Outcomes and Reversible with Mechanical Circulatory Support in Systolic Heart Failure. J. Am. Coll. Cardiol. 2016, 67, 291–299. [Google Scholar] [CrossRef]
- Ruiz, M.; Labarthe, F.; Fortier, A.; Bouchard, B.; Thompson Legault, J.; Bolduc, V.; Rigal, O.; Chen, J.; Ducharme, A.; Crawford, P.A.; et al. Circulating acylcarnitine profile in human heart failure: A surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H768–H781. [Google Scholar] [CrossRef] [Green Version]
- McCoin, C.S.; Knotts, T.A.; Adams, S.H. Acylcarnitines—old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol. 2015, 11, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Bloomgarden, Z. Diabetes and branched-chain amino acids: What is the link? J. Diabetes 2018, 10, 350–352. [Google Scholar] [CrossRef] [Green Version]
- Noland, R.C.; Koves, T.R.; Seiler, S.E.; Lum, H.; Lust, R.M.; Ilkayeva, O.; Stevens, R.D.; Hegardt, F.G.; Muoio, D.M. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J. Biol. Chem. 2009, 284, 22840–22852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muoio, D.M.; Noland, R.C.; Kovalik, J.-P.; Seiler, S.E.; Davies, M.N.; DeBalsi, K.L.; Ilkayeva, O.R.; Stevens, R.D.; Kheterpal, I.; Zhang, J.; et al. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 2012, 15, 764–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carre, J.E.; Singer, M. Cellular energetic metabolism in sepsis: The need for a systems approach. Biochim. Biophys. Acta 2008, 1777, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Singer, M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence 2014, 5, 66–72. [Google Scholar] [CrossRef]
- Ohtoshi, M.; Jikko, A.; Asano, M.; Uchida, K.; Ozawa, K.; Tobe, T. Ketogenesis during sepsis in relation to hepatic energy metabolism. Res. Exp. Med. 1984, 184, 209–219. [Google Scholar] [CrossRef]
- Brealey, D.; Singer, M. Mitochondrial Dysfunction in Sepsis. Curr. Infect. Dis. Rep. 2003, 5, 365–371. [Google Scholar] [CrossRef]
- Preiser, J.C.; Ichai, C.; Orban, J.C.; Groeneveld, A.B. Metabolic response to the stress of critical illness. Br. J. Anaesth. 2014, 113, 945–954. [Google Scholar] [CrossRef] [Green Version]
- Famularo, G.; De Simone, C.; Trinchieri, V.; Mosca, L. Carnitines and its congeners: A metabolic pathway to the regulation of immune response and inflammation. Ann. N. Y. Acad. Sci. 2004, 1033, 132–138. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Li, S.; Gao, D.; Jiang, Y. Function, Detection and Alteration of Acylcarnitine Metabolism in Hepatocellular Carcinoma. Metabolites 2019, 9, 36. [Google Scholar] [CrossRef] [Green Version]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.L.; Soeters, M.R.; Wüst, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turer, A.T. Using metabolomics to assess myocardial metabolism and energetics in heart failure. J. Mol. Cell Cardiol. 2013, 55, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Albert, C.L.; Tang, W.W. Metabolic Biomarkers in Heart Failure. Heart Fail. Clin. 2018, 14, 109–118. [Google Scholar] [CrossRef]
- Murashige, D.A.-O.; Jang, C.A.-O.; Neinast, M.A.-O.; Edwards, J.A.-O.; Cowan, A.A.-O.; Hyman, M.A.-O.; Rabinowitz, J.A.-O.; Frankel, D.A.-O.; Arany, Z.A.-O. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368. [Google Scholar]
- Edwards, I.R.; Aronson, J.K. Adverse drug reactions Adverse drug reactions: Definitions, diagnosis, and management. Lancet 2000, 356, 1255–1259. [Google Scholar] [CrossRef]
- Walker, D.K. The use of pharmacokinetic and pharmacodynamic data in the assessment of drug safety in early drug development. Br. J. Clin. Pharmacol. 2004, 58, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Dixit, R.; Riviere, J.; Krishnan, K.; Andersen, M.E. Toxicokinetics and physiologically based toxicokinetics in toxicology and risk assessment. J. Toxicol. Environ. Health Part B Crit. Rev. 2003, 6, 1–40. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.A.; Borgne-Sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef] [PubMed]
- Depakene [valproic acid]. Package Insert; Abbott Laboratories: Chicago, IL, USA, 2011. [Google Scholar]
- LAMPRENE [clofazimine]. Package Insert; Novartis Pharmaceuticals Corporation: East Hanover, NJ, USA, 2019. [Google Scholar]
- Retrovir [zidovudine]. Package Insert; GlaxoSmithKline: Research Triangle Park, NC, USA, 2008. [Google Scholar]
- Cisplatin. Package Insert; WG Critical Care, LLC.: Paramus, NJ, USA, 2019. [Google Scholar]
- DIPRIVAN [propofol]. Package Insert; Fresenius Kabi USA, LLC.: Lake Zurich, IL, USA, 2014. [Google Scholar]
- NEORAL [cyclosporine]. Package Insert; Novartis Pharmaceuticals Corporation: East Hanover, NJ, USA, 2009. [Google Scholar]
- Nishida, N.; Sugimoto, T.; Araki, A.; Woo, M.; Sakane, Y.; Kobayashi, Y. Carnitine metabolism in valproate-treated rats: The effect of l-carnitine supplementation. Pediatric Res. 1987, 22, 500–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurston, J.H.; Hauhart, R.E. Amelioration of adverse effects of valproic acid on ketogenesis and liver coenzyme a metabolism by cotreatment with pantothenate and carnitine in developing mice: Possible clinical significance. Pediatric Res. 1992, 31, 419–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opala, G.; Winter, S.; Vance, C.; Vance, H.; Hutchison, H.T.; Linn, L.S. The effect of valproic acid on plasma carnitine levels. Am. J. Dis. Child. 1991, 145, 999–1001. [Google Scholar] [CrossRef] [PubMed]
- Trexel, J.; Yoon, G.S.; Keswani, R.K.; McHugh, C.; Yeomans, L.; Banerjee, R.; Sud, S.; Sun, Y.; Rosania, G.R.; Stringer, K.A. Macrophage-Mediated Clofazimine Sequestration is Accompanied by a Shift in Host Energy Metabolism. J. Pharm. Sci. 2017, 106, 1162–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georges, B.; Galland, S.; Rigault, C.; Le Borgne, F.; Demarquoy, J. Beneficial effects of L-carnitine in myoblastic C2C12 cells: Interaction with zidovudine. Biochem. Pharmacol. 2003, 65, 1483–1488. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Leon-Monzon, M.E.; Bernardini, I.; Gahl, W.A.; Jay, C.A. Zidovudine-induced mitochondrial myopathy is associated with muscle carnitine deficiency and lipid storage. Ann. Neurol. 1994, 35, 482–487. [Google Scholar] [CrossRef]
- Heuberger, W.; Berardi, S.; Jacky, E.; Pey, P.; Krahenbuhl, S. Increased urinary excretion of carnitine in patients treated with cisplatin. Eur. J. Clin. Pharmacol. 1998, 54, 503–508. [Google Scholar] [CrossRef]
- Ikezaki, T.; Suzuki, K.; Kambara, K.; Inomata, M.; Okazawa, S.; Kajiura, S.; Miwa, T.; Tanabe, K.; Kashii, T. Relationship between Carnitine Pharmacokinetics and Fatigue in Patients Treated with Cisplatin-Containing Chemotherapy. Oncol. Res. Treat. 2017, 40, 42–45. [Google Scholar] [CrossRef]
- Wolf, A.; Weir, P.; Segar, P.; Stone, J.; Shield, J. Impaired fatty acid oxidation in propofol infusion syndrome. Lancet 2001, 357, 606–607. [Google Scholar] [CrossRef]
- Withington, D.E.; Decell, M.K.; Al Ayed, T. A case of propofol toxicity: Further evidence for a causal mechanism. Paediatr. Anaesth. 2004, 14, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.R.; Potter, F. Propofol infusion in children: When does an anesthetic tool become an intensive care liability? Paediatr. Anaesth. 2004, 14, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A.S.; Jyothirmayi, G.N.; Leevy, C.B.; Khalil, M.; DeAngelis, B.; Frank, O.; Baker, H. Effect of cyclosporine treatment on carnitine and myo-inositol in diabetic rats. Comp. Biochem. Physiol. 1992, 101, 151–153. [Google Scholar] [CrossRef]
- Wanner, C.; Schollmeyer, P.; Horl, W.H. Serum carnitine levels and carnitine esters of patients after kidney transplantation: Role of immunosuppression. Metabolism 1988, 37, 263–267. [Google Scholar] [CrossRef]
- Koch-Weser, J.; Browne, T.R. Drug therapy: Valproic acid. N. Engl. J. Med. 1980, 302, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Raskind, J.Y.; El-Chaar, G.M. The role of carnitine supplementation during valproic acid therapy. Ann. Pharmacother. 2000, 34, 630–638. [Google Scholar] [CrossRef]
- Coulter, D.L. Carnitine deficiency: A possible mechanism for valproate hepatotoxicity. Lancet 1984, 1, 689. [Google Scholar] [CrossRef]
- Millington, D.S.; Bohan, T.P.; Roe, C.R.; Yergey, A.L.; Liberato, D.J. Valproylcarnitine: A novel drug metabolite identified by fast atom bombardment and thermospray liquid. Clin. Chim. Acta 1985, 145, 69–76. [Google Scholar] [CrossRef]
- Brown, L.M.; Cupples, N.; Moore, T.A. Levocarnitine for valproate-induced hyperammonemia in the psychiatric setting: A case series and literature review. Ment. Health Clin. 2018, 8, 148–154. [Google Scholar] [CrossRef]
- Lheureux, P.E.R.; Hantson, P. Carnitine in the treatment of valproic acid-induced toxicity carnitine in VPA toxicity. Clin. Toxicol. 2009, 47, 101–111. [Google Scholar] [CrossRef]
- Jafarian, I.; Eskandari, M.R.; Mashayekhi, V.; Ahadpour, M.; Hosseini, M.J. Toxicity of valproic acid in isolated rat liver mitochondria. Toxicol. Mech. Methods 2013, 23, 617–623. [Google Scholar] [CrossRef]
- Cholo, M.C.; Steel, H.C.; Fourie, P.B.; Germishuizen, W.A.; Anderson, R. Clofazimine: Current status and future prospects. J. Antimicrob. Chemother. 2012, 67, 290–298. [Google Scholar] [CrossRef]
- Murashov, M.D.; Diaz-Espinosa, J.; Lalone, V.; Tan, J.W.Y.; Laza, R.; Wang, X.; Stringer, K.A.; Rosania, G.R. Synthesis and characterization of a biomimetic formulation of clofazimine hydrochloride microcrystals for parenteral administration. Pharmaceutics 2018, 10, 238. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, D.K.; Ellard, G.A.; Gammon, P.T.; Waters, M.F.R. Some observations on the pharmacology of clofazimine (B663). Am. J. Trop. Med. Hyg. 1974, 23, 1110–1115. [Google Scholar] [CrossRef]
- Baik, J.; Rosania, G.R. Molecular Imaging of Intracellular Drug-Membrane Aggregate Formation. Mol. Pharm. 2011, 8, 1742–1749. [Google Scholar] [CrossRef] [Green Version]
- Hastings, R.C.; Jacobson, R.R.; Trautman, J.R. Long-term clinical toxicity studies with clofazimine (B663) in leprosy. Int. J. Lepr. Other. Mycobact. Dis. 1976, 44, 287–293. [Google Scholar]
- Baik, J.; Stringer, K.A.; Mane, G.; Rosania, G.R. Multiscale distribution and bioaccumulation analysis of clofazimine reveals a massive immune system-mediated xenobiotic sequestration response. Antimicrob. Agents Chemother. 2013, 57, 1218–1230. [Google Scholar] [CrossRef] [Green Version]
- Yoon, G.S.; Sud, S.; Keswani, R.K.; Baik, J.; Standiford, T.J.; Stringer, K.A.; Rosania, G.R. Phagocytosed Clofazimine Biocrystals Can Modulate Innate Immune Signaling by Inhibiting TNFalpha and Boosting IL-1RA Secretion. Mol. Pharm. 2015, 12, 2517–2527. [Google Scholar] [CrossRef] [Green Version]
- Baik, J.; Rosania, G.R. Macrophages Sequester Clofazimine in an Intracellular Liquid Crystal-Like Supramolecular Organization. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Scruggs, E.R.; Dirks Naylor, A.J. Mechanisms of zidovudine-induced mitochondrial toxicity and myopathy. Pharmacology 2008, 82, 83–88. [Google Scholar] [CrossRef]
- Dykens, J.A.; Will, Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov. Today 2007, 12, 777–785. [Google Scholar] [CrossRef]
- Veronique, J.; Flandre, P.; Meiffredy, V.; Leturque, N.; Harel, M.; Aboulker, J.-P.; Yeni, P. Increased risk of lipoatrophy under stavudine in HIV-1-infected patients: Results of a substudy from a comparative trial. Aids 2002, 16, 2447–2454. [Google Scholar]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.M.; Kim, H.K.; Shim, W.; Anwar, M.A.; Kwon, J.W.; Kwon, H.K.; Kim, H.J.; Jeong, H.; Kim, H.M.; Hwang, D.; et al. Mechanism of Cisplatin-Induced Cytotoxicity Is Correlated to Impaired Metabolism Due to Mitochondrial ROS Generation. PLoS ONE 2015, 10, e0135083. [Google Scholar] [CrossRef] [Green Version]
- So, H.; Kim, H.; Lee, J.H.; Park, C.; Kim, Y.; Kim, E.; Kim, J.K.; Yun, K.J.; Lee, K.M.; Lee, H.Y.; et al. Cisplatin cytotoxicity of auditory cells requires secretions of proinflammatory cytokines via activation of ERK and NF-kappaB. J. Assoc. Res. Otolaryngol. 2007, 8, 338–355. [Google Scholar] [CrossRef] [Green Version]
- Jezek, P.; Hlavata, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef]
- Hockenberry, M.J.; Hooke, M.C.; Gregurich, M.; McCarthy, K. Carnitine plasma levels and fatigue in children/adolescents receiving cisplatin, ifosfamide, or doxorubicin. J. Pediatric Hematol./Oncol. 2009, 31, 664–669. [Google Scholar] [CrossRef]
- Marik, P. Propofol: Therapeutic Indications and Side-Effects. Curr. Pharm. Des. 2005, 10, 3639–3649. [Google Scholar] [CrossRef]
- Sahinovic, M.M.; Struys, M.M.R.F.; Absalom, A.R. Clinical Pharmacokinetics and Pharmacodynamics of Propofol. Clin. Pharmacokinet. 2018, 57, 1539–1558. [Google Scholar] [CrossRef] [Green Version]
- Cannon, M.L.; Glazier, S.S.; Bauman, L.A. Metabolic acidosis, rhabdomyolysis, and cardiovascular collapse after prolonged propofol infusion. J. Neurosurg. 2001, 95, 1053–1056. [Google Scholar] [CrossRef]
- Perrier, N.D.; Baerga-Varela, Y.; Murray, M.J. Death related to propofol use in an adult patient. Crit. Care Med. 2000, 28, 3071–3074. [Google Scholar] [CrossRef]
- Chan, K.; Truong, D.; Shangari, N.; O’Brien, P.J. Drug-induced mitochondrial toxicity. Expert Opin. Drug Metab. Toxicol. 2005, 1, 655–669. [Google Scholar] [CrossRef]
- Vollmer, J.P.; Haen, S.; Wolburg, H.; Lehmann, R.; Steiner, J.; Reddersen, S.; Fend, F.; Fallier-Becker, P. Propofol Related Infusion Syndrome: Ultrastructural Evidence for a Mitochondrial Disorder. Crit. Care Med. 2018, 46, e91–e94. [Google Scholar] [CrossRef]
- Kam, P.C.A.; Cardone, D. Propofol infusion syndrome. Anaesthesia 2007, 62, 690–701. [Google Scholar] [CrossRef]
- Schenkman, K.A.; Yan, S. Propofol impairment of mitochondrial respiration isolated perfused guinea pig hearts determined by reflectance spectroscopy. Crit. Care Med. 2000, 28, 172–177. [Google Scholar] [CrossRef]
- Sumi, C.; Okamoto, A.; Tanaka, H.; Nishi, K.; Kusunoki, M.; Shoji, T.; Uba, T.; Matsuo, Y.; Adachi, T.; Hayashi, J.I.; et al. Propofol induces a metabolic switch to glycolysis and cell death in a mitochondrial electron transport chain-dependent manner. PLoS ONE 2018, 13, e0192796. [Google Scholar] [CrossRef] [Green Version]
- Branca, D.; Roberti, M.S.; Vincenti, E.; Scutari, G. Uncoupling effect of the general anesthetic 2,6-diisopropylphenol in isolated rat liver mitochondria. Arch. Biochem. Biophys. 1991, 290, 517–521. [Google Scholar] [CrossRef]
- Rigoulet, M.; Devin, A.; Avéret, N.; Vandais, B.; Guérin, B. Mechanisms of inhibition and uncoupling of respiration in isolated rat liver mitochondria by the general anesthetic 2,6-diisopropylphenol. Eur. J. Biochem. 1996, 241, 280–285. [Google Scholar] [CrossRef]
- Branca, D.; Roberti, M.S.; Lorenzin, P.; Vincenti, E.; Scutari, G. Influence of the anesthetic 2,6-diisopropylphenol on the oxidative phosphorylation of isolated rat liver mitochondria. Biochem. Pharmacol. 1991, 42, 87–90. [Google Scholar] [CrossRef]
- Calne, R.Y.; White, D.J.; Thiru, S.; Evans, D.B.; McMaster, P.; Dunn, D.C.; Craddock, G.N.; Pentlow, B.D.; Rolles, K. Cyclosporin A in patients receiving renal allografts from cadaver donors. Lancet 1978, 2, 1323–1327. [Google Scholar] [CrossRef]
- NIH. National Institutes of Health Consensus Development Conference Statement: Diagnosis and treatment of attention-deficit/hyperactivity disorder (ADHD). J. Am. Acad. Child. Adolesc. Psychiatry 2000, 39, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Perez de Hornedo, J.; de Arriba, G.; Calvino, M.; Benito, S.; Parra, T. Cyclosporin A causes oxidative stress and mitochondrial dysfunction in renal tubular cells. Nefrologia 2007, 27, 565–573. [Google Scholar] [PubMed]
- Slikker, W. Biomarkers and their impact on precision medicine. Exp. Biol. Med. 2018, 243, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Puskarich, M.A.; Kline, J.A.; Krabill, V.; Claremont, H.; Jones, A.E. Preliminary Safety and Efficacy of L-carnitine Infusion for the Treatment of Vasopressor-Dependent Septic Shock. J. Parenter. Enter. Nutr. 2014, 38, 736–743. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.E.; Puskarich, M.A.; Shapiro, N.I.; Guirgis, F.W.; Runyon, M.; Adams, J.Y.; Sherwin, R.; Arnold, R.; Roberts, B.W.; Kurz, M.C.; et al. Effect of Levocarnitine vs Placebo as an Adjunctive Treatment for Septic Shock: The Rapid Administration of Carnitine in Sepsis (RACE) Randomized Clinical Trial. JAMA Netw. Open 2018, 1, e186076. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Disease | Proposed Mechanism | Subject | Biospecimen | Disease-Induced Alterations in Carnitine/Acylcarnitine Levels |
|---|---|---|---|---|
| Diabetes Mellitus |
| 1. Humans (T2D+complications) | 1. Serum | 1. 25% lower LC levels [40] |
| 2. Humans (T2D) | 2. Plasma | 2. Increased C2, SCAC, MCAC in patients with higher HbA1c [41] | ||
| 3. Humans (insulin resistant/obese) | 3. Serum | 3. Increased C3, C5, C6, C8:1 [42] | ||
| 4. Humans (T2D) | 4. Plasma | 4. Increased SCAC, MCAC, LCAC in T2D patients [22] | ||
| 5. Humans (T2D) | 5. Plasma | 5. LCAC most associated with developing T2D [43] | ||
| 6. Humans (gestational and T2D) | 6. Serum | 6. Strongest association with MCAC [44] | ||
| Sepsis/Septic shock |
| 1. Humans (sepsis) | 1. Plasma | 1. Increased C2, SCAC, MCAC in non-survivors [45,46] |
| 2. Humans (septic shock) | 2. Serum | 2. Increased LC, C2, C3, C8 in non-survivors [47] | ||
| 3. Humans (septic shock) | 3. Plasma | 3. Increased C2, C4 associated with 28-day mortality [48] | ||
| 4. Humans (ICU,60/90 with sepsis) | 4. Plasma | 4. Increased C3, C4, C5, C6 associated with 28-day mortality [49] | ||
| Cancer |
| 1. Humans (HCC) | 1. Serum | 1. Increased LC, LCAC; decreased SCAC, MCAC [50,51,52,53] |
| 2. Humans (breast) | 2. Plasma | 2. Increased C2 associated with disease risk [54] | ||
| 3. Humans (colorectal) | 3. Serum | 3. Predominantly increased SCAC, MCAC, LCAC [55] | ||
| 4. Humans (thyroid) | 4. Serum | 4. Increased MCAC, LCAC [56] | ||
| Heart Failure |
| 1. Humans (HF) | 1. Plasma | 1. Increased LCAC associated with worse disease severity [57] |
| 2. Humans (HF) | 2. Plasma | 2. Increased MCAC, LCAC associated with all-cause mortality and hospitalization [58] | ||
| 3. Humans (HFrEF) | 3. Plasma | 3. Increased C2, SCAC, MCAC, LCAC; C2 and MCAC associated with disease severity [59] |
| Drug Name | Valproic Acid (VPA) | Clofazamine (CFZ) | Zidovudine (ZDV) | Cisplatin (CSP) | Propofol | Cyclosporine (CyA) |
|---|---|---|---|---|---|---|
| Chemical Structure |  |  |  |  |  |  |
| Drug Class | antiepileptic | antibiotic | antiviral | anticancer | anesthetic | immunosuppressant |
| Mechanism of Pharmacological Action | Increases brain concentrations of gamma-aminobutyric acid (GABA), blocks voltage-gated ion channels, and inhibits histone deacetylase [83] | Inhibits mycobacterial growth and binds preferentially to mycobacterial DNA [84] | Inhibits HIV’s reverse transcriptase (RT) via DNA chain termination [85] | Binds to genomic DNA in the cell nucleus to form cross-links which trigger cytotoxic processes [86] | Increases GABA-mediated inhibitory function in the CNS [87] | Exact mechanism unknown, but thought to inhibit production and release of interleukin-2 [88] |
| Drug | Proposed Mechanism | Subject | Biospecimen | Drug-Induced Alterations in Carnitine/Acylcarnitine Levels |
|---|---|---|---|---|
| Valproic Acid |
| 1. Rats | 1. Serum, muscle, and urine | 1. Decreased LC (serum, muscle); increased pooled ACs and AC/LC ratio (serum, muscle); increased ACs (urine) [89] |
| 2. Mice | 2. Whole liver | 2. Decreased LC; increased AC/LC ratio [90] | ||
| 3. Humans (Pediatric) | 3. Plasma | 3. Decreased LC, increased AC/LC ratio [91] | ||
| Clofazimine |
| 1. Mice | 1. Urine, whole blood | 1. Decreased LC (urine); increased LC (whole blood) [92] |
| Zidovudine |
| 1. C2C12 cells 2. Humans | 1. Cells 2. Muscle biopsy | 1. Decreased LC [93] 2. Decreased LC [94] |
| Cisplatin |
| 1. Humans (Various cancers) | 1. Plasma, urine | 1. Increased LC (plasma); increased renal excretion of LC (urine) [95] |
| 2. Humans (Various cancers) | 2. Serum, urine | 2. Increased LC (serum, urine) [96] | ||
| Propofol |
| 1. Humans 2. Humans 3. Humans | 1. Serum 2. Plasma 3. Plasma | 1. Increased C3-DC, C5 [97] 2. Increased MCAC [98] 3. Increased C4 [99] |
| Cyclosporine |
| 1. Rats | 1. Urine, blood, liver, kidney, pancreas | 1. Decreased LC (urine, blood); increased LC (liver); no change (kidney, pancreas) [100] |
| 2. Humans | 2. Serum | 2. Increased LC, SCAC [101] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCann, M.R.; George De la Rosa, M.V.; Rosania, G.R.; Stringer, K.A. L-Carnitine and Acylcarnitines: Mitochondrial Biomarkers for Precision Medicine. Metabolites 2021, 11, 51. https://doi.org/10.3390/metabo11010051
McCann MR, George De la Rosa MV, Rosania GR, Stringer KA. L-Carnitine and Acylcarnitines: Mitochondrial Biomarkers for Precision Medicine. Metabolites. 2021; 11(1):51. https://doi.org/10.3390/metabo11010051
Chicago/Turabian StyleMcCann, Marc R., Mery Vet George De la Rosa, Gus R. Rosania, and Kathleen A. Stringer. 2021. "L-Carnitine and Acylcarnitines: Mitochondrial Biomarkers for Precision Medicine" Metabolites 11, no. 1: 51. https://doi.org/10.3390/metabo11010051
APA StyleMcCann, M. R., George De la Rosa, M. V., Rosania, G. R., & Stringer, K. A. (2021). L-Carnitine and Acylcarnitines: Mitochondrial Biomarkers for Precision Medicine. Metabolites, 11(1), 51. https://doi.org/10.3390/metabo11010051