
The mwtab Python Library for RESTful Access and Enhanced Quality Control, Deposition, and Curation of the Metabolomics Workbench Data Repository



Abstract
:1. Introduction
2. Results
2.1. Additional Functionality of the ‘mwtab’ Package Interface
2.2. Evaluation of the Metabolomics Workbench Repository
2.2.1. Analysis IDs with Files Missing from the Metabolomics Workbench
2.2.2. Analysis Files Which Could Not Be Parsed
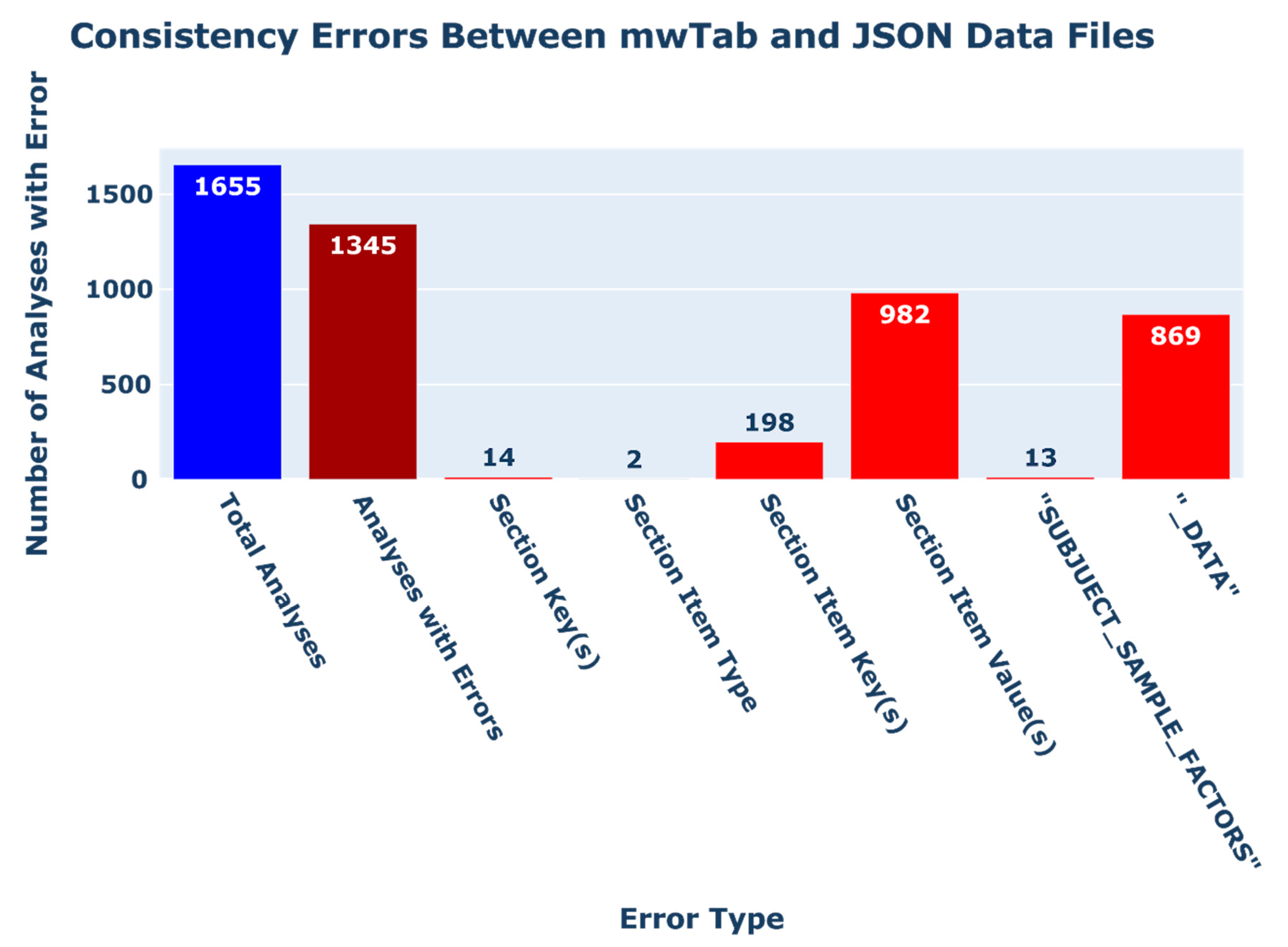
2.2.3. Consistency Errors between ‘mwTab’ and JSON Formatted Files
2.2.4. Validation Errors in ‘mwTab’ Formatted Files
2.2.5. Consistency Issues in ‘METABOLITES’ Section Metabolite Metadata Headings
2.3. Files Which Lack Data
3. Discussion
4. Methods
4.1. Updates to the ‘mwTab’ Format
4.2. Metabolomics Workbench JSON File Format
4.3. Overview of the Metabolomics Workbench REST Interface
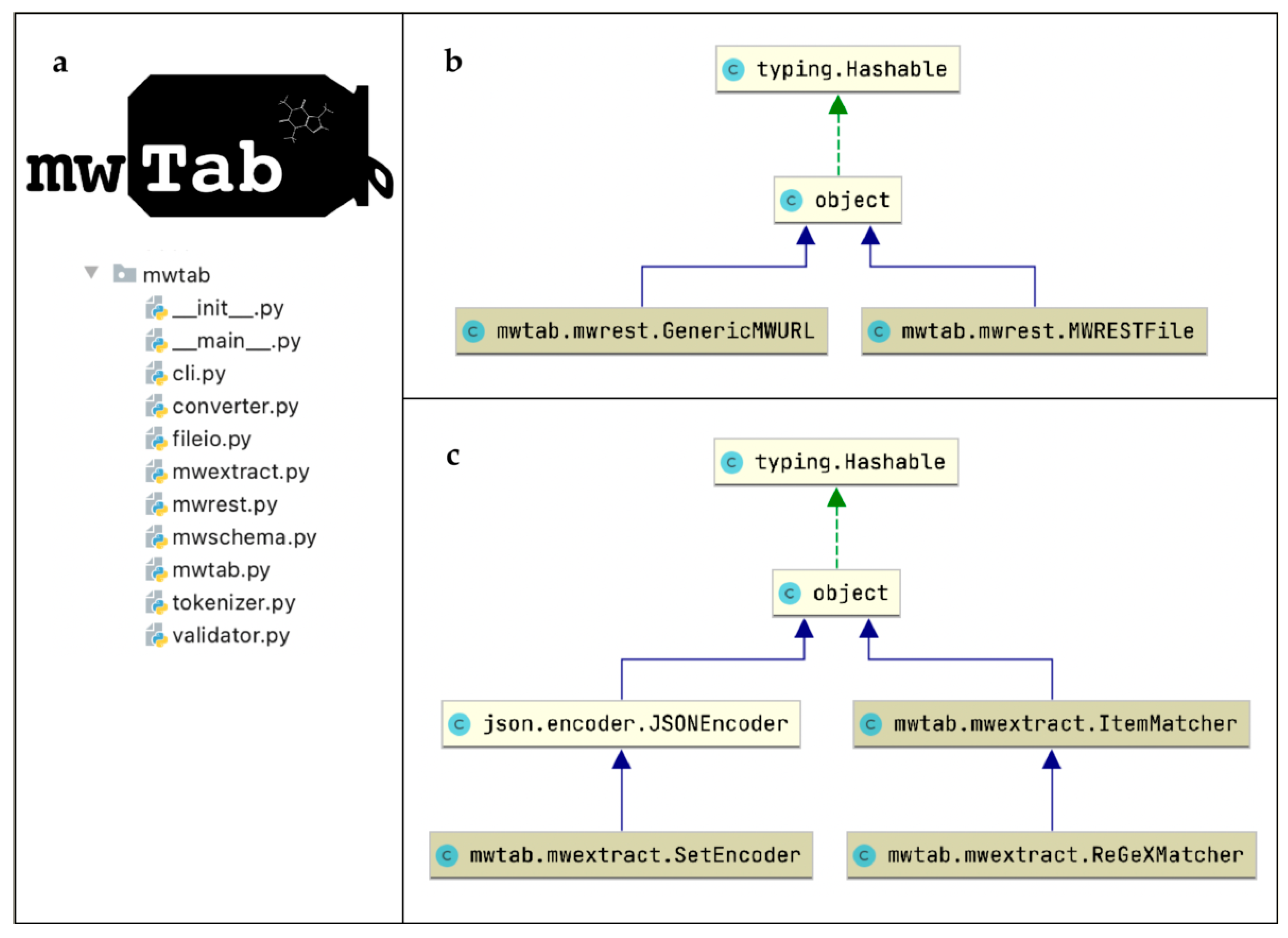
4.4. Package Implementation
4.5. Evaluation of the Metabolomics Workbench Repository
4.6. Updates to the ‘mwtab’ Package Documentation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, T. The Javascript Object Notation (Json) Data Interchange Format (No. RFC 8259). Technical Report. 2017. Available online: https://tools.ietf.org/html/rfc8259 (accessed on 11 March 2021).
- Crockford, D. Javascript Object Notation, RFC 4627; Internet Engineering Task Force: Fremont, CA, USA, 2006. [Google Scholar]
- Fielding, R. Representational state transfer. Architectural Styles and the Design of Network-Based Software Architectures. Ph.D. Thesis, University of California Irvine, Irvine, CA, USA, 2000. [Google Scholar]
- Smelter, A.; Moseley, H.N.B. A Python library for FAIRer access and deposition to the Metabolomics Workbench Data Repository. Metabolomics 2018, 14, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckhout, M.; Zielhuis, G.A.; Bredenoord, A.L. The FAIR guiding principles for data stewardship: Fair enough? Eur. J. Hum. Genet. 2018, 26, 931–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rossum, G. Python Programming Language. In Proceedings of the USENIX Annual Technical Conference 2007, Santa Clara, CA, USA, 17–22 June 2007; Volume 41, p. 36. [Google Scholar]
- Python Package Index. Available online: https://pypi.org/ (accessed on 1 January 2021).
- GitHub. Available online: https://github.com/ (accessed on 1 January 2021).
- Fiehn, O.; Robertson, D.; Griffin, J.; Van Der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C.; et al. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 175–178. [Google Scholar] [CrossRef]
- Salek, R.M.; Neumann, S.; Schober, D.; Hummel, J.; Billiau, K.; Kopka, J.; Correa, E.; Reijmers, T.; Rosato, A.; Tenori, L.; et al. Coordination of Standards in MetabOlomicS (COSMOS): Facilitating integrated metabolomics data access. Metabolomics 2015, 11, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Spicer, R.A.; Salek, R.; Steinbeck, C. A decade after the metabolomics standards initiative it’s time for a revision. Sci. Data 2017, 4, 1–3. [Google Scholar] [CrossRef]
- Rocca-Serra, P.; Salek, R.M.; Arita, M.; Correa, E.; Dayalan, S.; Gonzalez-Beltran, A.; Ebbels, T.M.D.; Goodacre, R.; Hastings, J.; Haug, K.; et al. Data standards can boost metabolomics research, and if there is a will, there is a way. Metabolomics 2016, 12, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.D.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; et al. BioMagResBank. Nucleic Acids Res. 2007, 36 (Suppl. S1), D402–D408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt Annotation Score. Available online: https://www.uniprot.org/help/annotation_score (accessed on 1 January 2021).
- Pundir, S.; Magrane, M.; Martin, M.J.; O’Donovan, C. The UniProt Consortium Searching and Navigating UniProt Databases. Curr. Protoc. Bioinform. 2015, 50, 1–27. [Google Scholar] [CrossRef] [Green Version]
- ReadTheDocs. Available online: https://readthedocs.org/ (accessed on 1 January 2021).
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Allison, P.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.V.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Chemin 2015, 7, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36 (Suppl. S1), D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sphinx: Python Documentation Generator. Available online: https://www.sphinx-doc.org/en/master/ (accessed on 1 January 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Usage | Example |
|---|---|
| Reading ‘mwTab’ Files From REST | # create first REST URL mwt_rest_url = mwtab.GenericMWURL({ “context”: “study”, “input_item”: “analysis_id”, “input_value”: “AN000002, “output_item”: “mwtab”, “output_format”: “txt”}).url # create second REST URL another_mwt_rest_url = next(mwtab.generate_mwtab_urls(“AN000003”)) # create a generator to call REST URLS and create MWTabFile objects mwt_generator = mwtab.read_files(mwt_rest_url, another_mwt_rest_url) |
| Extracting Metadata | # make a generator to create MWTabFile objects mwtabfile = next(fileio.read_files(“path-to-mwtabfile-dir”)) # extract metadata extracted_values = mwextract.extract_metadata(mwtabfile, “LAST_NAME”) |
| Extracting Metabolites | # make a generator to create MWTabFile objects mwtabfile_gen = fileio.read_files(“path-to-mwtabfile-dir”) # create matcher object matcher = mwextract.generate_matchers([(“PR:LAST_NAME”, “Smith”)]) # extract metabolites data metabolite_dict = mwextract.extract_metabolites(mwtabfile_gen, matcher) |
| Command | Description | Example |
|---|---|---|
| download | Download files through the Metabolomics Workbench REST API | $ mwtab download all $ mwtab download 1 $ mwtab download AN000001 $ mwtab download ST000001 --context=study \ --input-item=study_id --output-item=mwtab \ --output-format=json $ mwtab download C20H34O11 --context=compound \ --input-item=formula --output-item=all --output-format=txt |
| extract | Extract data or metadata from file(s) | $ mwtab extract metadata AN000001.txt./LAST_NAME \ CHROMATOGRAPHY_TYPE --to-format=csv $ mwtab extract metabolites file_dir/./PR:LAST_NAME \ Smith CH:CHROMATOGRAPHY_TYPE “Reversed phase” \ --to-format=json |
| File Format | Analysis ID |
|---|---|
| ‘mwTab’ | AN002380, AN002381, and AN002384 |
| JSON | AN000255, AN000404, AN000405, AN000410, AN000415, AN000436, AN000439, AN000444, AN000446, AN000450, AN000663, AN000665, AN000667, AN000871, AN001856, AN002131, AN002132, AN002133, AN002134, AN002135, AN002136, AN002137, AN002138, AN002141, AN002142, AN002145, AN002147, AN002148, AN002149, AN002150, AN002151, AN002152, AN002153, AN002154, AN002157, AN002158, AN002159, AN002160, AN002161, AN002162, AN002163, AN002164, AN002165, AN002166, AN002167, AN002168, AN002169, AN002170, AN002171, and AN002314 |
| Common Field Name | RegEx Pattern(s) | Example Matched Field Names |
|---|---|---|
| hmdb_id | r”(?i)[\s|\S]{,}(HMDB)” r”(?i)(Human Metabolome D)[\S]{,}” | HMDB ID (*representative) HMDB (*Representative ID) HMDB_ID … Total 14 Fields |
| inchi_key | r”(?i)(inchi)[\S]{,}” | Inchi_Key InChIKey InchiKey … Total 10 Fields |
| kegg_id | r”(?i)(kegg)$” r”(?i)(kegg)(\s|_)(i)” | KEGG KEGG I Kegg ID … Total 6 Fields |
| moverz | r”(?i)(m/z)” | m/z M/Z m/z rounded |
| moverz_quant | r”(?i)(moverz)(\s|_)(quant)” r”(?i)(quan)[\S]{,}(\s|_)(m)[\S]{,}(z)” | Quantified m/z quantitated mz Moverz Quant … Total 10 Fields |
| other_id | r”(?i)(other)(\s|_)(id)$” | Other ID Other_ID |
| pubchem_id | r”(?i)(pubchem)[\S]{,}” | PubChem CID Pubchem ID PubChem … Total 9 Fields |
| retention_index | r”(?i)(ri)$” r”(?i)(ret)[\s|\S]{,}(index)” | retention time index ri Retention index … Total 9 Fields |
| retention_time | r”(?i)(r)[\s|\S]{,}(time)[\S]{,}” | retention_times retention time index Retention Time … Total 20 Fields |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Powell, C.D.; Moseley, H.N.B. The mwtab Python Library for RESTful Access and Enhanced Quality Control, Deposition, and Curation of the Metabolomics Workbench Data Repository. Metabolites 2021, 11, 163. https://doi.org/10.3390/metabo11030163
Powell CD, Moseley HNB. The mwtab Python Library for RESTful Access and Enhanced Quality Control, Deposition, and Curation of the Metabolomics Workbench Data Repository. Metabolites. 2021; 11(3):163. https://doi.org/10.3390/metabo11030163
Chicago/Turabian StylePowell, Christian D., and Hunter N.B. Moseley. 2021. "The mwtab Python Library for RESTful Access and Enhanced Quality Control, Deposition, and Curation of the Metabolomics Workbench Data Repository" Metabolites 11, no. 3: 163. https://doi.org/10.3390/metabo11030163
APA StylePowell, C. D., & Moseley, H. N. B. (2021). The mwtab Python Library for RESTful Access and Enhanced Quality Control, Deposition, and Curation of the Metabolomics Workbench Data Repository. Metabolites, 11(3), 163. https://doi.org/10.3390/metabo11030163