
Development of a Microfluidic Platform for Trace Lipid Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
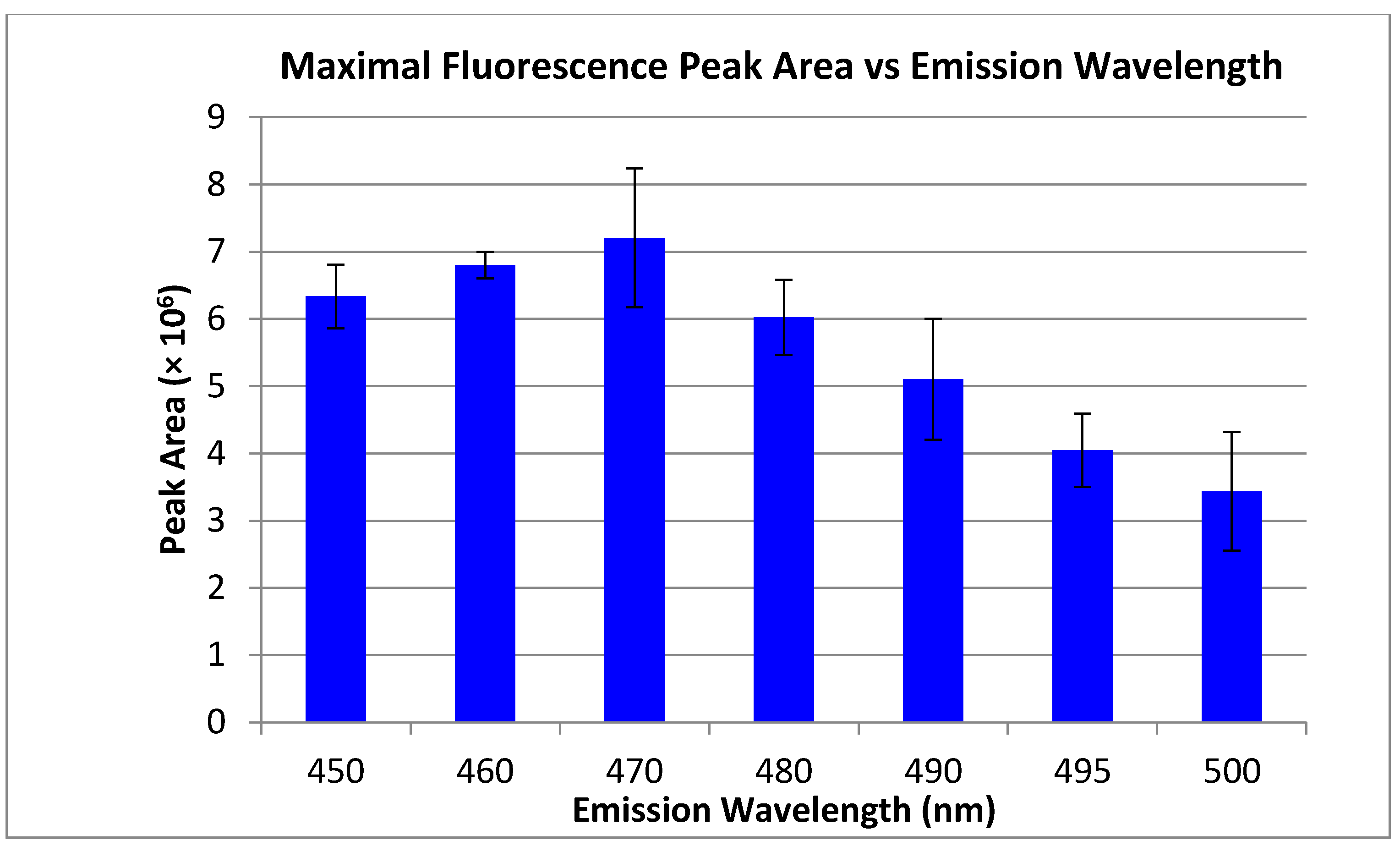
2.1. Fluorescent Tagging of Primary Amines
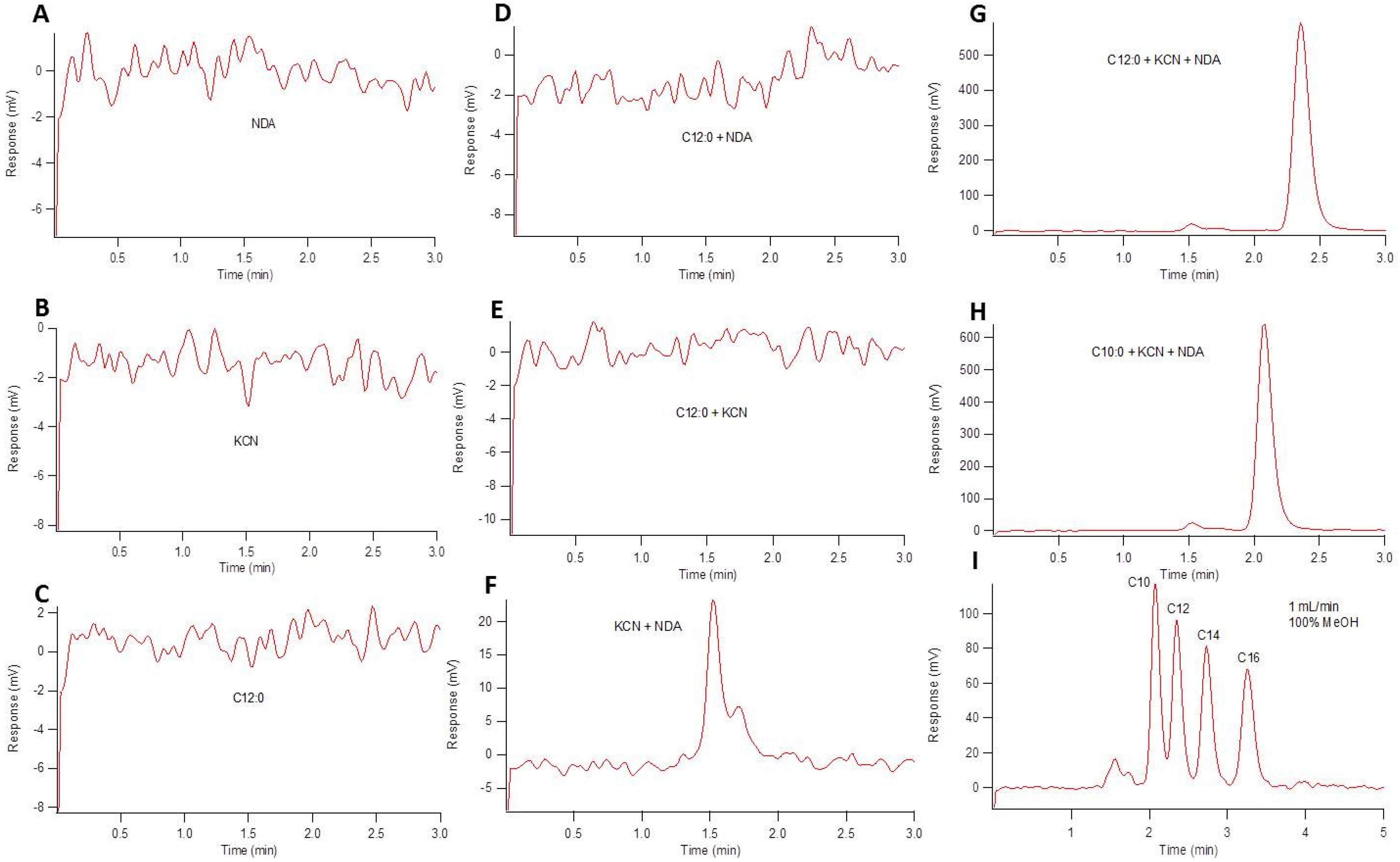
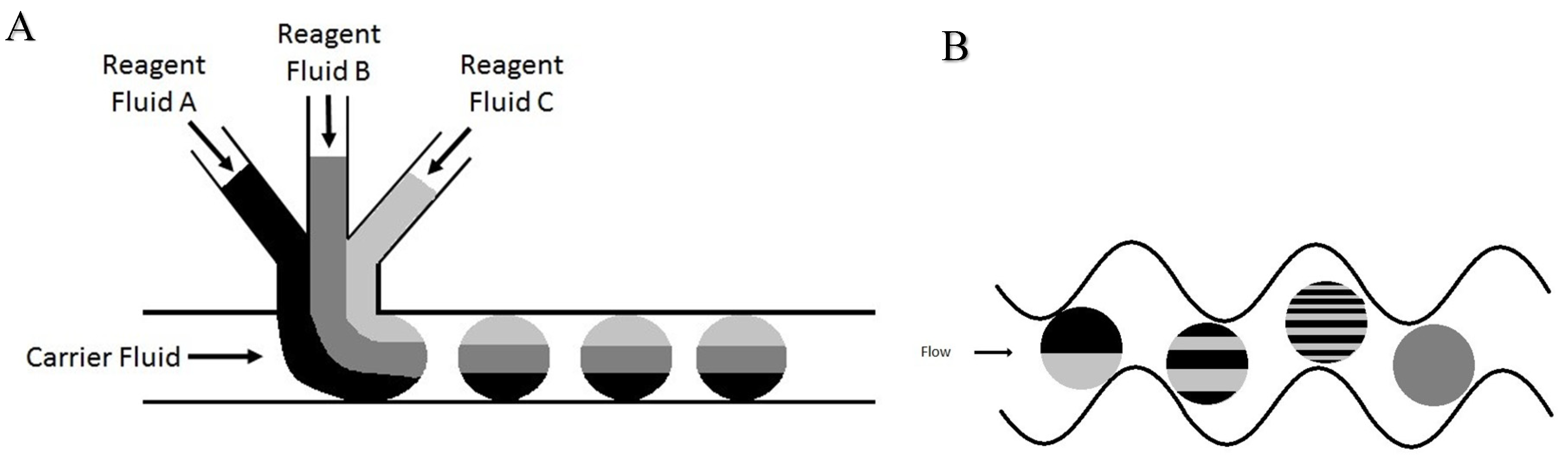
2.2. Segmented Flow Microfluidics
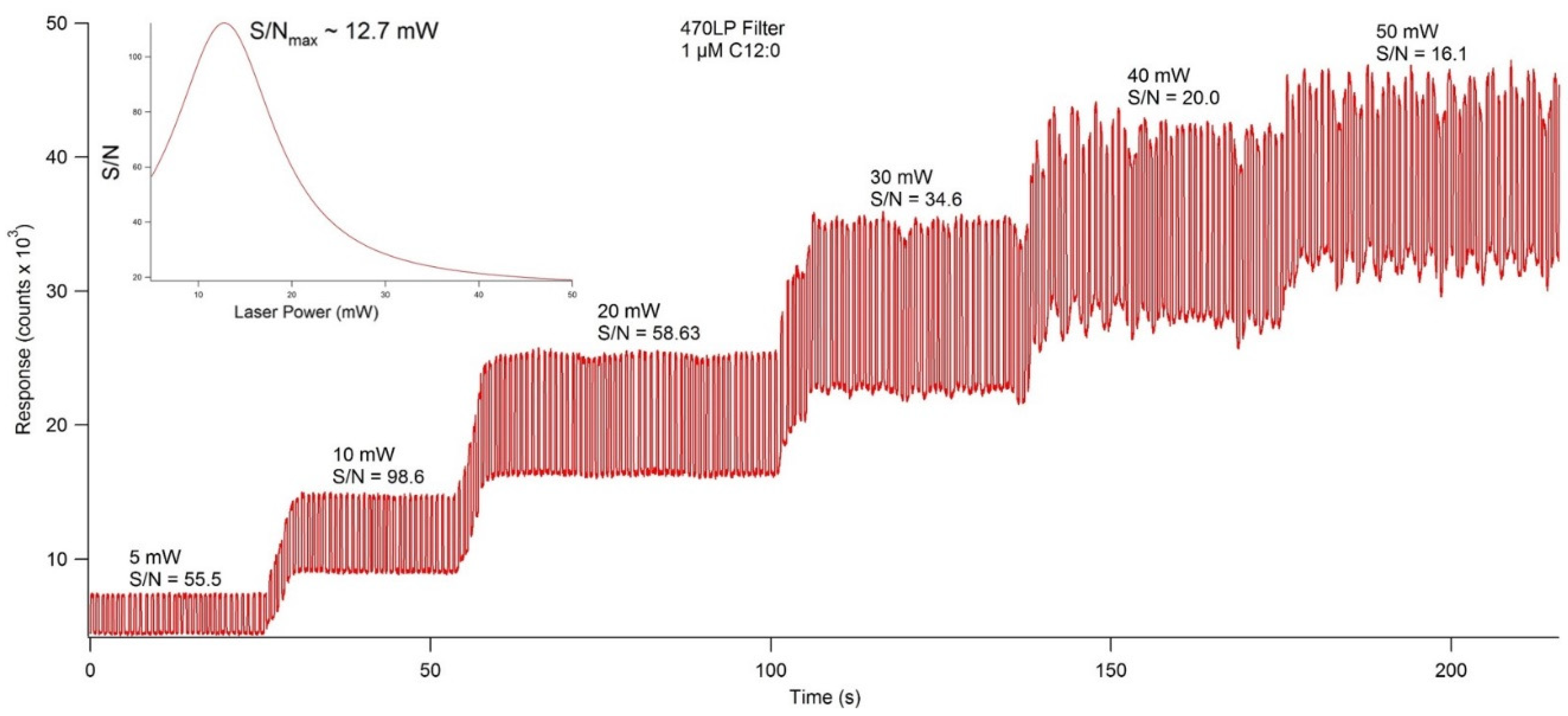
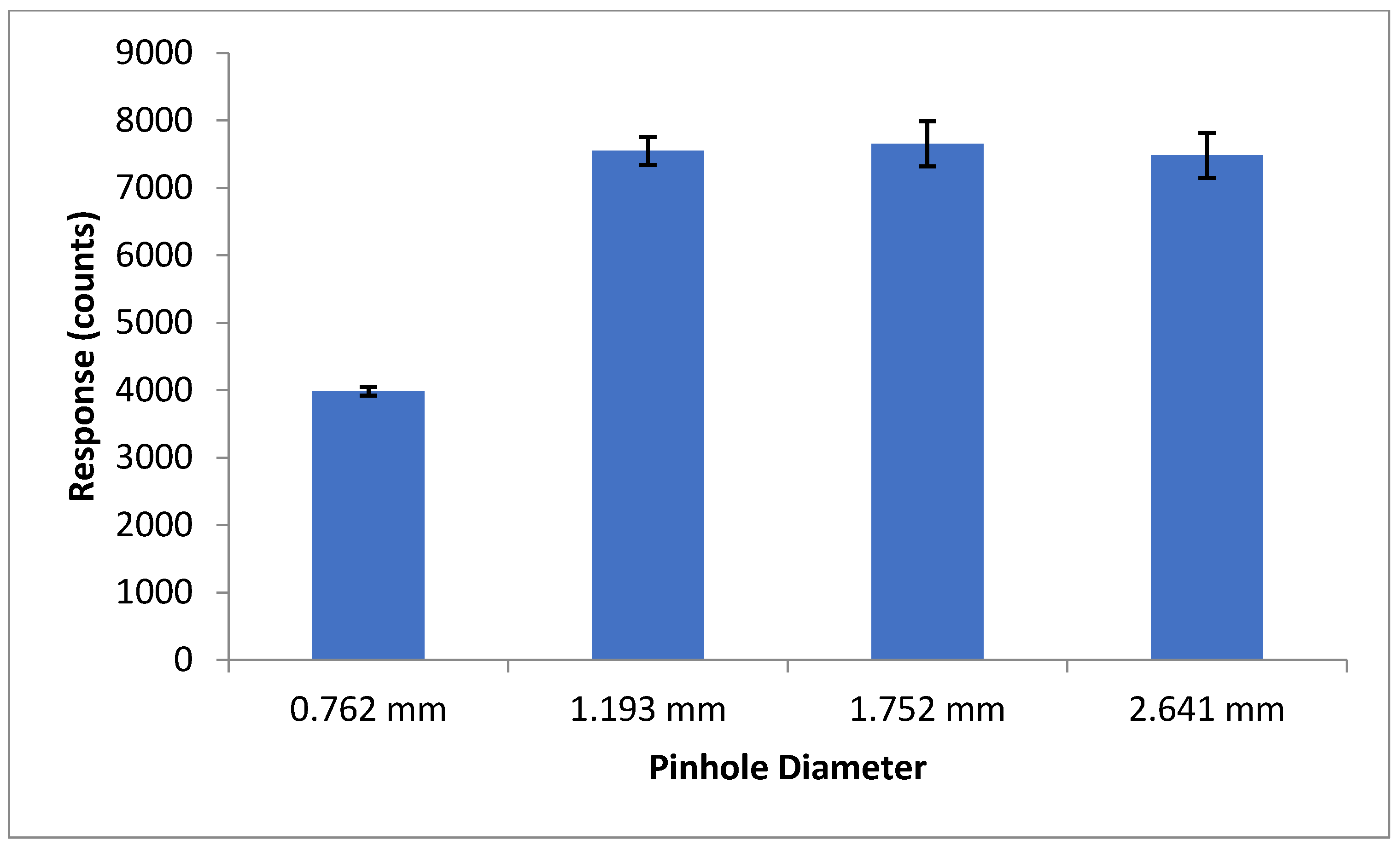
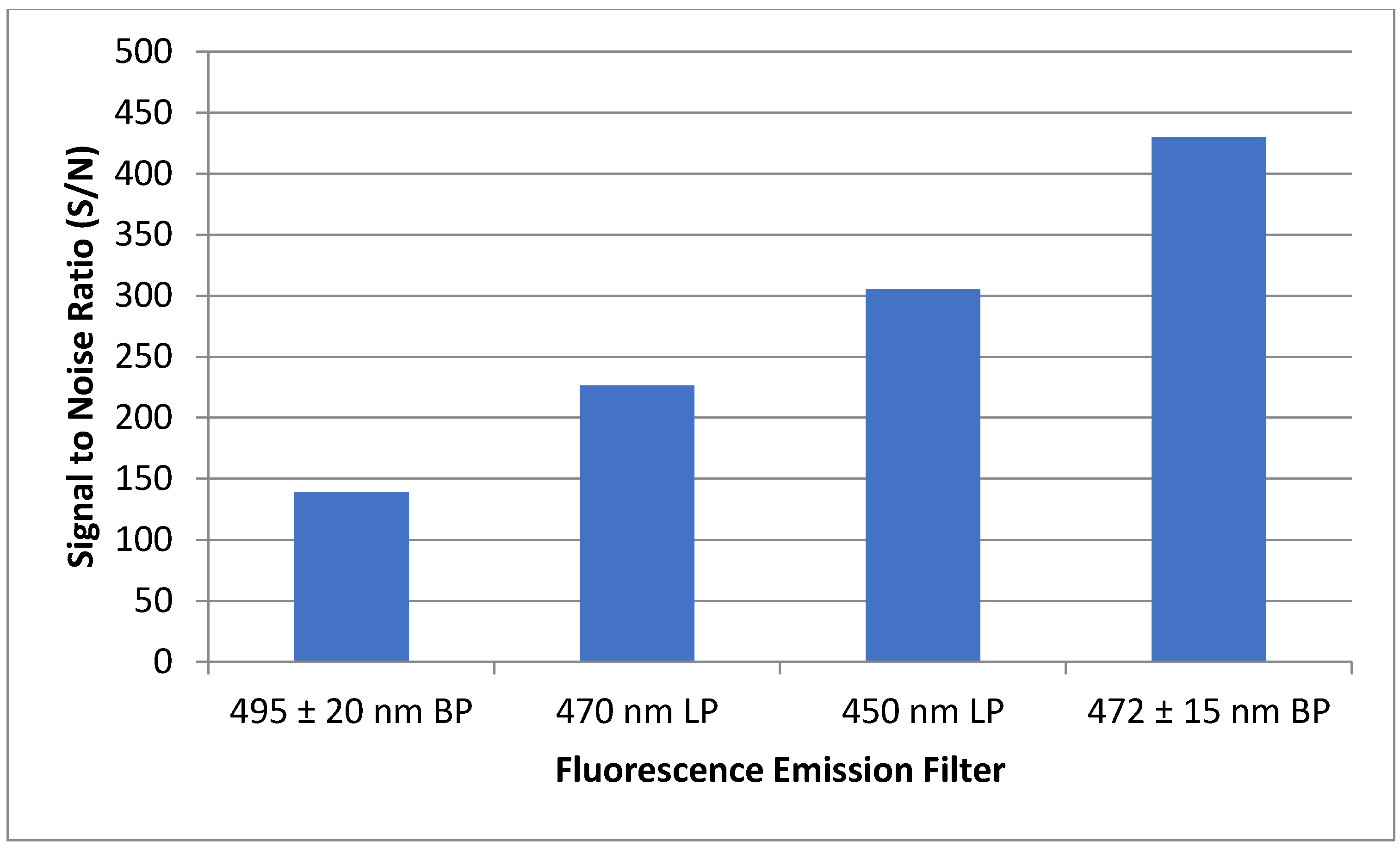
2.3. LIF Optical Detection System
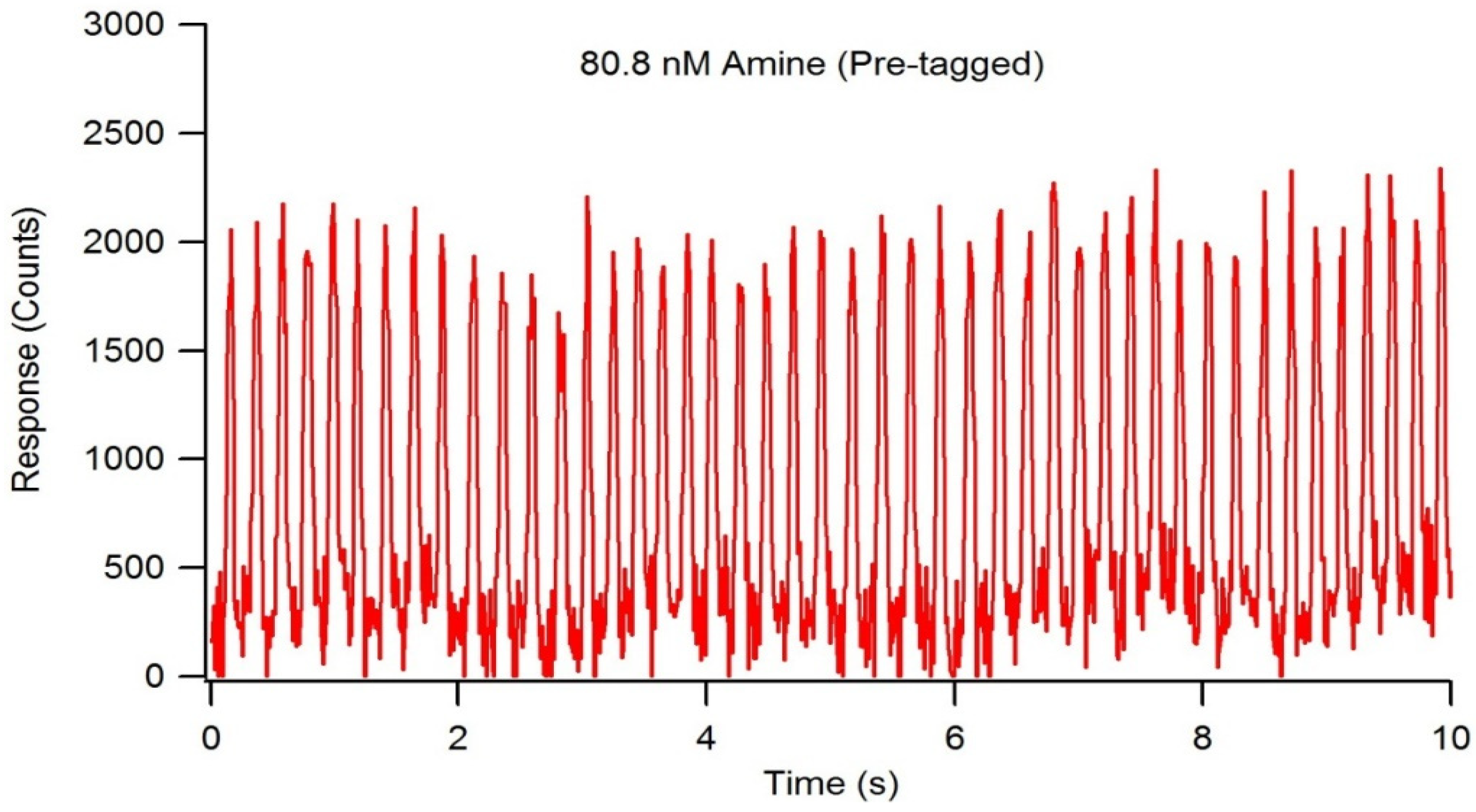
2.4. Droplet Formation and On-Chip Fluorescent Tagging
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Instrumentation
4.3. Fluorescent Labeling of Amines
4.4. Reversed Phase HPLC Separation of Primary Fatty Acid Amines
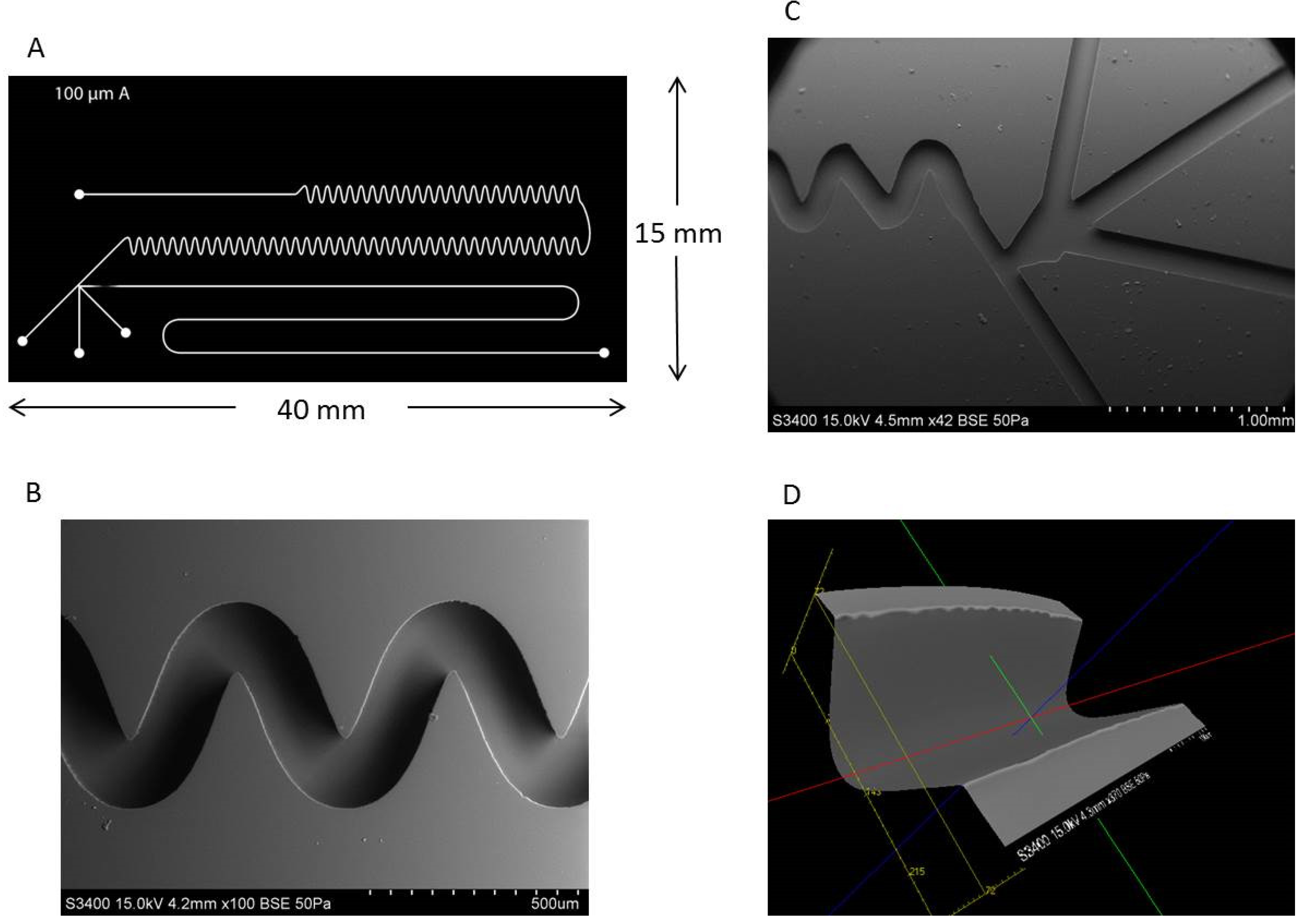
4.5. Microchip Fabrication
4.6. Scanning Electron Microscopy
4.7. Development of a Laser Induced Fluorescence Detection System
4.8. Droplet Based Microfluidic Flow
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Divito, E.B.; Cascio, M. Metabolism, physiology, and analyses of primary fatty acid amides. Chem. Rev. 2013, 113, 7343–7353. [Google Scholar] [CrossRef]
- Arafat, E.S.; Trimble, J.W.; Andersen, R.N.; Dass, C.; Desiderio, D.M. Identification of fatty acid amides in human plasma. Life Sci. 1989, 45, 1679–1687. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Prospero-Garcia, O.; Siuzdak, G.; Gilula, N.B.; Henriksen, S.J.; Boger, D.L.; Lerner, R.A. Chemical characterization of a family of brain lipids that induce sleep. Science 1995, 268, 1506–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, R.A.; Siuzdak, G.; Prospero-Garcia, O.; Henriksen, S.J.; Boger, D.L.; Cravatt, B.F. Cerebrodiene: A brain lipid isolated from sleep-deprived cats. Proc. Natl. Acad. Sci. USA 1994, 91, 9505–9508. [Google Scholar] [CrossRef] [Green Version]
- Cravatt, B.F.; Lerner, R.A.; Boger, D.L. Structure Determination of an Endogenous Sleep-Inducing Lipid, cis-9-Octadecanamide (Oleamide): A Synthetic Approach to the Chemical Analysis of Trace Quantities of a Natural Product. J. Am. Chem. Soc. 1996, 118, 580–590. [Google Scholar] [CrossRef]
- Hoi, P.M.; Hiley, C.R. Vasorelaxant effects of oleamide in rat small mesenteric artery indicate action at a novel cannabinoid receptor. Br. J. Pharmacol. 2006, 147, 560–568. [Google Scholar] [CrossRef]
- Sudhahar, V.; Shaw, S.; Imig, J.D. Mechanisms involved in oleamide-induced vasorelaxation in rat mesenteric resistance arteries. Eur. J. Pharmacol. 2009, 607, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Huitron-Resendiz, S.; Gombart, L.; Cravatt, B.F.; Henriksen, S.J. Effect of oleamide on sleep and its relationship to blood pressure, body temperature, and locomotor activity in rats. Exp. Neurol. 2001, 172, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Gonzalez, D.; Bonilla-Jaime, H.; Morales-Otal, A.; Henriksen, S.J.; Velazquez-Moctezuma, J.; Prospero-Garcia, O. Oleamide and anandamide effects on food intake and sexual behavior of rats. Neurosci. Lett. 2004, 364, 1–6. [Google Scholar] [CrossRef]
- Fedorova, I.; Hashimoto, A.; Fecik, R.A.; Hedrick, M.P.; Hanus, L.O.; Boger, D.L.; Rice, K.C.; Basile, A.S. Behavioral evidence for the interaction of oleamide with multiple neurotransmitter systems. J. Pharmacol. Exp. Ther. 2001, 299, 332–342. [Google Scholar]
- Huidobro-Toro, J.P.; Harris, R.A. Brain lipids that induce sleep are novel modulators of 5-hydroxytrypamine receptors. Proc. Natl. Acad. Sci. USA 1996, 93, 8078–8082. [Google Scholar] [CrossRef] [Green Version]
- Huidobro-Toro, J.P.; Valenzuela, C.F.; Harris, R.A. Modulation of GABAA receptor function by G protein-coupled 5-HT2C receptors. Neuropharmacology 1996, 35, 1355–1363. [Google Scholar] [CrossRef]
- Boger, D.L.; Patterson, J.E.; Jin, Q. Structural requirements for 5-HT2A and 5-HT1A serotonin receptor potentiation by the biologically active lipid oleamide. Proc. Natl. Acad. Sci. USA 1998, 95, 4102–4107. [Google Scholar] [CrossRef] [Green Version]
- Thomas, E.A.; Carson, M.J.; Neal, M.J.; Sutcliffe, J.G. Unique allosteric regulation of 5-hydroxytryptamine receptor-mediated signal transduction by oleamide. Proc. Natl. Acad. Sci. USA 1997, 94, 14115–14119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boger, D.L.; Patterson, J.E.; Guan, X.; Cravatt, B.F.; Lerner, R.A.; Gilula, N.B. Chemical requirements for inhibition of gap junction communication by the biologically active lipid oleamide. Proc. Natl. Acad. Sci. USA 1998, 95, 4810–4815. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Cravatt, B.F.; Ehring, G.R.; Hall, J.E.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. The sleep-inducing lipid oleamide deconvolutes gap junction communication and calcium wave transmission in glial cells. J. Cell Biol. 1997, 139, 1785–1792. [Google Scholar] [CrossRef] [Green Version]
- Hamberger, A.; Stenhagen, G. Erucamide as a modulator of water balance: New function of a fatty acid amide. Neurochem. Res. 2003, 28, 177–185. [Google Scholar] [CrossRef]
- Mitchell, C.A.; Davies, M.J.; Grounds, M.D.; McGeachie, J.K.; Crawford, G.J.; Hong, Y.; Chirila, T.V. Enhancement of neovascularization in regenerating skeletal muscle by the sustained release of erucamide from a polymer matrix. J. Biomater. Appl. 1996, 10, 230–249. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Masaki, T.; Itoh, F.; Kondo, K.; Sudo, K. Isolation of fatty acid amide as an angiogenic principle from bovine mesentery. Biochem. Biophys. Res. Commun. 1990, 168, 423–429. [Google Scholar] [CrossRef]
- Huang, J.K.; Jan, C.R. Linoleamide, a brain lipid that induces sleep, increases cytosolic Ca2+ levels in MDCK renal tubular cells. Life Sci. 2001, 68, 997–1004. [Google Scholar] [CrossRef]
- Wang, G.; Silva, J.; Dasgupta, S.; Bieberich, E. Long-chain ceramide is elevated in presenilin 1 (PS1M146V) mouse brain and induces apoptosis in PS1 astrocytes. Glia 2008, 56, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 1995, 80, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Nichols, K.K.; Ham, B.M.; Nichols, J.J.; Ziegler, C.; Green-Church, K.B. Identification of fatty acids and fatty acid amides in human meibomian gland secretions. Investig. Ophthalmol. Vis. Sci. 2007, 48, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Fontana, A. Anandamide, an endogenous cannabinomimetic eicosanoid: ’Killing two birds with one stone’. Prostaglandins Leukot Essent Fatty Acids 1995, 53, 1–11. [Google Scholar] [CrossRef]
- Bisogno, T.; Sepe, N.; De Petrocellis, L.; Mechoulam, R.; Di Marzo, V. The sleep inducing factor oleamide is produced by mouse neuroblastoma cells. Biochem. Biophys. Res. Commun. 1997, 239, 473–479. [Google Scholar] [CrossRef]
- Sultana, T.; Johnson, M.E. Sample preparation and gas chromatography of primary fatty acid amides. J. Chromatogr. A 2006, 1101, 278–285. [Google Scholar] [CrossRef]
- Hanus, L.O.; Fales, H.M.; Spande, T.F.; Basile, A.S. A gas chromatographic-mass spectral assay for the quantitative determination of oleamide in biological fluids. Anal. Biochem. 1999, 270, 159–166. [Google Scholar] [CrossRef]
- Brautigam, A.; Wesenberg, D.; Preud’homme, H.; Schaumloffel, D. Rapid and simple UPLC-MS/MS method for precise phytochelatin quantification in alga extracts. Anal. Bioanal. Chem. 2010, 398, 877–883. [Google Scholar] [CrossRef]
- Neususs, C.; Pelzing, M.; Macht, M. A robust approach for the analysis of peptides in the low femtomole range by capillary electrophoresis-tandem mass spectrometry. Electrophoresis 2002, 23, 3149–3159. [Google Scholar] [CrossRef]
- Divito, E.B.; Davic, A.P.; Johnson, M.E.; Cascio, M. Electrospray ionization and collision induced dissociation mass spectrometry of primary fatty acid amides. Anal. Chem. 2012, 84, 2388–2394. [Google Scholar] [CrossRef]
- Gee, A.J.; Groen, L.A.; Johnson, M.E. Ion trap mass spectrometry of trimethylsilylamides following gas chromatography. J. Mass Spectrom. 2000, 35, 305–310. [Google Scholar] [CrossRef]
- Carpenter, T.; Poore, D.D.; Gee, A.J.; Deshpande, P.; Merkler, D.J.; Johnson, M.E. Use of reversed phase HP liquid chromatography to assay conversion of N-acylglycines to primary fatty acid amides by peptidylglycine-alpha-amidating monooxygenase. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 809, 15–21. [Google Scholar] [CrossRef]
- Feng, L.; Johnson, M.E. Selective fluorescence derivatization and capillary electrophoretic separation of amidated amino acids. J. Chromatogr. A 1999, 832, 211–224. [Google Scholar] [CrossRef]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef]
- Duffy, D.C.; McDonald, J.C.; Schueller, O.J.; Whitesides, G.M. Rapid Prototyping of Microfluidic Systems in Poly(dimethylsiloxane). Anal. Chem. 1998, 70, 4974–4984. [Google Scholar] [CrossRef]
- Song, H.; Chen, D.L.; Ismagilov, R.F. Reactions in droplets in microfluidic channels. Angew. Chem. Int. Ed. Engl. 2006, 45, 7336–7356. [Google Scholar] [CrossRef] [Green Version]
- Stroock, A.D.; Dertinger, S.K.; Ajdari, A.; Mezic, I.; Stone, H.A.; Whitesides, G.M. Chaotic mixer for microchannels. Science 2002, 295, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Aref, H. Stirring by chaotic advection. J. Fluid Mech. 1984, 143, 1–21. [Google Scholar] [CrossRef]
- Song, H.; Bringer, M.R.; Tice, J.D.; Gerdts, C.J.; Ismagilov, R.F. Experimental test of scaling of mixing by chaotic advection in droplets moving through microfluidic channels. Appl. Phys. Lett. 2003, 83, 4664–4666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottino, J.M. Mixing and chemical reactions: A tutorial. Chem. Eng. Sci. 1994, 49, 4005–4027. [Google Scholar] [CrossRef]
- Beebe, D.J.; Mensing, G.A.; Walker, G.M. Physics and applications of microfluidics in biology. Annu. Rev. Biomed. Eng. 2002, 4, 261–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Tung, S.Y.; Lo, Y.S.; Huang, H.L.; Ko, C.H.; Wu, C.H. Sensitivity enhancement in the fluorometric determination of aliphatic amines using naphthalene-2,3-dicarboxaldehyde derivatization followed by vortex-assisted liquid-liquid microextraction. Talanta 2016, 152, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Manica, D.P.; Lapos, J.A.; Jones, A.D.; Ewing, A.G. Analysis of the stability of amino acids derivatized with naphthalene-2,3-dicarboxaldehyde using high-performance liquid chromatography and mass spectrometry. Anal. Biochem. 2003, 322, 68–78. [Google Scholar] [CrossRef]
- Becker, H.; Locascio, L.E. Polymer microfluidic devices. Talanta 2002, 56, 267–287. [Google Scholar] [CrossRef]
- Owen, M.J. Silicone Surface Fundamentals. Macromol. Rapid Commun. 2020, e2000360. [Google Scholar] [CrossRef]
- Madl, T.; Mittelbach, M. Quantification of primary fatty acid amides in commercial tallow and tallow fatty acid methyl esters by HPLC-APCI-MS. Analyst 2005, 130, 565–570. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davic, A.; Cascio, M. Development of a Microfluidic Platform for Trace Lipid Analysis. Metabolites 2021, 11, 130. https://doi.org/10.3390/metabo11030130
Davic A, Cascio M. Development of a Microfluidic Platform for Trace Lipid Analysis. Metabolites. 2021; 11(3):130. https://doi.org/10.3390/metabo11030130
Chicago/Turabian StyleDavic, Andrew, and Michael Cascio. 2021. "Development of a Microfluidic Platform for Trace Lipid Analysis" Metabolites 11, no. 3: 130. https://doi.org/10.3390/metabo11030130
APA StyleDavic, A., & Cascio, M. (2021). Development of a Microfluidic Platform for Trace Lipid Analysis. Metabolites, 11(3), 130. https://doi.org/10.3390/metabo11030130